

Abstract
Genome-wide network analysis is well implemented to study proteins of unknown function. Here, we effectively explored protein functions and the biological mechanism based on inferred high confident protein-protein interaction (PPI) network in cyanobacteria. We integrated data from seven different sources and predicted 1,997 PPIs, which were evaluated by experiments in molecular mechanism, text mining of literatures in proved direct/indirect evidences, and “interologs” in conservation. Combined the predicted PPIs with known PPIs, we obtained 4,715 no-redundant PPIs (involving 3,231 proteins covering over 90% of genome) to generate the PPI network. Based on the PPI network, terms in Gene ontology (GO) were assigned to function-unknown proteins. Functional modules were identified by dissecting the PPI network into sub-networks and analyzing pathway enrichment, with which we investigated novel function of underlying proteins in protein complexes and pathways. Examples of photosynthesis and DNA repair indicate that the network approach is a powerful tool in protein function analysis. Overall, this systems biology approach provides a new insight into posterior functional analysis of PPIs in cyanobacteria.
Cyanobacteria, the only known prokaryotes capable of oxygenic photosynthesis, are one of the most popular model organisms for photosynthesis, respiration, energy metabolism and regulatory function researches. Many studies have indicated that cyanobacteria could be applied in the wastewater treatment1, and significantly produce renewable energy source, like ethanol, biodiesel, hydrogen, etc.2,3,4. To date, our understanding to the molecular mechanisms underlying these biological functions is incomplete. For example, up to 60% of the proteins in Synechocystis sp. strain PCC 6803 are annotated as “unknown function” or “hypothetical protein”, although this organism is the first phototrophic organism fully sequenced and commonly selected in proteome analysis. To gain new insight into the important biological processes in cyanobacteria, protein-protein interaction (PPI) network construction and network-based protein function prediction are essential by providing a global understanding of protein relationships5,6. Experimental methods are focusing on genome-wide PPIs detection with yeast two-hybrid (Y2H) system and tandem affinity purification (TAP) coupled with mass spectroscopy5,7,8. Specifically, a Y2H screening system identified 3,236 interactions that provides new insight for gene function analyses in Synechocystis sp. strain PCC 68039. However, these experimental methods have their own limitations10. Firstly, they are labor- and time-intensive associated with high cost. Secondly, the experimental methods are prone to false positives. Thirdly, they are condition-specific and method-specific, which lead to a lower overlap even with the same species in the same platform.
Alternatively, computational methods have been widely used to effectively infer genome-wide PPIs and provide insight into protein properties in biological systems11,12,13. Such studies were also undertaken in Synechocystis sp. strain PCC 6803, such as SynechoNET database that integrated PPIs by domain information14 and InteroPORC database that inferred highly conserved PPIs15. However, the data from single source are bias in predicting PPIs, thus it is critical to integrate data computationally from multiple sources to construct high quality and coverage PPI network of an organism. For example, integration of multiple independent positive training datasets to predict PPIs can effectively reduce bias originally from single dataset by giving confidence scores for PPIs16,17. Also, in model plant Arabidopsis, integration of indirect evidences from multiple datasets by either Bayesian approach18 or support vector machine model19 has identified genome-wide PPIs with high reliability. Multiple datasets of indirect evidences to predict PPIs include genomic, evolutionary, domain, expression profiles and Gene Ontology (GO) information. Genomic context method contains gene neighborhood conservation, gene fusion and gene cluster. The assumption of gene fusion is that homologs of some interacting protein pairs in another species fuse into a single protein chain20,21. Gene neighborhood method presumes that the genes encoding interacting protein pairs are closely located and this closeness is conserved across different genomes22. Gene clusters assume that proteins, transcribed from a single functional unit (operon), are likely to have functional relation23. The evolutionary information, phylogenetic profile, assumes that functional related proteins are conserved in other organism24. Domain based information applies known interacting domains to predict potential protein interactions25. Besides, expression profiles and Gene Ontology (GO) annotation are also efficiently used to predict PPIs18.
Insights to the function of proteins and the mechanisms of biological processes can be gained by systematic analyses of large scale PPI network. A great number of studies predicted protein functions based on the assumption that functional similar proteins would cluster together in network and that interacting protein partners share similar function6. For example, the assignment of proteins to functional classes can be determined by simulated annealing method based on global optimization which minimizes the number of protein interactions among different functional classes26. This method solves the complicated computational problem resulting from global minimization from complex network and is the recommendatory method in global protein function prediction from PPI network.
In this work, we proposed a systematic approach to construct a high confident PPI network with predicted PPIs by integrating seven different datasets and known PPIs in Synechocystis sp. strain PCC 6803 (Fig. 1a). The quality of this network was evaluated by Y2H experiments, text mining and conserved interologs. We then conducted subsequent functional analysis based on the PPI network to deeply explore the annotation of function-unknown proteins, novel latent proteins in protein complexes and relative mechanisms of functional modules. Examples closely to biological processes of cyanobacteria were illustrated, suggesting that this systems biology approach is a powerful tool in PPI analysis of Synechocystis sp. strain PCC 6803.
Figure 1. Workflow of the study and the comparisons with other data.

(a) Workflow of protein-protein interaction prediction by Naïve Bayes integration and PPI network analyses. PPIs were predicted with seven different features: smallest shared biological process (SSBP), microarray gene expression profiles, domain interaction, phylogenetic profile, gene fusion, gene neighborhood and gene cluster. Using gold standard positive dataset (GSP) and gold standard negative dataset (GSN) sets as positive and negative data, we calculated the likelihood ratio (LR) for each PPI in each feature, and then integrated these LRs with Naïve Bayes approach. (b) The overlapped and unique PPIs predicted in this study and two other similar studies, InteroPORC and SynechoNET. (c) Evaluation and comparison of our prediction, InteroPORC and SynechoNET using an independent data, STRING scores.
Results
PPI prediction in Synechocystis sp. strain PCC 6803
To predict genome-wide PPIs in Synechocystis sp. strain PCC 6803, we first constructed gold standard datasets of PPIs. For gold standard positive dataset (GSP), we collected 2,718 known PPIs among 1,048 proteins confirmed by Y2H experiments9 (Supplementary Table S1). For the gold standard negative dataset (GSN) which means the protein pairs without interactions, we first generated protein pairs by collecting proteins from different cellular locations according to GO. After removing proteins with possible mobility or secretion, we obtained final GSN with 18,919 protein pairs (Supplementary Table S2).
