

Abstract
To investigate the specific target of PKA in the regulation of cell cycle progression and cell size we developed a new approach using the yeast strain GG104 bearing a deletion in adenylate cyclase gene and permeable to cAMP ( cyr1Δ, pde2Δ, msn2Δ, msn4Δ). In this strain the PKA activity is absent and can be activated by addition of cAMP in the medium, without any other change of the growth conditions. In the present work we show that the activation of PKA by exogenous cAMP in the GG104 strain exponentially growing in glucose medium caused a marked increase of cell size and perturbation of cell cycle with a transient arrest of cells in G1, followed by an accumulation of cells in G2/M phase with a minimal change in the growth rate. Deletion of CLN1 gene, but not of CLN2, abolished the transient G1 phase arrest. Consistently we found that PKA activation caused a transcriptional repression of CLN1 gene. Transcription of CLN1 is controlled by SBF and MBF dual-regulated promoter. We found that also the deletion of SWI4 gene abolished the transient G1 arrest suggesting that Swi4 is a target responsible for PKA modulation of G1/S phase transition. We generated a SWI4 allele mutated in the consensus site for PKA (Swi4S159A) and we found that expression of Swi4S159A protein in the GG104-Swi4Δ strain did not restore the transient G1 arrest induced by PKA activation, suggesting that Swi4 phosphorylation by PKA regulates CLN1 gene expression and G1/S phase transition.
Keywords: budding yeast, Cln1, Cyclic AMP, G1 cyclins, Mitosis, SBF
Introduction
In the budding yeast Saccharomyces cerevisiae the coordination between cell growth and cell cycle mostly takes place at a control step in the late G1 phase known as Start. 1 Start execution and entrance into S phase require synchronous transcriptional activation of several hundred genes, the so-called G1/S regulon.2 Genes of the G1/S regulon have been grouped according to the presence of binding sites for the transcription factors SBF (Swi4/Swi6), MBF (Mbp1/Swi6) or both. Both transcription factors are activated through Cln-dependent phosphorylation. Cln-Cdk-dependent phosphorylation of either 4 specific sites on Whi5 - out of 12 total Cdk phosphorylation sites - or phosphorylation of 4 sites on the Swi6 subunit of SBF3,4 is required to trigger transcription of the SBF-controlled genes of the G1/S regulon. Activation of MBF also requires Cdk1 activity, likely through phosphorylation of Swi6.5 At later times, MBF activity is down-regulated by the product of the NRM1 gene, encoding a protein related to Whi5.6,7 In early G1 cells Whi5 binds to SBF, inhibiting transcription. This complex regulatory mechanism results in an accurate timing of transcriptional activation of each gene. In different experimental conditions, CLN1 is the earliest activated gene, CLN2, being activated a few minutes later.2
At Start a critical cell size ( protein content per cell, Ps) is required for budding and entering S phase. 8 In S. cerevisiae, Ps increases proportionally with ploidy and is modulated by nutrients 9-12 and by the activity of the Ras/cAMP/PKA signaling pathway. 13-15 Hyper-activation (the RAS2Val-19 mutation) or a partial inhibition (the cdc25–1 temperature-sensitive mutation) of the Ras/cAMP/PKA pathway causes a marked adjustment of the critical cell mass required to enter into a new cell division cycle.13 Consistently, addition of cAMP to the medium of unperturbed exponentially growing yeast populations largely increases Ps and the cell size. 16 Indeed cAMP is able to influence also the exit from mitosis through the inhibition of the activation of APC and of the onset of anaphase,17 suggesting a regulatory role also for the cell size at mitosis. 18-20
The only known target for cAMP in yeast is the cAMP-dependent protein kinase A (PKA). The PKA is a hetero-tetramer composed of 2 catalytic subunits, redundantly encoded by TPK1, TPK2 and TPK3 and 2 regulatory subunits, encoded by BCY1. 21,22 cAMP is produced by adenylate cyclase (product of the CYR1 gene) that in turn is regulated by the small G-proteins Ras1 and Ras2. cAMP activates PKA by binding to the regulatory subunit, which results in the release of active catalytic subunits that then phosphorylate target proteins including enzymes of glycolysis and gluconeogenesis, transcriptional factors activated in stress condition, proteins for ribosomal biogenesis and enzymes involved in the metabolism of trehalose and glycogen. 23,24 Besides, the cAMP level in the cell is regulated also at the level of degradation by the low- and high-affinity phosphodiesterases, encoded by PDE1 and PDE2 respectively, which constitute a major feedback mechanism in the pathway. 25-28
Interestingly, the Ras/cAMP/PKA signaling pathway modulates the dependency of G1 cyclins on growth, explaining the previously described cAMP effect on critical cell size. In particular, there are evidences that PKA represses the transcription of CLN1 and CLN2, but not of CLN3 in yeast. 14,29,30 As a consequence, the Ras/cAMP/PKA pathway is involved in the control of cell cycle progression at G1 to S transition modulating the critical cell size required for entry into the S phase, although the molecular basis of the mechanism by which the increase in cAMP represses G1 cyclins are not known.8
Since cAMP and PKA activity are also normally required for growth, to better understand how PKA modulates cell cycle, avoiding the influence related to the general control of growth, we used the strain GG104 (cyr1Δ msn2Δ msn4Δ pde2Δ ), 31 in which PKA can be activated by exogenous cAMP without any change in the growth conditions. In this strain PKA activity is absent and is not required for growth, since the lack of adenylate cyclase, caused by deletion of CYR1 gene, does not allow cAMP synthesis. The deletion of genes encoding the transcription factors Msn2 and Msn4 allows this strain to bypass the lethality caused by deletion of CYR1 and to grow in absence of cAMP. Finally the deletion of PDE2 gene permits the use of exogenous cAMP to activate PKA.
In this paper, we show that the activation of PKA by exogenous cAMP in the cyr1Δ msn2Δ msn4Δ pde2Δ strain exponentially growing in glucose medium causes a marked increase of cell size and perturbation of cell cycle with a transient arrest of cells in G1, followed by an accumulation of cells in G2/M phase with a minimal change in the growth rate. Through deletion of several of the G1 regulatory genes, we found that the transient G1 arrest caused by PKA activation requires the expression of the CLN1 gene and that addition of cAMP causes an inhibition of CLN1 gene expression with a limited effect on CLN2 expression. Also the deletion of SWI4 abolished the transient G1 arrest as well as the mutation of the Swi4 protein in the consensus site for PKA phosphorylation (Swi4S159A), indicating that Swi4 is a major target of PKA-mediated cell size modulation in the G1/S transition.
Results
Addition of cAMP caused a marked increase in cell size and a transient G1/S arrest in the GG104 strain
In order to develop an experimental model that allows to discriminate between the requirement of a basal level of cAMP for cellular growth and a specific effect of PKA on cell size and on cell cycle progression, we used GG104 strain in which the activity of PKA is specifically dependent on the addition of extracellular cAMP. 31 First of all, we investigated the effect of exogenous cAMP on the growth parameters. Kinetic analyses showed that immediately after addition of 2mM cAMP to cells growing exponentially in YP medium supplemented with 2% glucose (cell density about 1.7–2 × 106 cells/ml) the increase in cell number was faintly affected, while a slight reduction was observed after 2 hours (Fig. 1A). cAMP treatment induces a massive increase in cell size (Fig. 1D and Supplementary Fig. S1) and causes a transient decrease in the percentage of budding cells (37%, while the control showed a budding index of 60%), followed by an increase to a value higher than the untreated culture (Fig. 1C). A small increase in growth rate was indeed observed when the accumulation of biomass was followed by the measurement of the OD at 600 nm (Fig. 1B). In fact while the doubling time (DT) for control culture was of 119 min (k = 0.35 h−1), the DT calculated after cAMP addition in the same experiment of Fig. 1 was of 105 min ( k = 0.39 h−1).
