

Abstract
The putative induction of adult β-cell regeneration represents a promising approach for the treatment of type 1 diabetes. Toward this ultimate goal, it is essential to develop an inducible model mimicking the long-lasting disease progression. In the current study, we have established a novel β-cell ablation mouse model, in which the β-cell mass progressively declines, as seen in type 1 diabetes. The model is based on the β-cell specific genetic ablation of the transcription initiation factor 1A, TIF-IA, essential for RNA Polymerase I activity (TIF-IAΔ/Δ). Using this approach, we induced a slow apoptotic response that eventually leads to a protracted β-cell death. In this model, we observed β-cell regeneration that resulted in a complete recovery of the β-cell mass and normoglycemia. In addition, we showed that adaptive proliferation of remaining β-cells is the prominent mechanism acting to compensate for the massive β-cell loss in young but also aged mice. Interestingly, at any age, we also detected β-like cells expressing the glucagon hormone, suggesting a transition between α- and β-cell identities or vice versa. Taken together, the TIF-IAΔ/Δ mouse model can be used to investigate the potential therapeutic approaches for type 1 diabetes targeting β-cell regeneration.
Keywords: β-cell proliferation, diabetes, insulin, islet of Langerhans, pancreatic β-cell, regeneration, TIF-IA
Abbreviations
- TIF-IA
Transcription Initiation Factor 1A
- RIP
Rat Insulin Promoter
- Pdx1
Pancreatic and duodenal homeobox 1
- Ngn3
Neurogenin 3
- Pax4
Paired box gene 4
- rDNA
ribosomal DNA
Introduction
As the one and only source of insulin production in the body, pancreatic β-cells play a pivotal role in the regulation of fuel metabolism. The presence of a sufficient number of functional glucose responsive β-cells is indispensable for normal glucose homeostasis.
It has been shown that the adult pancreatic tissue can regenerate in several species of mammals following, for instance, surgical insult or disease.1 This tissue has also the potential to increase its β-cell content in response to metabolic demand, as seen during pregnancy and in obesity.2 Identifying the cellular sources that can account for β-cell mass dynamics in different physiological and pathophysiological conditions could establish a ground for improvement of β-cell regeneration as a potential treatment of diabetes.
β-cell regeneration has been studied in several contexts, and it is concluded that the mechanism(s) contributing to regeneration greatly depends on the type and extent of injury or β-cell loss. Self-replication of pre-existing β-cell has been shown to represent the main mean of β-cell turnover in adult life but also in the context of β-cell regeneration induced by different types of pancreatic injury,3-6 as well as increased metabolic demands during pregnancy and in the obesity context.7,8 By means of lineage tracing, it was confirmed that after 70-80% β-cell ablation, proliferation of pre-existing insulin-positive cells is responsible for the complete regeneration of β-cells.9
The presence of stem/progenitor cells in the duct epithelium/lining and their contribution to endocrine cell neogenesis has been proposed by several studies dealing with pancreas injury models,10-12 as well as upon transient overexpression of cyclin D2/CDK4/GLP1.13,14 However, the contribution of duct cells to endocrine cell regeneration is challenged by additional lineage tracing experiments using different duct/centroacinar specific CreER lines, such as Hnf1B, Sox9, and Hes1.15-18
Interestingly, α−to-β-like cell conversion was shown to be the major mechanism underlying β-cell regeneration in condition of extreme β-cell loss19 and in a PDL (pancreatic duct ligation) model combined with alloxan-induced β-cell ablation.20 Moreover, in transgenic mice, the forced expression of Pax4 in α-cells, promotes their conversion into functional β-cells that counter chemically induced diabetes.21,22 Interestingly, the conversion of α-cells revealed their regenerative capacity, and the propensity of duct/duct lining, to contribute to β-cell neogenesis by epithelial mesenchymal transition mechanism.21
The existing data for progression of type 1 diabetes describe this disease as a chronic progressive autoimmune disorder, in which the loss of the β-cell mass occurs in a slow and gradual manner.23-25 Additionally, it is shown that the β-cell mass falls gradually over time in rodent models of type 1 diabetes. However, in all of the existing models, β-cell ablation occurs very rapidly within days after initial induction.6,9,19,20 To better understand the potential β-cell regeneration processes that might be induced in diabetic islets, it is important to use a model mimicking the slow progression and extent of β-cell loss seen in type 1 diabetes.