We predicted PPIs with seven different data: gene clusters23, microarray gene expression profiles12, smallest shared biological process (SSBP)12, gene fusion20,21, gene neighborhood20,21, gene co-occurrences or phylogenetic profile21,27 and domain interaction12,20. Using GSP and GSN sets as positive and negative data, we calculated the likelihood ratio (LR) for each PPI in each data source, and then integrated these LRs with Naïve Bayes approach to generate high confident PPIs with integrated LR scores and posterior odds (see method). Finally, we obtained 1,997 high confident PPIs involving 2,765 proteins (Supplementary Table S3).
Compared with two former studies on PPI prediction in Synechocystis sp. strain PCC 6803, we found a quite small overlap between the three datasets (Fig. 1b), indicating that different predictive method has its own inherent bias on PPI prediction, which is consistent with previous study28. To evaluate these different prediction datasets, we applied an independent data as benchmarks to evaluate the PPIs from different sources (Fig. 1c): the scores of PPIs in STRING database that contains global PPIs across nearly 100 fully sequenced genomes29. Our high confident PPIs were compared with three PPI datasets: an independent positive data in InteroPORC (182 PPIs), predicted PPIs in both InteroPORC and SynechoNET. The average score of our data (732.17) is more closer to that of the independent positive data (827.54) than that of predicted PPIs in other two databases (InteroPORC: 479.94; SynechoNET: 424.67). More remarkable, the performance of our result is better than the prediction in InteroPORC, although these independent positive data were used to infer PPIs in InteroPORC. All these results support the view that our prediction method is effective in generating high score PPIs. Furthermore, nearly half of our predicted PPIs are not contained in STRING database, suggesting the value of our data in novel PPI discovery.
Evaluation of high confident PPIs using experiments, text mining and interologs
To evaluate these high confident PPIs, we applied three approaches from different aspects: experiments in physiological interaction evidences, text mining of literatures in proved direct/indirect evidences and “interologs” in conservation evidences.
We first carried out experiments to verify high confident PPIs between critical protein complex members (Supplementary Table S4). In Synechocystis sp. strain PCC 6803, functional distinct multiple NADPH dehydrogenase (NDH-1) complexes are essential for CO2 uptake, cyclic electron transport around photosystem I and respiration. Therefore, we tested the interactions between 6 proteins related to the large-size NDH-1 complex (NDH-1L) by yeast two-hybrid method (Sll0519, NdhA; Sll0223, NdhB; Slr0261, NdhH; Sll0520, NdhI; Slr1280, NdhK; and Ssl1690, NdhO). The experimental results proved the interactions of NdhO-NdhI, NdhH-NdhK and NdhA-NdhB (Fig. 2). All of these PPIs located either in hydrophilic or hydrophobic NDH-1L sub-complex, but not between them, indicating the reliability of these predicted interactions. To investigate the physiological significance of the interaction among the NDH-1 subunits, we tested the protein expression levels of two subunits, NdhH and NdhA using the mutant strains defective in the PPI pair NdhK and NdhB, respectively, by immunodetection. By comparison with wild type (WT), the accumulation of NdhH in the NdhK defective mutant ∆ndhK decreased to more than half (Fig. 3a) and the NdhA in the NdhB defective mutant M55 lowered to nearly 1/4 (Fig. 3b), suggesting the dependence of the PPI pair in protein accumulation. Further, we compared the activity of NDH-1-dependent cyclic electron flow around PSI (NDH-CET) via analyzing the post-illumination increase in chlorophyll fluorescence after termination of actinic light. NDH-CET was completely inactivated in M55, being consistent with previous studies30 and partly in ∆ndhK (Fig. 3c), indicating the PPI pairs of NDH-1 are involved in regulation of the NDH-CET activity. It is worth noting that predicted protein protein interaction through integrative bioinformatics approaches include both of physical protein or domain interactions and the functional associations between potentially interacting proteins31. For example, the product of gene slr0815 was predicted to interact with NdhL (Ssr1386; Supplemental Table S3), and deletion of slr0815 almost completely impaired NDH-CET activity (Fig. 4a–c), being consistent with the results of ndhL-deleted mutant (∆ndhL)30.
Figure 2. Yeast two-hybrid system for the interaction identification among Ndh subunits.
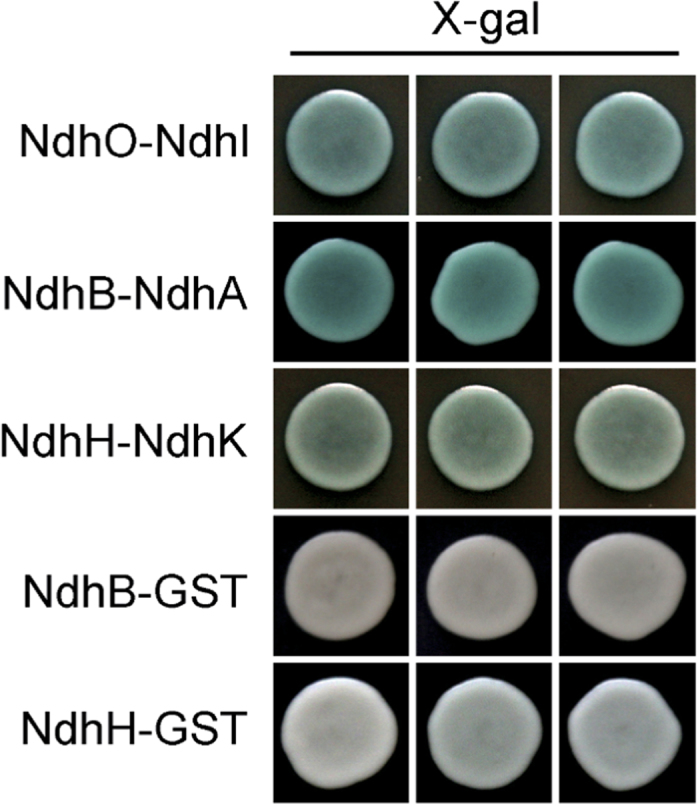
NdhB, NdhH and NdhO were constructed into bait vectors, whereas NdhA, NdhI, NdhK and GST were constructed into prey vectors, respectively. Subsequently, they were transformed into the yeast strain EGY48. Transformed yeast was dropped onto X-gal medium. Blue precipitate represents accumulated β-galactosidase activity resulting from the activation of the lacZ reporter gene by protein-protein interaction. The induction plate was incubated at 30 °C for 22 h and then photographed. The interactions of NdhO-NdhI, NdhB-GST and NdhH-GST were assayed as one positive and two negative controls, respectively. At least six independent experiments were performed, and the result of one representative is shown. See Supplementary Figure S1 for full views of yeast two-hybrid results.
Figure 3. Functional analysis of PPI protein pairs.