Figure 1.
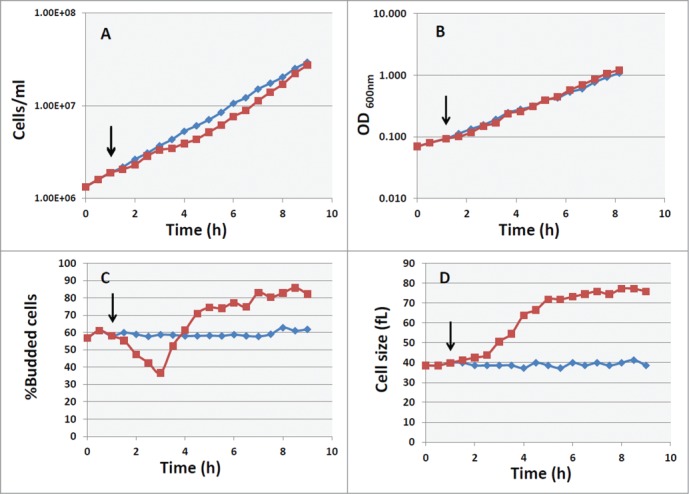
Effect of cAMP on growth and cell size in cyr1Δ msn2Δ msn4Δ pde2Δ mutant. Asynchronous culture of the GG104 strain growing exponentially (1.7–2 × 106 cells/ml) in YPD medium was splitted into 2 fractions and 2mM cAMP was added to one of them (▪), while the other was the untreated control (♦). The black arrows indicate the time of cAMP addition. Samples were harvested every 30 minutes to measure the cell number (panel A), the absorbance at 600 nm (panel B), the percentage of budded cells (panel C) and the mean cell size (panel D).
The increase in cell size was directly related to an increase of protein content per cell, as demonstrated by protein distribution analyzed by flow cytometry (Fig. 2, upper panel). The effect of cAMP on the cell cycle progression was also detected from DNA distribution analysis (Fig. 2,lower panel). Shortly after PKA activation a small increase of cells in G1 was evident, followed by an accumulation of cells in the G2+M phase. When a new steady state was reached (after 5 hrs), a large fraction of cells was in S+G2 phase, in agreement with the 75–80% of budded cells. From flow cytometry data we calculated the percentage of cells in G1 phase and the average protein content/cell expressed as channel number (Table 1). Besides, using a model for age distribution of budding yeast population, developed several years ago in our laboratory, 9,32 we also estimated the length of budded phase, (TB) of the population, as well as the protein content at budding (Ps) and the protein content at cell division (Pm) (Table 1).
Figure 2.
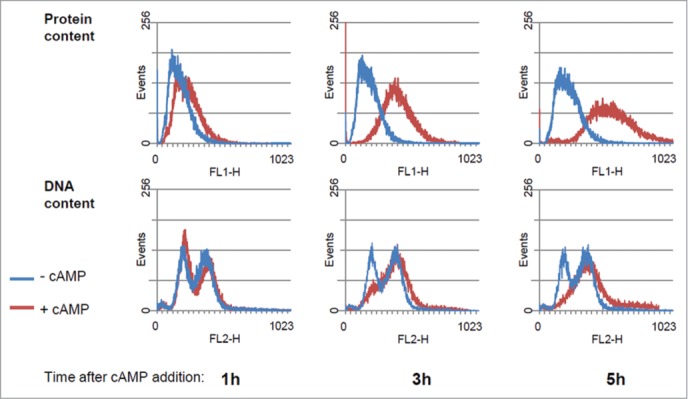
Protein and DNA distribution of GG104 cells. Histograms of protein and DNA distributions of control cultures and of cultures treated with 2 mM cAMP collected at the indicated time after cAMP addition. Yeast cells were collected fixed with 70% ethanol, stored at 4°C, and subsequently processed for flow cytometry, using a Becton Dickinson FACStarPlus. Fluorescein isothiocianate (FITC) for protein staining and Propidium Iodide (PI) for DNA staining were used. Approx 30.000 events were analyzed for each sample. Plot generation and analysis were performed with WinMDI2.9 software.
Table 1.
Growth and Cell cycle parameters for GG104 strain growing without cAMP and 5 hours after addition of cAMP.
| −cAMP | +cAMP | |
|---|---|---|
| K(h-1) | 0.35 | 0.39* |
| %G1 | 32 | 17 |
| %B | 60 | 76 |
| TB(min) | 80 | 95 |
| P(channel) | 251 | 472 |
| Ps | 216 | 363 |
| Pm | 346 | 639 |
The exponential growth rate constant ( k, h−1) was calculated from the increase of OD at 600 nm between 1 and 7 hrs after cAMP addition. The % of G1 cells was calculated from histograms of DNA distributions acquired by flow-cytometry after staining with Propidium iodide, using a software based on the method of Slater.49 TB, the length of budded phase was calculated using the formula TB = DT × log(FB+1) / log2.9 P (channel number) is the average of the protein distributions acquired by flow cytometry after staining with Fluoresceine isothiocyanate.9 Ps, the relative protein content (channel number) at budding, was estimated knowing the relevant cell cycle parameters of the population using a model for yeast population and a software previously developed in our laboratory, 9,32 while the value of Pm (protein content at cell division) was calculated with the formula: Pm = Ps ( 1+ FB).50
A direct determination of cell size at budding (Vs) and at cell division (Vm) was also performed after staining of cells with DAPI (Fig. 3A), as described in the Materials and Method section. In fact the cell size at budding can be estimated by the size of mothers with small buds, while the cell size at division can be measured on binucleate or mitotic cells. As shown in Fig. 3B, the size at budding increased immediately after addition of cAMP, while the cell size at cell division started to increase with a delay of at least 30 min. The cells size at budding reached a new steady-state value after 90 min, while the cell size at division continued to increase and reached a steady state value after 210 min (3 hrs and half). When a new steady-state was reached the cell size at budding increased 2 fold, while the size at cell division increased a little more than 2 fold, in good agreement with the increase of Ps and Pm values calculated from protein distributions.
Figure 3.