Transcription initiation factor 1A TIF-IA, the mammalian homolog of yeast Rrn3p,26 interacts with RNA polymerase I and is essential for rDNA transcription.27 Genetic inactivation of TIF-IA perturbs nucleolar integrity and consequently, leads to cell cycle arrest and apoptosis mediated by a p53-dependent pathway.28 Targeted deletion of TIF-IA in adult mouse hippocampus neurons induces a protracted neuronal degeneration, whereas in embryonic neural progenitors, it triggers a rapid apoptosis.29,30 These observations suggested that the conditional ablation of TIF-IA could be used as a genetic tool to induce a protracted suicide response in slowly dividing or post-mitotic cells. Here, we report the development of a novel inducible β-cell loss model, based on the conditional ablation of TIF-IA, in which β-cell death occurs gradually and over a longer period of time compared to previously used animal models.31 Moreover, we show that this model is suitable to investigate the source and mechanisms of β-cell regeneration.
Results
The loss of TIF-IA induces an efficient and specific β-cell ablation
In order to generate an inducible model of β-cell loss, we crossed the TIF-IAfl/fl mouse line with RipCreERT2 transgenic animals (Fig. 1A). In the resulting double transgenic mice (TIF-IAΔ/Δ), TIF-IA gene is specifically deleted in insulin-producing β-cells upon systemic administration of tamoxifen. We also crossed a Rosa26YFP/YFP reporter line with TIF-IAΔ/Δ mice to specifically label and trace insulin-producing cells following TIF-IA deletion (YFP::TIF-IAΔ/Δ Fig. 1A). Ablation of β-cells was induced only once by tamoxifen treatment for 7 d. Loss of TIF-IA in pancreatic β-cells resulted in the progressive appearance of dying TUNEL-positive cells in pancreatic islets (Fig. 1B). The number of TUNEL-positive cells per islet was significantly higher in the mutant mice in comparison to control littermates and it increased over time. This was expected based on the fact that the consequences of TIF-IA perturbation were found to be slower in postmitotic neurons in comparison with fast dividing cells.30 To further characterize the β-cell ablation model, the YFP-labeled and unlabeled β-cells were quantified at different time points following tamoxifen injection (Fig. 1C and 1D). Two weeks after injection, 95% of β-cells were found YFP+ in YFP::TIF-IA+/+ controls, while the labeled fraction was slightly lower in mutant littermates. In both cases, YFP labeling was specific of β-cells and absent in non-β endocrine cells. This outlines the robustness and specificity of recombination induced by tamoxifen injection. Following Cre recombination, 88% of mutant β-cells were YFP labeled (Fig. 1C and D), suggesting that such amount of insulin-producing cells lost TIF-IA expression and eventually died. This is consistent with the finding that the YFP-marked fraction decreased over time so that 3 months post injection only few labeled β-cells could be detected in some islets (Fig. 1C and 1D). The gradual shift from YFP+ β-cells to YFP− β-cells indicates a loss of β-cells but also their continuous replacement through regeneration from unlabeled cells. We conclude that the massive and gradual β-cell loss observed in TIF-IAΔ/Δ model can mimic the slow-progression and extent of β-cell loss observed in type 1 diabetes and could thereby provide a useful tool to monitor β-cells regeneration.
Figure 1.