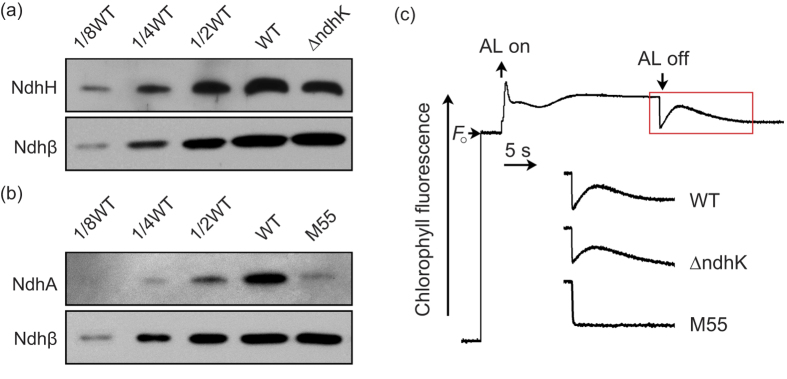
(a) Immunodetection of Ndh subunits in thylakoid membranes from the WT (including indicated serial dilutions) and ∆ndhK strains. Immunoblotting was performed with antibodies against NdhH subunit. Lanes were loaded with thylakoid membrane proteins corresponding to 1 μg chlorophyll a and ATPβ was used as a loading control. See Supplementary Figure S2 for full views of western blots. (b) Immunodetection of Ndh subunits in thylakoid membranes from the WT (including indicated serial dilutions) and M55 strains. Immunoblotting was performed with antibodies against NdhA subunit. Lanes were loaded with thylakoid membrane proteins corresponding to 1 μg chlorophyll a and ATPβ was used as a loading control. See Supplementary Figure S2 for full views of western blots. (c) Monitoring of NDH-CET activity using chlorophyll fluorescence analysis. The top curve shows a typical trace of chlorophyll fluorescence in the WT Synechocystis sp. strain PCC 6803. The chlorophyll a concentration was adjusted to 10 μg mL−1 before measurement. Cells were exposed to AL (620 nm; 45 μmol photons m−2s−1) for 30 s. AL was turned off, and the subsequent change in the chlorophyll fluorescence level was monitored as an indicator of NDH-CET activity.
Figure 4. slr0815 gene deletion mutation and its effect on NDH-CET.
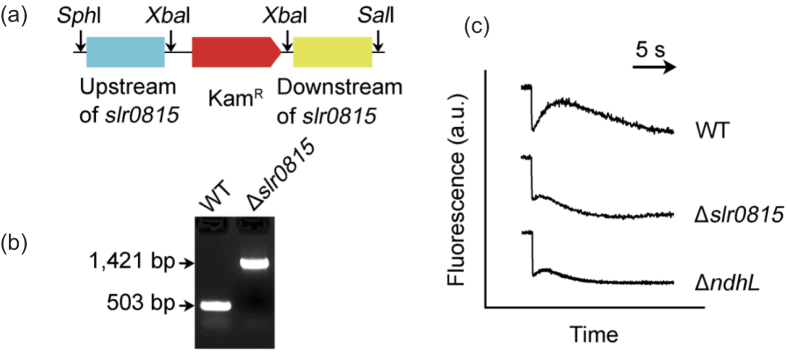
(a) Construction of plasmid used to generate the slr0815 deletion mutant (∆slr0815). (b) PCR segregation analysis of the ∆slr0815 mutant using the slr0815-G and slr0815-H primer sequences (Supplemental Table S4). See Supplementary Figure S3 for full views of DNA electrophoresis gels. (c) Monitoring of NDH-CET activity by chlorophyll fluorescence. Two-day-old cells were exposed to actinic light (45 μmol photons m−2s−1) for 30 sec. After illumination, the subsequent transient change in chlorophyll fluorescence was monitored as an indication of NDH-CET activity. a.u., arbitrary units.
We then evaluated the high confident PPIs by mining published literature evidences of double-mutant phenotypes and Y2H results (Supplementary Table S5). Totally, 6 and 19 PPIs were verified by double-mutants and Y2H results with significance (p < 1e–4) that were computed by probability according to a hypergeometric model15. On one hand, we examined whether double-mutants lose normal function compared with the WT, which suggests intermolecular interactions between proteins32. For instance, in NDH-1 family, the predicted interaction between Slr0331 (NdhD1) and Slr1291 (NdhD2) is consistent with the double mutant ∆ndhD1/ndhD2 (∆D1/D2), which loses capability to grow under photoheterotrophic conditions and exhibits low respiration rate33. Another predicted pair of NDH-1 family members, Sll1733 (NdhD3) and Sll0027 (NdhD4), is also verified by their double mutant, ∆ndhD3/ndhD4 (∆D3/D4), which grows slowly in air and cannot uptake CO234. As for the photosystem II (PSII) complex, under situation of absence of glucose, the double mutant ∆psbO/psbV strain lost the ability to grow35, supporting the high confident PPI, Sll0258 (PsbV) and Sll0427 (PsbO). Moreover, high confident PPI between Sll0258 (PsbV) and Sll1194 (PsbU) was verified by double deletion mutant ∆psbU/psbV, which cannot grow in the environment of absence Ca2+ or Cl−, compared with the ∆psbU and WT strains36. Similarly, experimental evidence for the ∆cpcG1/cpcG2 double mutant also confirmed the predicted functional linkage between Slr2051 (CpcG1) and Sll1471 (CpcG2)37. On the other hand, text mining of Y2H experiments provides direct evidence of the high confident PPIs. For example, Y2H experiments verified two PPIs between PSII complex members: Sll1867 (PsbA3, photosystem II D1 protein) and Slr1311 (PratA, photosynthesis and respiration PSII)38; Sll0698 (Hik33, two-component sensor histidine kinase) and Ssl3451 (SipA, hypothetical protein)39. In addition, the formation of a reaction center pre-complex with D1 and Psbl40 also supported our prediction: Sml0001(Psbl, PSII reaction center PsbI protein) interacts with Sll1867, Slr1311 and Slr1181 (all these three proteins are PSII proteins). As for PSI complex members, experimental results of physical interactions proved the reliability of two PPIs: Slr0737 (PsaD) and Slr1655 (PsaL), Ssr2831 (PsaE) and Sll0819 (PsaF)41.