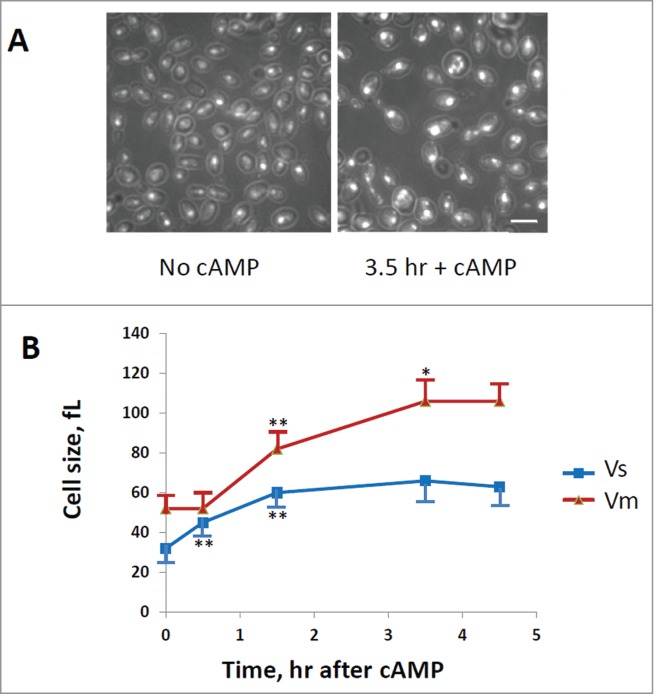
Evaluation of cells size at budding (Vs) and at cell division (Vm). GG104 cells exponentially growing were treated with 2 mM cAMP, stained with DAPI and photographed with a fluorescence microscope. Panel A: sample image of the cells growing without cAMP and after 3.5 hr of cAMP treatment. The image is taken in light transmission merged with DAPI fluorescence. Panel B: Cell size at budding (Vs) and at cell division (Vm) measured on the photographs taken at different times after cAMP addition. The average size of at least 30 cells with small buds (Vs) and of at least 30 binucleate cells (Vm) is reported. Error bars indicate Standard Deviation. *p < 0.05, **p < 0.01 is related to a comparison with the preceding mean value.
In conclusion, using our experimental model in which PKA is activated “on demand” and its activity is not required for growth, we confirmed that PKA modulates the dependence on growth of cell cycle events increasing the growth required to trigger both S and M-phase events, as expected for the CLN repression 14 and for the inhibition of G2+M cell cycle step by PKA activation. 15,17-19
Transcriptional response to PKA activation
To study the early response to activation of PKA, a transcriptome analysis of GG104 strain was performed one hour after addition of cAMP. The experiment was carried out as described in the Materials and Methods section and data handling with AMDA software showed that a few genes were significantly modulated by PKA. In particular, 55 genes were downregulated, while 30 genes were up-regulated in a statistically significant way (p < 0.05). Among these, none of the main G1/S transition regulators appeared to be significantly affected. Most of the genes differentially regulated by addition of cAMP can be classified for their involvement in glucose metabolism and signaling, lipid metabolism, mitochondrial function, heat-shock stress and transcriptional regulation (Tables S1 and S2).
Transcription factor analysis of the down-regulated genes brought us to identify 2 major clusters: the first, regulated by Hsf1 and counting HSP26, HSP12, XBP1, TMA10; the second, regulated by Adr1/Cat8 and including genes involved in glucose metabolism and respiration (HXT5, HXK1, GLK1, FBP1). Among upregulated genes, some were controlled by Fkh1, such as CHA1, HXT1 and JJJ3, while others, implicated in rRNA processing and ribosome biogenesis, were controlled by Fhl1 (GFD2, REX4, TOD6, FAF1). Interestingly, our data suggest that a new negative feedback mechanism operating at a transcriptional level might regulate the Ras/cAMP/PKA pathway. In particular, besides the down regulation of the cAMP-dependent protein kinase catalytic subunit TPK1, our transcriptional analysis showed a down regulation of GPG1, reported to code for a gamma subunit of the heterotrimeric G protein Gpa2 33 and an up regulation of RGS2, a negative regulator of glucose-induced cAMP signaling mediated by Gpa2, 34,35 suggesting a down regulation of the Gpr1/Gpa2 system.
CLN1 mediates the cAMP-induced G1/S transient arrest
With the aim to identify specific molecular targets operating downstream PKA, we analyzed the effect of cAMP addition in yeast mutants made in the GG104 background, carrying deletions in genes coding for proteins regulating the progression of Start. We deleted the G1 cyclin genes, CLN1, CLN2, CLN3 or the transcriptional repressor WHI5 gene. 36,37 All strains carrying a different gene deletion grew with a comparable rate in glucose medium, but as expected they showed differences in cell size and in the fraction of budded cells (Table S3). The growth kinetics, budding index and cell size were also evaluated after addition of cAMP. Our results showed that only when CLN1 gene was deleted, the G1/S transient arrest observed in the control strain after addition of cAMP was abolished, as clearly indicated by the lack of the decrease in the fraction of budded cells (Fig. 4A). This lack of response was confirmed by direct measurements of Vs and Vm in cln1Δ strain after cAMP addition (Fig. 5), where it is evident that the increase of cell size was caused mainly by the increase of Vm. In fact an increase of cell size was still present in cln1Δ strain but in a reduced amount in comparison with the control GG104 strain (an increase of 42% after 5 hrs of cAMP addition in comparison with 87% of control) and could be related to an effect of PKA activity on mitosis. 17 Most interestingly, although CLN1 and CLN2 encode proteins that are highly homologous to each other and are often considered to be co-regulated, the loss of Cln2 did not cause a different response to addition of cAMP in comparison with the control strain (Fig. 4B). Consequently, Cln1, but not Cln2, appears to be a specific target of the Ras/cAMP/PKA pathway. No relevant difference was found in the response to addition of cAMP for the other deletion mutant strains, as shown for cln3Δ and whi5Δ strains (Figs. S2 and S3).
Figure 4.
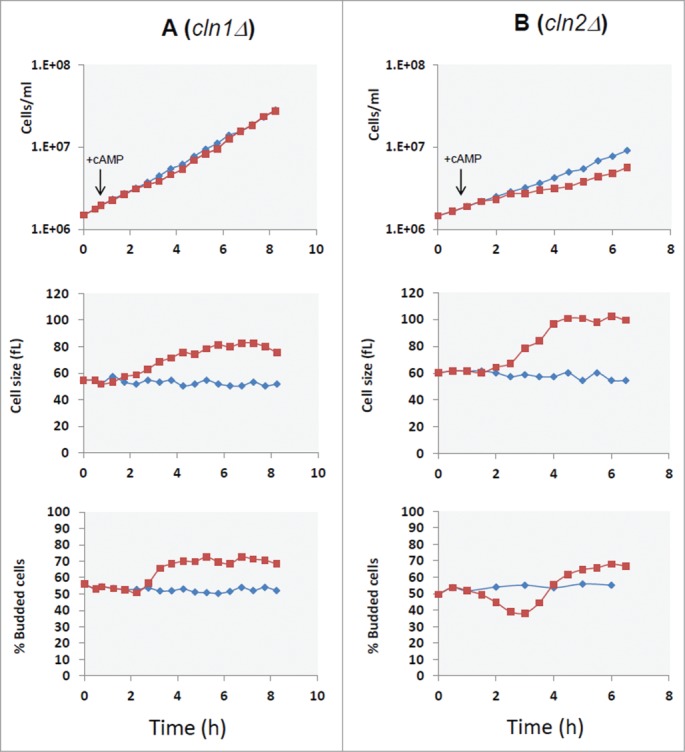
CLN1 but not CLN2 is required for G1 delay induced by cAMP addition. Asynchronous cultures of the GG104 cln1Δ (A) and GG104 cln2Δ (B) strains growing exponentially (1.7–2 × 106 cells/ml) in YPD medium were splitted into 2 fractions and 2mM cAMP was added to one of them (▪), while the other was the untreated control (♦). Samples were harvested every 30 minutes to assay cell number, cell size and budding index.
Figure 5.