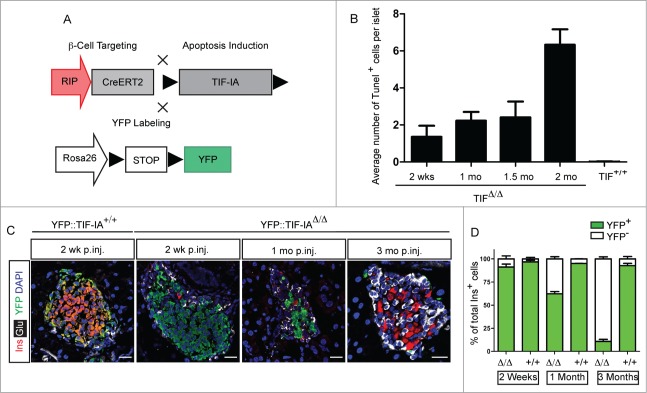
Generation of TIF-IAΔ/Δ mouse model to study β-cell regeneration. (A) β-cell specific deletion of TIF-IA is used to induce apoptosis. YFP labeling marks the recombined cells. (B) Apoptotic β-cells are detected in the islet of TIF-IAΔ/Δ mice. (C) Representative immunostaining of insulin (red), glucagon (gray), and YFP (green) in the pancreata of TIF-IA+/+ and TIF-IAΔ/Δ mice at different time points after tamoxifen injection. (D) Quantification of insulin and YFP expressing cells shows 95% YFP-labeling upon Cre-mediated recombination. In TIF-IAΔ/Δ mutants, YFP-labeled cells are lost gradually, and replaced with non-labeled cells (D). mo p.inj.: months post injection. Scale bar: 25 μm. Values are presented by mean ± SEM. N >3 animals for each experimental condition.
Upon massive protracted β-cells are fully regenerated
To monitor β-cell regeneration, one-month old mice were injected with tamoxifen for 7 d. Ten days after the arrest of tamoxifen administration, blood glucose levels raised in TIF-IAΔ/Δ mice to reach their highest level after one month (Fig. 2A). A slow and gradual decrease of hyperglycemia was subsequently observed. Importantly, after 8 months, glycemia went back to normal levels and remained within normal ranges for longer than one month. Consistently, we detected a continuous increase in the number of insulin-expressing cells following initial ablation (Fig. 2B and C). Eight months after tamoxifen induction, the number of β-cells appeared similar to wild type controls and islet structure was indistinguishable from wild type islets (Fig. 2C). This was further confirmed by circulating insulin measurement showing a drastic decrease in TIF-IAΔ/Δ mice one month post-injection. Eight months post-injection, insulin concentration in TIF-IAΔ/Δ mice was found comparable to age-matched controls (Fig. 2D). Glut2 and MafA staining showed that the majority of insulin+ cells, after recovery, displayed a mature β-cell phenotype and were able to release insulin and maintain normoglycemia (Fig. S1). Altogether, our results show that, upon TIF-IA loss, the β-cell mass can be spontaneously regenerated in adult mice following the progressive loss of 95% of β-cells. β-like cell regeneration leads to a complete recovery from long-lasting and severe hyperglycemia in mice.
Figure 2.
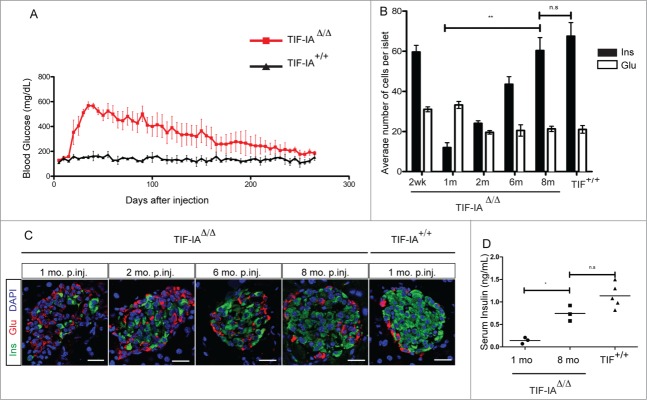
β-cell ablation and regeneration in TIF-IAΔ/Δ mice. (A) Massive β-cell ablation leads to development of severe and long-lasting hyperglycemia in TIF-IAΔ/Δ mice. Mutant mice gradually recover from hyperglycemia. (B and C) Immunostaining and quantification of insulin (green) and glucagon (red) expressing cells in the pancreata of TIF-IA+/+ and TIF-IAΔ/Δ mice at different time points post injection (A). β-cell number is increased gradually in mice injected at one-month (B). (D) Measurement of blood insulin level confirms the changes in functional β-cell number. mo p.inj.: months post injection. Scale bar: 25 μm. Values are presented by mean ± SEM. P-value <0.05 : *, P-value <0.01: **. N >3 animals for each experimental condition.