Last, we utilized the “conserved interologs” of PPIs from 25 organisms to verify the interactions between conserved proteins, based on the assumption that interactions between ortholog pairs are conserved across different organisms42. On average, 17.7% of predictions were verified in interologs across the 25 species (Supplementary Figure S4) and the union of all conserved PPIs is 449. For example, one predicted interaction pair, Slr0543 (TrpB, tryptophan synthase subunit beta) and slr0966 (trpA, tryptophan synthase subunit alpha), has the related physical interacting interolog in E.coli, NP_415777 with NP_41577643. Similarly, another predicted interaction pair, Slr1199 (MutL, DNA mismatch repair protein) and sll1165 (MutS, DNA mismatch repair protein) has the corresponding experimental confirmed interolog in E. coli, NP_418591 and NP_41721344. Interologs could also suggest that some predicted PPIs form complexes, such as Sll1260 (Rps2, 30S ribosomal protein S2) with Ssl3432 (Rps19, 30S ribosomal protein S19), and Sll1260 with Sll1097 (Rps7, 30S ribosomal protein S7), both confirmed between their orthologs NP_011859 with NP_014435, and NP_011859 with NP_012647 in S. Cerevisae45. Actually, NP_011859 is mitochondrial 37S ribosomal protein MRP4, NP_014435 represents mitochondrial 37S ribosomal protein S19 and NP_014435 means mitochondrial 37S ribosomal protein S7. The three proteins are all ribosomal subunits involving in cellular process of translation.
Overall, the above evaluation suggests the reliability of our high confident PPIs that could be used in further analysis. Since GSP is considered as known confirmed PPIs, we combined them with our high confident PPIs, making final 4,715 protein pairs involving 3,231 proteins after eliminating redundancy to construct PPIs network (Fig. 5) for subsequently PPI network analysis and unknown protein function annotation.
Figure 5. 19 sub-networks in PPI network.

The PPI network (4,715 protein pairs involving 3,231 proteins) was constructed with high confident predicted PPIs and GSP (known confirmed PPIs). The whole PPI network was partitioned by the molecular complex detection algorithm (MCODE) with the default parameter in Cytoscape into 19 dense protein sets, which were extended by adding the first-layer neighbor proteins in PPI network to generate the final 19 sub-networks that were marked with numbers.
GO terms assignment of proteins with unknown function
Traditional homology method employed protein function alignment from one protein to its cousins, which were assumedly descended from the same ancestor to roughly assign functions of almost 40%–70% protein in a genome46. However, there are extremely less proteins with unknown function, which have homological proteins in proximal model species. In Synechocystis sp. strain PCC 6803, based on the existing protein annotation nearly 60% proteins are of unknown function, so we applied a non-homology method, simulated annealing algorithm, to annotate the proteins of unknown function based on PPI network (see method).
Totally, we annotated 1,391 proteins with 160 GO terms in biological process, 1,518 proteins with 10 GO terms in cellular component and 1,366 proteins with 55 GO terms in molecular function (Supplementary Table S6). Those function-unknown proteins were mainly assigned for metabolic process, carbohydrate biosynthetic process, catalytic activity and transporter activity. For instance, one hypothetical protein (Sll1252) was predicted to be involved in ion trans-membrane transporter activity (GO:0015075), monovalent inorganic cation trans-membrane transporter activity (GO:0015077). Recently, Sll1252 was reported closely related to redox sensing of the plastoquinone pool to balance the photosynthetic electron flow and cope with global environmental stresses47. Another hypothetical protein (Sll0822) was predicted involving in regulation of gene expression (GO:0010468), nitrogen compound metabolic process (GO:0006807), cellular response to stimulus (GO:0051716), stress (GO:0006950), and extracellular stimulus (GO:0009991). Experiment result suggests that this protein is involved in the regulation of nitrogen uptake systems and acts as a repressor, or as part of a repressor complex48.
Identification and function annotation of function modules in PPI network
Function modules are important in revealing biological mechanism of complex PPI network. Well defined and annotated function modules could be achieved by extracting sub-network (a form of module which is effective in sparse topological relationship among proteins rather than merely pair) and pathway (a group of proteins with similar functions which cooperate with each other in related biological processes) from PPI network. Therefore, we identified sub-networks annotated by pathway enrichment analysis to define function modules for further analysis.
We first partitioned the whole PPI network by the molecular complex detection algorithm (MCODE) with the default parameter in Cytoscape49, and got 19 dense protein sets, which were extended by adding the first-layer neighbor proteins in PPI network to generate the final 19 sub-networks (Supplementary Table S7). We then undertook pathway enrichment analysis for each sub-network. 98 pathways were obtained from KEGG, most of which are involved in metabolic pathway (syn01100), biosynthesis of secondary metabolites (syn01110), ABC transporters (syn02010) and porphyrin and chlorophyll metabolism (syn00860). Through pathway enrichment analysis, 19 sub-networks were significantly enriched in 5 pathways, including metabolic pathways (syn01100), photosynthesis (syn00195), two-component system (syn02020) and photosynthesis-antenna proteins (syn00196). For example, sub-network 4 was enriched in photosynthesis-antenna proteins. And this sub-network includes 15 proteins, Slr2051 (CpcG), Sll1051 (CpcF), Slr2067 (ApcA), Slr1459 (ApcF), Sll1577 (CpcB), Slr1986 (ApcB), Ssr3383 (ApcC), Slr1878 (CpcE), Sll1580 (CpcC), Sll1579 (CpcC), Ssl3093 (CpcD), Sll1471 (CpcG), Sll1578 (CpcA), Sll0928 (ApcD) and Slr0335 (ApcE). These proteins are phycobili-proteins serving for external antenna proteins for PSII. Another example is sub-network 10 that was enriched in the two-component system pathway (syn02020). Two of sub-network 10 members, Sll1229 (Hik41) and Sll1228 (Hik4), were speculated to participate in the current two-component system in KEGG. In fact, both of the two proteins have been annotated as two-component hybrid sensor and regulator in Cyanobase.
Mining PPI mechanism in function modules
Based on annotated function-unknown proteins and well-defined function modules, we could efficiently mining PPI mechanism. Focusing on purine metabolism and carbon dioxide, we deeply explored biological mechanism based on function modules.
The first example is sub-network 14. In this sub-network, six function unknown proteins (Slr1660, Slr1658, Slr1657, Sll0537, Slr1659 and Sll0536) were assigned to GO terms: catalytic activity (GO:0003824), cellular ketone metabolic process (GO:0042180), regulation of cellular process (GO:0050794), ligase activity (GO:0016874) and hydrolase activity (GO:0016787). These proteins with unknown function have no homologs in model organisms, so we explored their functions by transferring the function of proteins with the same GO terms from other species rather than sequence similarity. In E. coli, we found seven functional similar proteins (b0480, bifunctional UDP-sugar hydrolase/5'-nucleotidase; b0522, N5-carboxyaminoimidazole ribonucleotide synthase; b1849, phosphoribosylglycinamide formyltransferase 2; b3397, adenosine nucleotide hydrolase; b4005, phosphoribosylglycinamide synthetase phosphoribosylamine-glycine ligase; b4006, fused IMP cyclohydrolase/phosphoribosylaminoimidazolecarboxamide formyltransferase; and b4213, 2'-3'-cyclic-nucleotide 2'-phosphodiesterase), which were enriched in pathway purine metabolism (eco00230). By mapping same purine metabolism in E.coli and Synechocystis sp. PCC6803, we found two enzymes in purine metabolism pathway that could have the potential function of six proteins with unknown function in Synechocystis sp. PCC6803 (see Supplementary Figure S5).