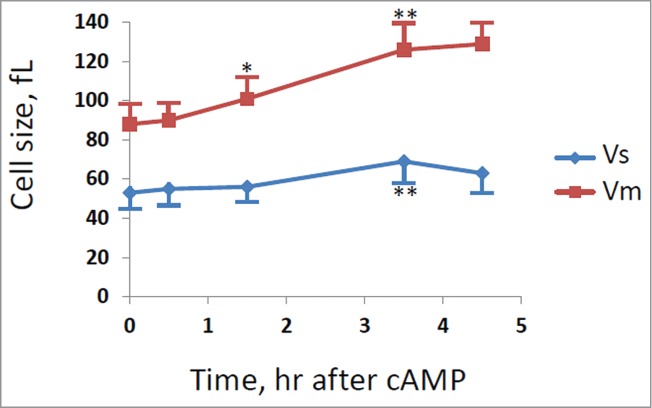
Cell size at budding and at cell division for cln1Δ strain. GG104 cln1Δ cells exponentially growing were treated with 2 mM cAMP, stained with DAPI and photographed with a fluorescence microscope. Cell size at budding (Vs) and at cell division (Vm) were measured on the images taken at different times after cAMP addition. The average size of at least 30 cells with small buds (Vs) and of at least 30 binucleate cells (Vm) is reported. Error bars indicate Standard Deviation. * p < 0.05, ** p < 0.01 is related to a comparison with the preceding mean value.
To further characterize PKA-mediated control on CLN1, we performed a real-time PCR. GG104 cell were grown to exponential phase and samples were harvested before and after addition of cAMP. Our data showed that cAMP (and consequently PKA activity) regulated CLN1 expression at the transcriptional level, since a 50% reduction of Cln1 mRNA was measured 30 minutes after addition of cAMP and lasted for at least 2 hours. Instead, CLN2 mRNA level appeared to be less influenced 30 minutes after addition of cAMP and recovered faster at longer times (Fig. 6), in agreement with results shown above indicating that transient G1 arrest was only dependent upon the presence of the CLN1 gene.
Figure 6.
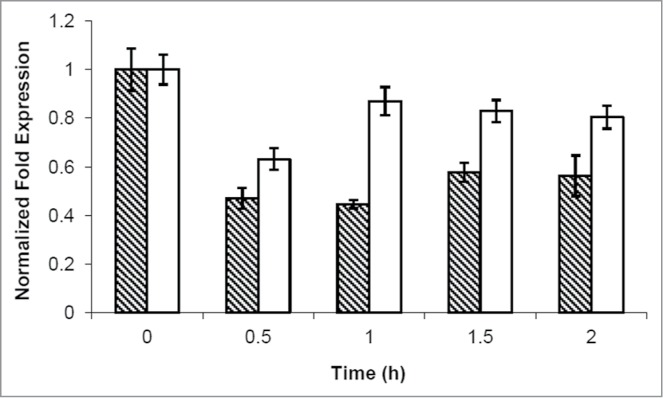
cAMP addition caused a CLN1 repression. Quantitative RealTime-PCR assays on CLN1 (hatched bars) and CLN2 (white bars) mRNA prepared from samples obtained during kinetic growth analysis, collected at the indicated time after cAMP addition. Graphic represents the mean values of 3 independent experiments, error bars indicate standard deviations.
Two promoter elements are reported to activate G1/S –specific transcription in S. cerevisiae. SCB elements (Swi4,6 cell cycle box; CACGAAA) are activated by the Swi4-Swi6 complex and MCB elements (MluI cell cyle box; ACGCGTNA) are activated by the Mbp1-Swi6 complex. 2 However, the major UAS activity associated to CLN1 promoter is limited to a 106-bp fragment containing 3 MCB-like core elements, depending primarily upon Swi4 for maximal expression and responsible for approximately 75% of G1-specific expression of the controlled gene.38 Conversely, SCB-like sequences within the CLN1 promoter play little or no role in transcription of the gene. 38 In addition also the regulation of cell size by glucose was found to be specifically exerted via repression of the CLN1 promoter through the fragment containing the 3 MCB-like elements 39 and it is known that cAMP treatment activates the elements of the Ras/cAMP/PKA pathway that plays the prominent role in responding to changes in glucose availability. The response to glucose was indeed partially suppressed by inactivation of SWI4 gene.39
Swi4-DNA binding protein is a specific PKA target at G1/S transition
To elucidate the involvement of the DNA binding protein Swi4 in the regulation of CLN1 gene expression exerted by PKA, we deleted the SWI4 gene in the GG104 strain and we investigated the effect of exogenous cAMP addition on the growth parameters. Kinetic analysis performed on the strain lacking the Swi4 protein showed similarities with GG104 cln1Δ strain (Table S3) and when treated with 2mM cAMP the profile of budding cells fraction revealed the loss of G1/S transient arrest normally associated with the drop of the budded cell fraction (Fig. 7A).
Figure 7.

GG104 swi4Δ mutant does not show G1/S transient arrest after PKA activation. Asynchronous cultures of the GG104 swi4Δ growing in YPD medium, GG104 swi4Δ [YCplac111-SWI4S159A], GG104 swi4Δ [YCplac111-SWI4] strains growing exponentially in selective medium were splitted into 2 fractions and 2mM cAMP was added to one of them (▪), while the other was the untreated control (Δ). Samples were harvested every 30 minutes to assay cell number, cell size and budding index.
Since G1/S-specific transcription of CLN1 requires Swi4 and its deletion causes the absence of the cAMP-induced transient arrest in late G1, we considered the hypothesis that Swi4 might be a target of PKA. Notably, PhosphoGRID (http://www.phosphogrid.org/), a database of experimentally verified in vivo phosphorylation sites, indicates a PKA phosphorylation site in the sequence of Swi4, on serine 159. To study whether CLN1 repression determined by PKA was mediated by the Swi4 phosphorylation, we generated a SWI4 allele mutated in the consensus site for PKA phosphorylation where serine residue at position 159 was replaced by alanine (Swi4S159A). Then a centromeric vector YCplac111 bearing either the non-phosphorylable SWI4S159A allele or the wild-type SWI4 gene was used to transform the GG104 swi4Δ strain. Cells expressing Swi4S159A that could not be phosphorylated by PKA exhibited only a very small transient G1/S arrest when cAMP was added. In fact, only a small decrease of budded cells was observed 2 hours after cAMP addition (from 66 to 63%) (Fig. 7B). On the other hand, the expression of the wild-type Swi4 re-establishes a marked cAMP-mediated effect on budding and on the increase of cell size (Fig. 7C). These results support our hypothesis that Swi4 protein phosphorylation on the serine 159 is one of the key mechanism by which PKA controls CLN1 expression.
Discussion
In yeast the cAMP signaling pathway is involved in the control of cell cycle progression at G0/G1 and G2 phase 17,40,41 and in nutritional modulation of the critical cell size (Ps) required for entry in the S phase. 2,13,42,43 Baroni et al. 14 and Tokiwa et al.29 showed that upon either glucose or cAMP addition, transcription of the G1 cyclin CLN1 (and to a lesser extend CLN2) is inhibited, whereas CLN3 transcription is unaffected. Later, Flick et al.39 mapped this inhibition to the CLN1 promoter. Inhibition of downstream G1 cyclin transcription was found not only during transient size resetting, but also played a role during steady-state growth on glucose. However the details of the connection between PKA activity and G1 cyclins expression are not clearly understood. 8 The matter is also complicated by the fact that PKA is normally required for growth and its activity is influenced by the growth conditions, and therefore it is difficult to put in evidence a specific regulation mediated by PKA avoiding the influence of growth condition and nutrient sensing.