The second example is sub-network 1, which was extremely enriched with photosynthesis, especially photosystem II and the biological process of carbon dioxide. Based on PPIs network, we found a key protein, Slr1740 (AppA, oligopeptide binding protein of ABC transporter) structuring a highly important linkage between modules of photosynthesis and carbon dioxide, suggesting the potential function of Slr1740 that relates carbon dioxide uptake to photosystem II (see Fig. 6a). In fact, ABC transporters are critical membrane proteins responsible for transport of manganese (Mn, composing functional Mn cluster in the photosystem II)50 and HCO3− (accumulated in carbon dioxide uptake systems)51.
Figure 6. Examples of functional linkages in PPI network.

(a) Protein-protein interactions in functional linkage of CO2 uptake and photosystem II. Sub-network 1 is enriched in photosynthesis, especially photosystem II and carbon dioxide. A key protein, slr1740 (appA, oligopeptide binding protein of ABC transporter) structured a highly important linkage between modules of photosynthesis and carbon dioxide, suggesting the potential function of slr1740 in carbon dioxide uptake and photosystem II. Blue nodes, proteins involved in the photosystem II process; green nodes, proteins involved in CO2 uptake; pink nodes, first neighbor of CO2 uptake proteins; yellow node, appA, oligopeptide binding protein of ABC transporter. (b) Protein-protein interactions in biological processes of photosystem II reaction center subunits under UV irradiation. Copies of FtsH (slr1390 and slr1640) separately interacting with their own relative proteins could both link with sml0003 (PS II reaction center M protein), which also interacts with D1 and D2 protein. D1 and D2 proteins then interact with cytochrome proteins, and finally affect slr0008 (CtpA, Carboxyl-terminal processing protease) by a series of PPIs. Blue nodes, FtsH copies; red node photosystem II reaction center M protein; green nodes, protein members of D1 and D2; yellow nodes, cytochrome proteins; pink node, CtpA.
Discussion
Here we present a comprehensive systems biology approach of integrating data from different sources, identifying high confident PPIs and undertaking a series downstream functional analyses based on PPI network to investigate protein function, especially proteins with unknown function. This method is verified to be efficient in predicting PPIs and understanding the biological mechanism of proteins, protein complexes and their molecular interactions.
Our work has advantages in data source, integrating method, PPIs qualities and PPI network analysis. Firstly, we applied seven different sources data including not only domain information, but also genome context, evolutionary and function information. Secondly, we applied Naïve Bayes approach to statistically integrate results by providing PPI probabilities. In addition, to enhance the power of prediction, we applied GSP and GSN to appraise the performance of each protein pairs with quantitative score. Thirdly, our high confident PPIs have higher coverage in genome and have been evaluated by different evidences. Specifically, we predicted 1,997 high confident PPIs involving 2,765 proteins, the reliability of which was confirmed by Y2H experiments, and assessed by text mining and conserved interologs. Furthermore, we tested the physiological significance of the PPI pairs of NDH-1 (Fig. 3). Fourthly, we constructed PPI network, and carried out subsequent functional analysis based on the PPI network to infer function of uncharacterized proteins and explore the mechanism of PPIs. As the result, functional modules not only strengthen the reliability of high confident PPIs, but also present underlying functional relationships between PPIs, particularly for protein family members in a certain pathway or cell complex (component).
In contrast, previous studies only predicted information-bias PPIs. For example, Kim et al. predicted less high confident PPIs (1,591 pairs involving 509 proteins) with simply combining domain biased information together14, and only 3 of their high confident PPIs overlap with yeast two-hybrid experimental results9; Michaut et al. only applied interologs to infer PPIs, making the prediction highly biased toward conservation and dependent on its unique data source15.
Besides informative examples in results, we take one more intriguing instance to demonstrate the application of our result by explaining biological processes of photosystem II reaction center subunits (both D1 and D2 proteins) under UV irradiation. Microorganisms, such as cyanobacteria, possess a range of compounds that absorb UV so that they are proposed to function as sunscreen52. Energy transfer, tetrapyrrole synthesis nitrate and ammonium uptake and cell differentiation would be negatively affected by UV light. UV-B irradiation results in the loss of steady state oxygen evolution and a parallel loss of the photosystem II reaction center subunits53. Additionally, in parallel with the transcripts of D1-processing protease (CtpA) and D1-degrading FtsH protease, wholly biosynthesis processing and degradation of D1 are coordinated54. By mining the whole biosynthesis and degradation of D1 protein pathway in our PPI network, we found a comparative intact path with UV irradiation (Fig. 6b). Copies of FtsH (Slr1390 and Slr1640) through separately interacting with their own relative proteins, could both link with Sml0003 (PS II reaction center M protein), which also interacts with D1 and D2 protein. D1 and D2 proteins then interact with cytochrome proteins, and finally affect Slr0008 (CtpA, Carboxyl-terminal processing protease) by a series of PPIs.
For future work, we will focus on high confident PPIs that have not been proved by existing evidence and try to explore their underlying biological function, such as Sll0654 (PhoA) and Sll0656 (NucH) in sub-network 17, Sll0320 (probable ribonuclease D) and Sll0319 (periplasmic protein, function unknown) in sub-network 15, Sll0431, Sll0651, Sll0201, Sll0700 (all are putative transposases) in sub-network 9.
As discussed above, our systems biology approach is efficient in predicting PPIs. And the resulting PPI network includes plenty of validated functional linkages among protein pairs and sub-networks, which could be viewed as a novel resource to investigate the potential mechanisms of PPIs in Synechocystis sp. PCC6803. This systems biology approach provides the first step to explore functional linkage of cellular network, and is expected to extent the coverage and accuracy of the PPIs accompanying with more experimental data in the future.
Materials and Methods
Data preparation and features calculation
In gene expression, we first analyzed 20 microarrays datasets (GSE10708, GSE11970, GSE14410, GSE16162, GSE1695, GSE21133, GSE24882, GSE27406, GSE3682, GSE3703, GSE3715, GSE3716, GSE3717, GSE3755, GSE4019, GSE4604, GSE4606, GSE4613, GSE5391 and GSE9577) which were collected from GEO. Pearson correlation co-efficiencies were computed for each dataset to find out co-expressed gene pairs by cor function in R. We then maintained only three datasets (GSE4613, GSE37482 and GSE1695) where co-expression was positively correlated with interacting proteins. The three datasets were time course response to dehydration/desiccation and UV irradiation (GSE4613), response to inorganic carbon limitation (GSE1695) and transcriptomic response of Synechocystis 6803 encapsulated in silica gel (GSE37482).
GO annotation were collected from Gene Ontology Consortium55 to compute smallest shared biological process (SSBP) for each protein pair, assuming that protein pairs having the same GO term annotation should be more likely to interact with each other.
The genome context methods (gene fusion, gene neighborhood, phylogenetic profile and gene cluster) were analyzed by InPrePPI28. Domain data were obtained from DOMINE database (Database of Protein Domain Interactions, http://domine.utdallas.edu/cgi-bin/Domine) and Pfam database (http://pfam.sanger.ac.uk/).
Orthologs of 23 organisms were derived from InParanoid database (http://inparanoid.sbc.su.se/cgi-bin/index.cgi). For two species, E. coli and S. cerevisae with no data in InParanoid database, orthologs were obtained with reciprocal BLASTP between proteins with E-value < 1e-10. PPIs data of these species were downloaded from Database of Interacting Proteins (DIP, http://dip.doe-mbi.ucla.edu) and STRING (http://string-db.org/).
Data integration method: Naïve Bayes approach
This approach, depending on the prior odds of protein-protein pairs, computed the likelihood ratio of each individual approach (or non-redundancy) and integrated them into a global likelihood. According to basic Bayes rules, we obtained eventual scores which represent the probability of each protein pair.
The prior odds were defined as equation (1). Here, P(pos) was the probability of a pair of proteins sharing an interaction and P(neg) was the probability of finding a non-interacting protein pairs.
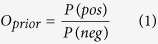 |
The likelihood ratio of individual approach was defined as equation (2), where fi was a pair of proteins and n was the number of all possible protein pairs in each feature. Pr( f1…fn|GSP) was the probability based on the condition of GSP and so was it with Pr( f1…fn|GSN). When each element in the model was considered as independent (or non-redundancy), the global likelihood ratio could be calculated easily as the product of several individual likelihood ratio as equation (3).
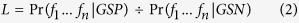 |
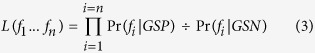 |
According to Bayes rule, the posterior odds could be computed according to equation (4) and equation (5).
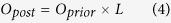 |
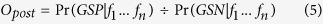 |
Equation (5) could be recognized as ultimate result of predicted protein-protein interaction. It meant, based on known condition of pairs of proteins, the probability belonging to GSP versus the probability belonging to GSN. The higher score they had, the more probability they might interact. We defined high confident PPIs as posterior odds larger than 5, which was stricter than previously defined posterior odds cutoff17.
Yeast two-hybrid analysis
Several pairs of protein interactions were validated by yeast two-hybrid experiments. The encoding sequences of these proteins were amplified using primers listed in (Supplementary Table S4). The PCR products were digested with EcoRI/XhoI and unidirectionally inserted into pJG45 and pB42AD plasmids to construct baits and preys, respectively. Combinations of bait, prey and the reporter vector pSH18-34 were co-transformed into yeast strain EGY48 according to previously described procedures. The selection of transformants and the analysis of the galactosidase were performed as the former described procedure56.
Isolation of crude thylakoid membranes
The cell cultures (800 mL) were harvested at the logarithmic phase (A730 = 0.6–0.8) and washed twice by suspending in 50 mL of fresh BG-11 medium, and the thylakoid membranes were isolated according to Gombos et al.57 with some modifications as follows. The cells suspended in 5 mL of disruption buffer (10 mM HEPES-NaOH, 5 mM sodium phosphate, pH7.5, 10 mM MgCl2, 10 mM NaCl, and 25% glycerol (v/v)) were supplemented by zirconia/silica beads and broken by vortexing 15 times at the highest speed for 20 s at 4 °C with 5 min cooling on ice between the runs. The crude extract was centrifuged at 5,000 × g for 5 min to remove the glass beads and unbroken cells. By further centrifugation at 20,000 × g for 30 min, we obtained crude thylakoid membranes from the precipitation.
Electrophoresis, immunoblotting and chlorophyll fluorescence
SDS-PAGE of Synechocystis sp. strain PCC 6803 crude thylakoid membranes was carried out on 12% polyacrylamide gel with 6 M urea as described earlier58.
For immunoblotting, the proteins were electrotransferred to a polyvinylidene difluoride (PVDF) membrane (Immobilon-P; Millipore, Bedford, MA) and detected by protein-specific antibodies using an ECL assay kit (Amersham Pharmacia, NJ) according to the manufacturer’s protocol. The NDH-1 complexes were detected using the antibodies against NdhA and NdhH, respectively, which were previously raised in our laboratory59. Antibody against ATPβ was purchased from Agrisera Co. (Cännäs, Sweden).
The transient increase in chlorophyll fluorescence after actinic light turned off was monitored as described59.
Construction of Δslr0815 mutant
Δslr0815 mutant was constructed as follows. The upstream and downstream regions of an unknown encoding gene slr0815 were amplified by PCR creating appropriate restriction sites. A DNA fragment encoding a kanamycin resistance (KamR) cassette was also amplified by PCR creating XbaI sites using appropriate PCR primers, slr0815-C and slr0815-D (Supplemental Table S4). These three PCR products were ligated into the MCS of pUC19 (Fig. 4a) and was used to transform the WT cells of Synechocystis sp. strain PCC 6803 to generate the Δslr0815 mutant. The transformants were spread on agar plates containing BG-11 medium and kanamycin (10 μg mL−1) buffered at pH 8.0, and the plates were incubated in 2% (v/v) CO2 in air under illumination by fluorescent lamps at 40 μmol photons m–2s–1. The mutated slr0815 in the transformants was segregated to homogeneity (by successive streak purification) as determined by PCR amplification analysis (Fig. 4b).
Function annotations with GO terms
GO terms were used to infer the annotations of proteins with unknown function. We selected the 5th upper GO terms as the level for proper functional annotation. Simulated annealing algorithm was applied to detect proteins with unknown function based on association rules with the whole network26. With the simulated annealing algorithm, each protein in the network would be assigned several GO terms with simulated frequency. Partial assignments were caused by randomly selected processes rather than true functional annotation. To minimize the false assignments, 10% of total results were randomly selected to compute a background noise and simulate the process with 100 times repeat. At last, GO terms, whose frequencies were larger than the background noise, were selected and viewed as true function annotation to proteins with unknown function.
Pathway and GO enrichment analysis
Pathway data of Synechocystis sp. strain PCC 6803 was downloaded from KEGG (http://www.genome.jp/kegg/). With the annotation of gene and its participated pathway, using the hypergeometric distribution method, we compared the pathway information with genes in each sub-network and identified statistic significant function of each sub-network. One protein complex was selected to be enriched by one certain pathway if any pathway mapped to the complex with adjusted p-value <0.05. The statistical analysis was applied with R. The whole process of GO enrichment analysis was executed by the online tools AmiGO.