To investigate the effects of PKA activation just on cell cycle and on the modulation of the critical cell size, here we used an experimental system in which cAMP addition does not affect the metabolism or the growth conditions. We used the GG104 strain (cyr1Δ pde2Δ msn2Δ msn4Δ) 31 in which the growth is no more dependent upon PKA activity. In this mutant strain the activity of PKA is close to zero (since there is no cAMP inside the cell) and it can be activated by the addition of extracellular cAMP. Indeed we have evidence that PKA is activated within few minutes after addition of cAMP (Martegani et al. unpublished results). The addition of 2 mM cAMP to glucose growing cells did not caused dramatic changes in gene expression, in metabolism and in the growth rate (measured as increase of OD of the culture) and caused a small perturbation of the increase in cell number. A transient drop of the fraction of budded cells is observed following cAMP addition, to reach a new steady state at a higher value after about 5 hrs. The average cell size of the population started to increase about 1 hr after the addition of cAMP, reaching a new steady-state value after about 5 hrs, and this is also related to a marked increase of the cell size both at budding (calculated Ps and measured Vs) and at cell division (Pm and Vm).
The increase of cell size during G1/S phase transition mediated by PKA activation is associated to a specific down-regulation of CLN1. Deletion of CLN1, but not of CLN2, fully reverts the effects of PKA on critical cell size at budding. In addition the real-time PCR performed on samples collected after PKA activation shows a strong and long lasting repression of CLN1 gene expression.
Consequently, despite the high sequence identity between CLN1 and CLN2, this study describes a circumstance in which these 2 genes are independently regulated and play a different role. These data partially disagree with previous works relating the inhibition of G1/S transition to G1 cyclins genes repression 14,29, however the different complexity of the model system and the different experimental conditions may explain the different results. In addition, here we show that in a cln2Δ strain expressing only Cln1, that we indicate to be the cyclin subjected to PKA regulation, the cell cycle control is more prominent: cln2Δ strain presents a longer transient inhibition and larger cell size after cAMP treatment than wild type.
Specific involvement of CLN1 in cell size control exerted by glucose has been previously reported. 39 The promoter region involved in glucose-dependent modulation of CLN1 expression includes multiple sequence related to MCB-like elements, normally target sites for MBF, but indeed acting as binding site for SBF and responsible for 75% of G1 specific expression of CLN1.38,39 In agreement with this findings, also the deletion of SWI4 gene abolished the transient G1/S arrest induced by cAMP, indicating that Swi4 may be a target of PKA. We mutated the single PKA phospho-acceptor site within the Swi4 DNA binding protein to alanine. As shown in the present work, GG104 cells expressing only Swi4S159A behave as a strain deleted in CLN1, indicating that cAMP-induced transient inhibition of G1/S transition and increase of Ps are mediated largely by PKA phosphorylation of Swi4. The residual effect on cell size of Swi4S159A is likely due to the well known effect of PKA on cell division 17-19 and is similar to that observed in cln1Δ and in swi4Δ strains. In fact, while in GG104 cells the increase in cells size after 5 hrs of cAMP is about 85% and a similar value was observed for mutants with deletion in CLN3, CLN2, WHI5 genes, this increase was reduce to 45% in mutants with a deletion in CLN1 or in SWI4 genes. As shown in Fig. 5 this increase is related only to a 45% increase of size at cell division (Vm) at least when measured in a cln1Δ strain.
The specific involvement of Cln1 and not of Cln2 in the modulation of cell size at budding in response to PKA activation, which has been shown to involve Swi4, is so far unexplained.
Although both CLN1 and CLN2 are SBF-MBF dual regulated genes,2 they have distinct time course of activation,2 thereby indicating the presence of reproducible differences in their transcriptional regulation. It is possible that chromatin structure and chromatin remodelling could play a significant role to explain this different regulation.44
A cell size increase induced by a delay in the exit from mitosis was observed also during a nutritional shift up from ethanol to glucose.15 The 2 conditions share a common molecular mechanism, given that addition of glucose to cells growing in a poor medium stimulates the increase of cAMP level that activates PKA. 36,45 PKA activation plays a role in blocking the degradation of Securin (required to metaphase to anaphase transition) and of Clb2 and Ase1 (required for mitotic exit). 17,20 In fact PKA phosphorylates Cdc20 46 which, following this modification, inactivates APC thereby delaying the exit from mitosis, which requires the degradation of Clb2 and Ase1. During this delay the cellular growth continues, yielding a substantial increase of cell size of newborn daughter cells.
According to this idea, our data reported in Fig. 3B suggest that there should be a time window in which the daughter cells reach a size even bigger than mothers (between 1 and 1.5 hs after cAMP addition). For example a mother cell that starts budding just before cAMP was added (time zero) with a size of about 32 fL, will divide with a Vm of about 75–80 fL.
Materials and Methods
Strains, plasmids and growth conditions
Yeast strains used in this work are listed in Table 2. Deletion and epitope tagging of yeast genes were made using polymerase chain reaction (PCR)-based strategy as previously described.47 PCR were performed with REDAccuTaq (Sigma-Aldrich) enzyme. All the primers used in this study are listed in Table S4. The Swi4–9myc tagged protein was obtained by in-locus 3′; in-frame insertion of SWI4 gene with a 9myc-hphR epitope sequence amplified from pYM20 plasmid (Euroscarf). To delete either CLN1, or CLN2 or CLN3 or WHI5 a PCR-cassette containing URA3 gene amplified from plasmid YCp50 was used. To delete SWI4, a PCR-cassette containing URA3 gene from Kluyveromyces lactis (KlURA3), which complements ura3 mutation in Saccharomyces cerevisiae, was amplified from plasmid pBKlURA3,48 (a generous gift from Simonetta Piatti, University of Milan-Bicocca). Yeast strains were grown at 30°C in rich medium containing 1% yeast extract and 2% peptone (Biolife), supplemented with 2% glucose (YPD). Selective medium was prepared with Synthetic complete media (SD) contained 2% glucose, 6.7 g/L YNB w/o aminoacids (Becton and Dickinson Italia, Buccinasco) and the proper selective drop-out CSM (MP Biomedicals). When required the culture was splitted into 2 parts and cAMP (Sigma-Aldrich) was added at a final concentration of 2 mM to one of them. Yeast growth was followed either by measuring the cell/ml with a Coulter Counter (Coulter mod. Z2) or by measuring the absorbance at 600 nm with a spectrophotometer. The population doubling time (DT) expressed in minutes was calculated as: DT = (ln2)/ k × 60, where k (h−1) is the constant of exponential growth.9 The percentage of budded cells was determined by counting the number of cells with buds under a microscope (at least 300 cells) after a mild sonication. The length of budded phase (TB) was calculated from the fraction of budded cell (FB) by the formula TB = DT × log(FB+1) / log2.9 Cell volume distributions were acquired with a Coulter Counter Channelyzer 256. Mean cell volume was calculated from the cell volume distributions, calibrated with latex beads, using the formula Vm = ( Σni × ci / Σni) F, where ni is the number of cells contained in the ci channel and F is a calibration factor.9 A representative experiment is presented for each strain.
Table 2.