Additional Information
How to cite this article: Lv, Q. et al. Genome-wide protein-protein interactions and protein function exploration in cyanobacteria. Sci. Rep. 5, 15519; doi: 10.1038/srep15519 (2015).
Supplementary Material
Acknowledgments
This work was supported by the National 973 Key Basic Research Program (2013CB127000); the National Natural Science Foundation of China (31171264, 31000590); Research Fund for the Doctoral Program of Higher Education of China (20120076110006); the Supercomputer Center of East China Normal University and the Science and Technology Commission of Shanghai Municipality (11DZ2260300). We are grateful to Dr. Wenzhong Xiao from Stanford Genome Technology Center for his valuable suggestions for the manuscript improvement.
Footnotes
Author Contributions T.S. designed the bioinformatics workflow, W.M. and F.L. executed biological experiments, H.L., J.L. and C.Z. predicted PPIs and analyzed data, Q.L. and H.W. prepared figures and wrote the manuscript, T.S. finalized the manuscript.
References
- Martins J., Peixe L. & Vasconcelos V. M. Unraveling cyanobacteria ecology in wastewater treatment plants (WWTP). Microb. Ecol. 62, 241–256 (2011). [DOI] [PubMed] [Google Scholar]
- Carrieri D., Ananyev G., Garcia Costas A. M., Bryant D. A. & Dismukes G. C. Renewable hydrogen production by cyanobacteria: Nickel requirements for optimal hydrogenase activity. Int. J. Hydrogen Energy 33, 2014–2022 (2008). [Google Scholar]
- Quintana N., Van der Kooy F., Van de Rhee M. D., Voshol G. P. & Verpoorte R. Renewable energy from Cyanobacteria: energy production optimization by metabolic pathway engineering. Appl. Microbiol. Biotechnol. 91, 471–490 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse O., Rupprecht J., Mussgnug J. H., Dismukes G. C. & Hankamer B. Photosynthesis: a blueprint for solar energy capture and biohydrogen production technologies. Photochem. Photobiol. Sci. 4, 957–970 (2005). [DOI] [PubMed] [Google Scholar]
- Stelzl U. et al. A human protein-protein interaction network: a resource for annotating the proteome. Cell 122, 957–968 (2005). [DOI] [PubMed] [Google Scholar]
- Sharan R., Ulitsky I. & Shamir R. Network-based prediction of protein function. Mol. Syst. Biol. 3, 88 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S. et al. A map of the interactome network of the metazoan C. elegans. Science 303, 540–543 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T. et al. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl. Acad. Sci. USA 98, 4569–4574 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S. et al. A large-scale protein protein interaction analysis in Synechocystis sp. PCC6803. DNA Res. 14, 207–216 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitre S. et al. Computational methods for predicting protein-protein interactions. Adv. Biochem. Eng. Biotechnol. 110, 247–267 (2008). [DOI] [PubMed] [Google Scholar]
- von Mering C. et al. Comparative assessment of large-scale data sets of protein-protein interactions. Nature 417, 399–403 (2002). [DOI] [PubMed] [Google Scholar]
- Rhodes D. R. et al. Probabilistic model of the human protein-protein interaction network. Nat. Biotechnol. 23, 951–959 (2005). [DOI] [PubMed] [Google Scholar]
- Zhong W. & Sternberg P. W. Genome-wide prediction of C. elegans genetic interactions. Science 311, 1481–1484 (2006). [DOI] [PubMed] [Google Scholar]
- Kim W. Y. et al. SynechoNET: integrated protein-protein interaction database of a model cyanobacterium Synechocystis sp PCC 6803. BMC Bioinformatics 9 Suppl 1, S20 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaut M. et al. InteroPORC: automated inference of highly conserved protein interaction networks. Bioinformatics 24, 1625–1631 (2008). [DOI] [PubMed] [Google Scholar]
- Yu J. & Finley R. L. Combining multiple positive training sets to generate confidence scores for protein–protein interactions. Bioinformatics 25, 105–111 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen R. et al. A Bayesian networks approach for predicting protein-protein interactions from genomic data. Science 302, 449–453 (2003). [DOI] [PubMed] [Google Scholar]
- Cui J. et al. AtPID: Arabidopsis thaliana protein interactome database--an integrative platform for plant systems biology. Nucleic. Acids. Res. 36, D999–1008 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M., Shen X. & Chen X. PAIR: the predicted Arabidopsis interactome resource. Nucleic. Acids. Res. 39, D1134–1140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte E. M. et al. Detecting protein function and protein-protein interactions from genome sequences. Science 285, 751–753 (1999). [DOI] [PubMed] [Google Scholar]
- Enright A. J., Iliopoulos I., Kyrpides N. C. & Ouzounis C. A. Protein interaction maps for complete genomes based on gene fusion events. Nature 402, 86–90 (1999). [DOI] [PubMed] [Google Scholar]
- Dandekar T., Snel B., Huynen M. & Bork P. Conservation of gene order: a fingerprint of proteins that physically interact. Trends Biochem. Sci. 23, 324–328 (1998). [DOI] [PubMed] [Google Scholar]
- Bowers P. M. et al. Prolinks: a database of protein functional linkages derived from coevolution. Genome Biol. 5, R35 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vert J. P. A tree kernel to analyse phylogenetic profiles. Bioinformatics 18 Suppl 1, S276–284 (2002). [DOI] [PubMed] [Google Scholar]
- Deng M., Mehta S., Sun F. & Chen T. Inferring domain-domain interactions from protein-protein interactions. Genome Res. 12, 1540–1548 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez A., Flammini A., Maritan A. & Vespignani A. Global protein function prediction from protein-protein interaction networks. Nat. Biotechnol. 21, 697–700 (2003). [DOI] [PubMed] [Google Scholar]
- Pellegrini M., Marcotte E. M., Thompson M. J., Eisenberg D. & Yeates T. O. Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc. Natl. Acad. Sci. USA 96, 4285–4288 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J. et al. InPrePPI: an integrated evaluation method based on genomic context for predicting protein-protein interactions in prokaryotic genomes. BMC Bioinformatics 8, 414 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Mering C. et al. STRING: a database of predicted functional associations between proteins. Nucleic. Acids. Res. 31, 258–261 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H., Endo T., Schreiber U., Ogawa T. & Asada K. Electron Donation from Cyclic and Respiratory Flows to the Photosynthetic Intersystem Chain is Mediated by Pyridine Nucleotide Dehydrogenase in the Cyanobacterium Synechocystis PCC 6803. Plant Cell Physiol. 33, 1233–1237 (1992). [Google Scholar]
- Shoemaker B. A. & Panchenko A. R. Deciphering protein-protein interactions. Part II. Computational methods to predict protein and domain interaction partners. PLoS Comp. Biol. 3, e43 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman E. R., Dall’Acqua W., Braden B. C. & Mariuzza R. A. Analysis of binding interactions in an idiotope-antiidiotope protein-protein complex by double mutant cycles. Biochemistry 36, 49–56 (1997). [DOI] [PubMed] [Google Scholar]
- Ohkawa H., Pakrasi H. B. & Ogawa T. Two types of functionally distinct NAD(P)H dehydrogenases in Synechocystis sp strain PCC6803. J. Biol. Chem. 275, 31630–31634 (2000). [DOI] [PubMed] [Google Scholar]
- Shibata M. et al. Distinct constitutive and low-CO2-induced CO2 uptake systems in cyanobacteria: genes involved and their phylogenetic relationship with homologous genes in other organisms. Proc. Natl. Acad. Sci. USA 98, 11789–11794 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J. R., Burnap R. L. & Inoue Y. An independent role of cytochrome c-550 in cyanobacterial photosystem II as revealed by double-deletion mutagenesis of the psbO and psbV genes in Synechocystis sp. PCC 6803. Biochemistry 34, 12661–12668 (1995). [DOI] [PubMed] [Google Scholar]
- Shen J. R., Ikeuchi M. & Inoue Y. Analysis of the psbU gene encoding the 12-kDa extrinsic protein of photosystem II and studies on its role by deletion mutagenesis in Synechocystis sp. PCC 6803. J. Biol. Chem. 272, 17821–17826 (1997). [DOI] [PubMed] [Google Scholar]
- Kondo K., Geng X. X., Katayama M. & Ikeuchi M. Distinct roles of CpcG1 and CpcG2 in phycobilisome assembly in the cyanobacterium Synechocystis sp. PCC 6803. Photosynthesis Res. 84, 269–273 (2005). [DOI] [PubMed] [Google Scholar]
- Klinkert B. et al. PratA, a periplasmic tetratricopeptide repeat protein involved in biogenesis of photosystem II in Synechocystis sp PCC 6803. J. Biol. Chem. 279, 44639–44644 (2004). [DOI] [PubMed] [Google Scholar]
- Sakayori T., Shiraiwa Y. & Suzuki I. A Synechocystis homolog of SipA protein, Ssl3451, enhances the activity of the histidine kinase Hik33. Plant Cell Physiol. 50, 1439–1448 (2009). [DOI] [PubMed] [Google Scholar]
- Dobakova M., Tichy M. & Komenda J. Role of the PsbI protein in photosystem II assembly and repair in the cyanobacterium Synechocystis sp PCC 6803. Plant Physiol. 145, 1681–1691 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q., Armbrust T. S., Guikema J. A. & Chitnis P. R. Organization of Photosystem I Polypeptides (A Structural Interaction between the PsaD and PsaL Subunits). Plant Physiol. 106, 1057–1063 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walhout A. J. M. et al. Protein interaction mapping in C-elegans using proteins involved in vulval development. Science 287, 116–122 (2000). [DOI] [PubMed] [Google Scholar]
- Rowlett R. et al. Mutations in the Contact Region between the α and β Subunits of Tryptophan Synthase Alter Subunit Interaction and Intersubunit Communication. Biochemistry 37, 2961–2968 (1998). [DOI] [PubMed] [Google Scholar]
- Mendillo M. L. et al. A conserved MutS homolog connector domain interface interacts with MutL homologs. Proc. Natl. Acad. Sci. USA 106, 22223–22228 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin A. C. et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. FASEB J. 16, A523–A523 (2002). [DOI] [PubMed] [Google Scholar]
- Eisenberg D., Marcotte E. M., Xenarios I. & Yeates T. O. Protein function in the post-genomic era. Nature 405, 823–826 (2000). [DOI] [PubMed] [Google Scholar]
- Inoue-Kashino N. et al. S4 Protein Sll1252 Is Necessary for Energy Balancing in Photosynthetic Electron Transport in Synechocystis sp. PCC 6803. Biochemistry 50, 329–339 (2011). [DOI] [PubMed] [Google Scholar]
- Lieman-Hurwitz J. et al. A cyanobacterial AbrB-like protein affects the apparent photosynthetic affinity for CO2 by modulating low-CO2-induced gene expression. Environ. Microbiol. 11, 927–936 (2009). [DOI] [PubMed] [Google Scholar]
- Bader G. D. & Hogue C. W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics 4, 2 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartsevich V. V. & Pakrasi H. B. Molecular identification of an ABC transporter complex for manganese: analysis of a cyanobacterial mutant strain impaired in the photosynthetic oxygen evolution process. EMBO J. 14, 1845–1853 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badger M. R. & Price G. D. CO2 concentrating mechanisms in cyanobacteria: molecular components, their diversity and evolution. J. Exp. Bot. 54, 609–622 (2003). [DOI] [PubMed] [Google Scholar]
- Gao Q. & Garcia-Pichel F. Microbial ultraviolet sunscreens. Nat. Rev. Microbiol. 9, 791–802 (2011). [DOI] [PubMed] [Google Scholar]
- Sass L., Spetea C., Mate Z., Nagy F. & Vass I. Repair of UV-B induced damage of photosystem II via de novo synthesis of the D1 and D2 reaction centre subunits in Synechocystis sp. PCC 6803. Photosynthesis Res. 54, 55–62 (1997). [Google Scholar]
- Huang L. X., McCluskey M. P., Ni H. & LaRossa R. A. Global gene expression profiles of the cyanobacterium Synechocystis sp strain PCC 6803 in response to irradiation with UV-B and white light. J. Bacteriol. 184, 6845–6858 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris M. A. et al. The Gene Ontology (GO) database and informatics resource. Nucleic. Acids. Res. 32, D258–261 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNellis T. W., Torii K. U. & Deng X. W. Expression of an N-terminal fragment of COP1 confers a dominant-negative effect on light-regulated seedling development in Arabidopsis. Plant Cell 8, 1491–1503 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gombos Z., Wada H. & Murata N. The recovery of photosynthesis from low-temperature photoinhibition is accelerated by the unsaturation of membrane lipids: a mechanism of chilling tolerance. Proc. Natl. Acad. Sci. USA 91, 8787–8791 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli U. K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 (1970). [DOI] [PubMed] [Google Scholar]
- Ma W. & Mi H. Expression and activity of type 1 NAD(P)H dehydrogenase at different growth phases of the cyanobacterium, Synechocystis PCC 6803. Physiol. Plant. 125, 135–140 (2005). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.