Yeast strains used in this study
| Genotype | ||
|---|---|---|
| StrainsGG104 | MATa W303 with pde2::TRP1 cdc35::KanMX2 msn2::HIS3 msn4::TRP1 | SourceRoosen et al.31 |
| GG104 cln1Δ | GG104 cln1::URA3 | This study |
| GG104 cln2Δ | GG104 cln2::URA3 | This study |
| GG104 cln3Δ | GG104 cln3::URA3 | This study |
| GG104 whi5Δ | GG104 whi5::URA3 | This study |
| GG104 swi4Δ | GG104 swi4::KlURA3 | This study |
| GG104 Swi4–9myc | GG104 SWI4–9myc::Hph | This study |
| GG104 Swi4WT | GG104 swi4Δ [YCplac111-SWI4WT] | This study |
| GG104 Swi4S159A | GG104 swi4Δ [YCplac111-SWI4S159A] | This study |
Flow cytofluorimetric analysis
A total of 2×107 cells collected at the indicated time before and after addition of cAMP were fixed with 70% ethanol, stored at 4°C, and subsequently processed for flow cytometry, using a Becton Dickinson FACStarPlus equipped with a Argon ion laser (488 nm emission). DNA and protein analyses were performed essentially as described previously. 43 Samples were resuspended in 1 ml of PBS, containing 2 mg/ml RNAse (Roche) and incubated over night at 37°C. Fluorescein isothiocianate (FITC) for protein staining and Propidium Iodide (PI) for DNA staining were used. 30.000 events were analyzed for each sample. Plot generation and analysis were performed with WinMDI2.9 software (http://facs.scripps.edu/software.html). The fraction of cells in G1 was estimated from DNA histograms using the method of Slater et al.49 The value of PS (protein content at budding) was estimated knowing the relevant cell cycle parameters of the population using a model for yeast population and a software previously developed in our laboratory,9,33 while the value of Pm (protein content at cell division) was calculated with the formula: Pm = Ps ( 1+ FB).50
4′,6-Diamidino-2-phenylindole (DAPI) staining
Cells were collected before and after addition of cAMP, fixed with 70% ethanol, stained with 0.04 µg/ml of DAPI for 10 minutes, washed with distilled water and resuspended in 50% glycerol solution. Images were acquired with a Nikon Eclipse E600 fluorescence microscope using a DAPI filter, recorded digitally using a Leica DC 350F camera, and processed using Adobe Photoshop (Adobe Systems, Inc.). The size of cells was obtained by measurement of the lengths of major and minor axes of suitable cells on the digital images (using ImageJ software) and considering a yeast cell as a prolate spheroid. 51
RNA extraction and qReal-time PCR
5×108 cells/sample were collected by filtration and rapidly frozen at −80°C. They were resuspended in LETS buffer (200 mM LiCl, 20 mM EDTA, 20 mM Tris, 0.4% SDS) and lysed with glass beads. RNA was prepared through phenol:chloroform:isoamyl alcohol extraction followed by precipitation with NaCl (0.3 M) at −80°C. 40 μg of RNA were treated with 3 units of DNase I recombinant, RNase-free (Roche) for 1 h at 37°C, followed by a phenol:chloroform:isoamyl extraction. Reverse transcription of 0.5 μg of mRNAs was carried out with iScript cDNA Synthesis Kit (Bio-Rad). Quantitative Real-time PCR for CLN1 and CLN2 gene expression was performed with Sso Fast EvaGreen Supermix (Bio-Rad) using the following primers: CLN1-FW and CLN1-RE, CLN2-FW and CLN2-RE, ACT1-FW and ACT1-RE (Table S4). Three technical replicates for every sample of the 2 biological replicates were tested. The obtained data were normalized on ACT1 reference gene and organized with CFX Manager software (Bio-Rad).
Protein extraction and Western blot analysis
2×108 cells/sample were harvested, resuspended in ice-cold Lysis buffer (200 mM Hepes, 5 M NaCl, 75 mM MgCl2, 10% Nonidet) containing Complete EDTA-free Protease Inhibitor Cocktail (Roche) and lysed with glass beads. Protein concentration was determined using the Bio-Rad protein assay. SDS–PAGE was performed with 10% polyacrylamide gel. Western blot was performed according to standard procedures using nitrocellulose membrane (Protran). c-Myc (9E11) mouse monoclonal antibody (Santa Cruz Biotechnology) diluted 1:1000 and the secondary antibody conjugated to horseradish peroxidase (dilution 1:5000) were used. The signals were detected by chemioluminescence, using Protein Detection System (GeneSpin).
Site Directed Mutagenesis
Mutagenesis of Swi4 at codon 159 was performed using Phusion High-Fidelity DNA Polymerase (New England Biolabs) and plasmid p2713 52 as template with the following PCR program: 2 min 98°C, 12 times [10 sec 98°C, 20 sec 68°C, 5,5 min 68°C], 10 min 68°C. Primers Swi4S159A-FW and Swi4S159A-RE were used (Table S4). After PCR mutagenesis was verified by sequencing analysis, SalI/PvuII mutated fragment was ligated to PvuII/SacI fragment obtained from plasmid p2713 and subcloned into the yeast expression vector YCplac111 (YCplac111-SWI4S159A). Plasmid expressing the wild type counterpart (YCplac111-SWI4WT) was prepared subcloning SalI/SacI fragment from p2713 into YCplac111.
Gene expression analysis
Cells were grown to 1.7–2 × 106 cells/ml in YPD and subsequently the culture was divided into 2 fractions. 2mM cAMP was added to one fraction. Both fractions were grown for another hour and then 3 ×108 cells/sample were harvested. Cells were resuspended in LETS buffer (200 mM LiCl, 20 mM EDTA, 20 mM Tris, 0.4% SDS) and lysed with glass beads. Total RNA was prepared through phenol:chloroform:isoamyl alcohol extraction followed by precipitation with NaCl (0.3 M) at −80°C. Biotinylated cRNA was prepared and fragmented according to the Affymetrix protocol and analyzed by Agilent Bioanalyzer, before and following fragmentation. Fragmented cRNAs were hybridized to the Affymetrix Genechip, Yeast Genome 2.0 including probe sets for 5841 of the 5845 genes present in S. cerevisiae. Microarray data analysis was performed with the AMDA software.53 The identification of differentially expressed genes (DEGs) with a threshold p-value equal or under of 0.05 was addressed using Rank Product (RP).54
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Joris Winderickx for providing GG104 strain, Simonetta Piatti for PKBlURA3 plasmid and David Levin for p2713 plasmid bearing the wild type SWI4 gene.
Funding
This work was partially supported by CE project UniCellSys and by the Program SysBioNet, Italian Roadmap research Infrasctructure.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Pringle JR, Hartwell LH. The Saccharomyces cerevisiae cell cycle In: Strathern JN, Jones EW, Broach JR, editors The molecular biology of the yeast Saccharomyces cerevisiae: life cycle and inheritance. Cold Spring Harbor: New York, NY, USA: Cold Spring Harbor Laboratory; 1981; 97-142. [Google Scholar]
- 2.Eser U, Falleur-Fettig M, Johnson A, Skotheim JM. Commitment to a Cellular Transition Precedes Genome-wide Transcriptional Change. Mol Cell 2011; 43: 515-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wagner MV, Smolka MB, de Bruin RAM, Zhou H, Wittenberg C, Dowdy SF. Whi5 regulation by site specific CDK-phosphorylation in Saccharomyces cerevisiae. PLoS One 2009; 4:e4300; PMID:19172996; http://dx.doi.org/ 10.1371/journal.pone.0004300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hasan MM, Brocca S, Sacco E, Spinelli M, Papaleo E, Lambrughi M, Alberghina L, Vanoni M. A comparative study of Whi5 and retinoblastoma proteins: from sequence and structure analysis to intracellular networks. Front. Physiol. 2014; 4:315; PMID:24478706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang H, Carey LB, Cai Y, Wijnen H, Futcher B. Recruitment of Cln3 cyclin to promoters controls cell cycle entry via histone deacetylase and other targets. PloS Biol 2009; 7:e1000189; PMID:19823669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Bruin RAM, Kalashnikova TI, Chahwan C, McDonald HW, Wohlschlegel J, Yates J, Russel P, Wittenberg C. Constraining G1-specific transcription to late G1 phase: the MBF-associated corepressor Nrm1 acts via negative feedback. Mol Cell 2006; 23:483-96; PMID:16916637 [DOI] [PubMed] [Google Scholar]
- 7.Ferrezuelo F, Colomina N, Futcher B, Aldea M. The transcriptional network activated by Cln3 cyclin at the G1-to-S transition of the yeast cell cycle. Genome Biol 2010; 11:R67; PMID:20573214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turner JJ, Ewald JC, Skotheim JM. Cell size control in yeast. Curr Biol 2012; 22:R350-R359; PMID:22575477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vanoni M, Vai M, Popolo L, Alberghina L. Structural heterogeneity in populations of the budding yeast Saccharomyces cerevisiae. J Bacteriol 1983; 156:1282-91; PMID:6358196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neufeld TP, Edgar BA. Connections between growth and the cell cycle. Curr Opin Cell Biol 1998; 10:784-90; PMID:9914170 [DOI] [PubMed] [Google Scholar]
- 11.Rupes I. Chacking cell size in yeast. Trends Genet 2002; 18:479-85; PMID:12175809 [DOI] [PubMed] [Google Scholar]
- 12.Wells WA. Does size matter? J Cell Biol 2002; 158:1156-59; PMID:12356860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baroni MD, Martegani E, Monti P, Alberghina L. Cell size modulation by CDC25 and RAS2 genes in Saccharomyces cerevisiae. Mol Cell Biol 1989; 9:2715-23; PMID:2548086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baroni MD, Monti P, Alberghina L. Repression of growth-regulated G1 cyclin expression by cyclic AMP in budding yeast. Nature 1994; 371:339-45; PMID:8090203; http://dx.doi.org/ 10.1038/371339a0 [DOI] [PubMed] [Google Scholar]
- 15.Alberghina L, Smeraldi C, Ranzi BM, Porro D. Control by nutrients of growth and cell cycle progression in budding yeast, analysed by double-tag flow cytometry. J Bacteriol 1998; 180:3864-72; PMID:9683483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baroni MD, Monti P, Marconi G, Alberghina L. cAMP-mediated increase in the critical cell size required for the G1 to S transition in Saccharomyces cerevisiae. Exp Cell Res 1992; 201:299-306; PMID:1322313 [DOI] [PubMed] [Google Scholar]
- 17.Anghileri P, Branduardi P, Sternieri F, Monti P, Visintin R, Bevilacqua A, Alberghina L, Martegani E, Baroni MD. Chromosome separation and exit from mitosis in budding yeast: dependence on growth revealed by cAMP-mediated inhibition. Exp Cell Res 1999; 250(2):510-23. [DOI] [PubMed] [Google Scholar]
- 18.Irniger S, Baumer M, Braus GH. Glucose and Ras activity influence the ubiquitin ligases APC/C and SCF in Saccharomyces cerevisiae. Genetics 2000; 154:1509-21; PMID:10747049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bolte M, Dieckhoff P, Krause C, Braus GH, Irniger S. Synergistic inhibition of APC/C by glucose and activated Ras proteins can be mediated by each of the Tpk1- 3 proteins in Saccharomyces cerevisiae. Microbiology 2003; 149:1205-16; PMID:12724382; http://dx.doi.org/ 10.1099/mic.0.26062-0 [DOI] [PubMed] [Google Scholar]
- 20.Alberghina L, Coccetti P, Orlandi I. Systems biology of the cell cycle of Saccharomyces cerevisiae: From network mining to system-level properties. Biotecnol Adv 2009; 27:960-78. [DOI] [PubMed] [Google Scholar]
- 21.Toda T, Cameron S, Sass P, Zoller M, Scott JD, McMullen B, Hurwitz M, Krebs EG, Wigler M. Cloning and characterization of BCY1, a locus encoding a regulatory subunit of the cyclic AMP-dependent protein kinase in Saccharomyces cerevisiae. Mol Cell Biol 1987; 7:1371-77; PMID:3037314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Toda T, Cameron S, Sass P, Zoller M, Wigler M. Three different genes in S. cerevisiae encode the catalytic subunits of the cAMP-dependent protein kinase. Cell 1987; 50:277-87; PMID:3036373; http://dx.doi.org/ 10.1016/0092-8674(87)90223-6 [DOI] [PubMed] [Google Scholar]
- 23.Zaman S, Lippman SI, Zhao X, Broach JR. How Saccharomyces responds to nutrients. Annu Rev Genet 2008; 42:27-81; PMID:18303986 [DOI] [PubMed] [Google Scholar]
- 24.Smets B, Ghillebert R, De Snijder P, Binda M, Swinnen E, De Virgilio C, Winderickx J. Life in the midst of scarcity: adaptations to nutrient availability in Saccharomyces cerevisiae. Curr Genet 2010; 56:1-32; PMID:20054690 [DOI] [PubMed] [Google Scholar]
- 25.Sass P, Field J, Nikawa J, Toda T, Wigler M. Cloning and characterization of the high-affinity cAMP phosphodiesterase of Saccharomyces cerevisiae. Proc Natl Acad Sci USA 1986; 83:9303-07; PMID:3025832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nikawa J, Sass P, Wigler M. Cloning and charachterization of the low-affinity cyclic AMP phosphodiesterase gene of Saccharomyces cerevisiae. Mol Cell Biol 1987; 7:3629-36; PMID:2824992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilson RB, Renault G, Jacquet M, Tatchell K. The pde2 gene of Saccharomyces cerevisiae is allelic to rca1 and encodes a phosphodiesterase which protects the cell from extracellular cAMP. FEBS Lett 1993; 325:191-95; PMID:8391474 [DOI] [PubMed] [Google Scholar]
- 28.Ma P, Wera S, Van Dijck P, Thevelein JM. The PDE1-encoded low-affinity phosphodiesterase in the yeast Saccharomyces cerevisiae has a specific function in controlling agonist-induced cAMP signaling. Mol Biol Cell 1999; 10:91-104; PMID:9880329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tokiwa G, Tyers M, Volpe T, Futcher B. Inhibition of G1 cyclin activity by the Ras/cAMP pathway in yeast. Nature 1994; 371:342-45; PMID:8090204; http://dx.doi.org/ 10.1038/371342a0 [DOI] [PubMed] [Google Scholar]
- 30.Hall DD, Markwardt DD, Parviz F, Heideman W. Regulation of the Cln3-Cdc28 kinase by cAMP in Saccharomyces cerevisiae. EMBO J 1998; 17:4370-78; PMID:9687505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roosen J, Engelen K, Marchal K, Mathys J, Griffioen G, Cameroni E, Thevelein JM, De Virgilio C, De Moor B, Winderickx J. PKA and Sch9 control a molecular switch important for the proper adaptation to nutrient availability. Mol Microbiol 2005; 55:862-80; PMID:15661010 [DOI] [PubMed] [Google Scholar]
- 32.Martegani E, Vanoni M, Delia D. A computer algorithm for the analysis of protein distribution in budding yeast. Cytometry 1984; 5:81-5; PMID:6365484; http://dx.doi.org/ 10.1002/cyto.990050112 [DOI] [PubMed] [Google Scholar]
- 33.Harashima T, Heitman J. The Galpha protein Gpa2 controls yeast differentiation by interacting with kelch repeat proteins that mimic Gbeta subunits. Mol Cell 2002; 10(1): 163-73; PMID:12150916; http://dx.doi.org/ 10.1016/S1097-2765(02)00569-5 [DOI] [PubMed] [Google Scholar]
- 34.Versele M, de Winde JH, Thevelein JM. A novel regulator of G protein signalling in yeast, Rgs2, downregulates glucose-activation of the cAMP pathway through direct inhibition of Gpa2. EMBO J 1999; 18(20): 5577-91; PMID:10523302; http://dx.doi.org/ 10.1093/emboj/18.20.5577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rolland F, de Winde JH, Lemaire K, Boles E, Thevelein JM, Winderickx J. Glucose-induced cAMP signalling in yeast requires both a G-protein coupled receptor system for extracellular glucose detection and a separable hexose kinase-dependent sensing process. Mol. Microbiol. 2000; 38:348-58; PMID:11069660; http://dx.doi.org/ 10.1046/j.1365-2958.2000.02125.x [DOI] [PubMed] [Google Scholar]
- 36.Costanzo M, Nishikawa JL, Tang X, Millman JS, Schub O, Breitkreuz K, Dewar D, Rupes I, Andrews B, Tyers M. CDK activity antagonizes Whi5, an inhibitor of G1/S transcription in yeast. Cell 2004; 117:899-913; PMID:15210111; http://dx.doi.org/ 10.1016/j.cell.2004.05.024 [DOI] [PubMed] [Google Scholar]
- 37.de Bruin RA, McDonald WH, Kalashnikova TI, Yates J 3rd, Wittenberg C. Cln3 activates G1-specific transcription via phosphorylation of the SBF bound repressor Whi5. Cell 2004; 117:887-98; PMID:15210110; http://dx.doi.org/ 10.1016/j.cell.2004.05.025 [DOI] [PubMed] [Google Scholar]
- 38.Partridge JF, Mikesell GE, Breeden LL. Cell cycle-dependent transcription of CLN1 involves Swi4 binding to MCB-like elements. J Biol Chem 1997; 272:9071-7; PMID:9083033; http://dx.doi.org/ 10.1074/jbc.272.14.9071 [DOI] [PubMed] [Google Scholar]
- 39.Flick K, Chapman-Shimshoni D, Stuart D, Guaderrama M, Wittenberg C. Regulation of cell size by glucose is exerted via repression of the CLN1 promoter. Mol Cell Biol 1998; 18:2492-501; PMID:9566870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Drebot MA, Barnes CA, Singer RA, Johnston GC. Genetic assessment of stationary phase for cells of the yeast Saccharomyces cerevisiae. J Bacteriol 1990; 172, 3584-89; PMID:2163381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thevelein JM. The Ras-adenylate cyclase pathway and cell cycle control in Saccharomyces cerevisiae. Antonie van Leeuwenhoek 1992; 62, 109-130; PMID:1444331; http://dx.doi.org/ 10.1007/BF00584466 [DOI] [PubMed] [Google Scholar]
- 42.Mitsuzawa H. Responsiveness to exogenous cAMP of Saccharomyces cerevisiae strain conferred by naturally occurring alleles of PDE1 and PDE2. Genetics 1993; 135:321-26; PMID:8243997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Belotti F, Tisi R, Martegani E. The N-terminal region of the Saccharomyces cerevisiae RasGEF Cdc25 is required for nutrient-dependent cell-size regulation. Microbiology 2006; 152:1231-46; PMID:16549685; http://dx.doi.org/ 10.1099/mic.0.28683-0 [DOI] [PubMed] [Google Scholar]
- 44.Stillman D. Dancing the cell cycle two-step: regulation of yeast G1-cell-cycle genes by chromatin structure. Trends in Bioch. Sci. 2013; 38:467-475; http://dx.doi.org/ 10.1016/j.tibs.2013.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Colombo S, Ma P, Cauwenberg L, Winderickx J, Crauwels M, Teunissen A, Nauwelaers D, de Winde JH, Gorwa MF, Colavizza D, Thevelein JM. Involvement of distinct G-proteins, Gpa2 and Ras, in glucose- and intracellular acidification- induced cAMP signalling in the yeast Saccharomyces cerevisiae. EMBO J. 1998; 17:3326-41; PMID:9628870; http://dx.doi.org/ 10.1093/emboj/17.12.3326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Searle JS, Schollaert KL, Wilkins BJ, Sanchez Y. The DNA damage checkpoint and PKA pathways converge on APC substrates and Cdc20 to regulate mitotic progression. Nature Cell Biol 2004; 6:138-145; PMID:14743219; http://dx.doi.org/ 10.1038/ncb1092 [DOI] [PubMed] [Google Scholar]
- 47.Knop M, Siengers K, Pereira G, Zachariae W, Winson B, Nasmyth K, Schiebel E. Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast 1999; 15:963-72; PMID:10407276; http://dx.doi.org/ 10.1002/(SICI)1097-0061(199907)15:10B%3c963::AID-YEA399%3e3.0.CO;2-W [DOI] [PubMed] [Google Scholar]
- 48.Fraschini R, Formenti E, Lucchini G, Piatti S. Budding Yeast Bub2 Is Localized at Spindle Pole Bodies and Activates the Mitotic Checkpoint via a Different Pathway from Mad2. J Cell Biol 1999; 145:979-91; PMID:10352016; http://dx.doi.org/ 10.1083/jcb.145.5.979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Slater ML, Sharrow SO, Gart JJ. Cell cycle of Saccharomyces cerevisiae in population growing at different rates. Proc Natl Acad Sci USA 1977; 74:3850-56; PMID:333447; http://dx.doi.org/ 10.1073/pnas.74.9.3850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mariani L, Martegani E, Alberghina L. Yeast population models for monitoring and control of biotechnical processes. IEE Proc. 1986; 133:PT-D, 210-216; http://dx.doi.org/ 10.1049/ip-d.1986.0035 [DOI] [Google Scholar]
- 51.Martegani E, Porro D, Ranzi BM, Alberghina L. Involvement of a cell size control mechanism in the induction and maintenance of oscillations in continuous cultures of budding yeast. Biotechnology and Bioengineering 1990; 36:453-59; PMID:18595101; http://dx.doi.org/ 10.1002/bit.260360504 [DOI] [PubMed] [Google Scholar]
- 52.Truman AW, Kim KY, Levin DE. Mechanism of Mpk1 mitogen-activated protein kinase binding to the Swi4 transcriptional factor and its regulation by a novel caffeine-induced phosphorylation. Mol Cell Biol 2009; 29:6449-61; PMID:19805511; http://dx.doi.org/ 10.1128/MCB.00794-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pelizzola M, Pavelka N, Foti M, Ricciardi-Castagnoli P. AMDA: an R package for the automated microarray data analysis. BMC Bioinformatics 2006; 7:335; PMID:16824223; http://dx.doi.org/ 10.1186/1471-2105-7-335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Breitling R, Armengaud P, Amtmann A, Herzyk P. Rank Products:A simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments. FEBS Letter 2004; 57383-92 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.