Adaptive β-cell proliferation contributes to β-cell regeneration in TIF-IAΔ/Δcmice
We then queried the cellular source of neo-generated β-like cells in TIF-IAΔ/Δ mice. To address this question, we searched for different regenerative mechanisms. First, we verified whether the self-replication of β-cells was altered upon tamoxifen induction. We therefore measured the β-cell proliferation rate as a fraction of Ki67-expressing cells in YFP− β-cells in TIF-IAΔ/Δ (both Cre-escaper and newly-formed β-cells) as compared to wild type animals. The β-cells proliferation rate appeared significantly higher in the mutants at different time points following β-cell ablation (Fig. 3A). A three-fold increase, as compared to controls, remained almost constant at any given age, as reported also by other.32 Consistently, a long-term BrdU labeling analysis (initiated 2 weeks after tamoxifen treatment and continued for 6 weeks) showed a significantly elevated BrdU incorporation in TIF-IAΔ/Δ β-cells compared to their wild type counterparts (Fig. 3B and C) confirming that, using a cumulative approach, neo-generated β-cells went through a proliferative phase. However, BrdU-labeled β-cells could be formed either by self-replication of preexisting β-cells or they could derive from other cells having undergone replication prior to their conversion into β-cells. These findings are consistent with previous reports3-6 showing that increased proliferation of pre-existing β-cells can compensate, at least partially, for a massive β-cell loss, as found in the TIF-IAΔ/Δ model.
Figure 3.

Increased β-cell proliferation rate upon massive β-cell loss in TIF-IAΔ/Δ mice. (A) Quantification of Ki67-expressing β-cells shows higher proliferative fraction of β-cells in TIF-IAΔ/Δ mice compared to the wild types at different time points following β-cell loss. (B and C) Quantification of BrdU labeled β-cells shows 2-fold increase in the BrdU labeled β-cell population in TIF-IAΔ/Δ mice compared to the wild types after two months BrdU treatment. mo.: months post injection. Scale bar: 50 μm. Values are presented by mean ± SEM. P-value <0.05: *, P-value <0.01 : **. N >3 animals for each experimental condition.
Pancreatic α-cells are considered as another cellular source that contributes to β-cell mass increase upon extreme β-cell loss.19,20 Interestingly, upon TIF-IA deletion in β-cells, a progressive increase in the α-cell number was noted (Fig. 2B and C). In fact, α-cells quickly became the major cell types in the islets while β-cells died. Subsequently, this elevated number of α-cells appeared attenuated as the number of β-cells increased. This points to a potential role for α-cells in the β-cell mass replenishment in response to hyperglycemia, or β-cell injury. We found several evidences that may support α-cell contribution to the β-cell regeneration occurring in the TIF-IAΔ/Δ model. Indeed, immunohistochemical analyses revealed the presence of some cells co-expressing insulin and glucagon (bi-hormonal cells) in the islets of TIF-IAΔ/Δ mice at different time points following β-cell loss (Fig. 4A and B). Such cells could very well be transitioning from an α- to a β-cell identity.19 Interestingly, further characterization of these bi-hormonal cells indicated that they expressed both Arx and Pdx1, which are α- and β-cell specific transcription factors, respectively (Fig. 4C). C-peptide, a side product of pre-insulin cleavage, could also be detected in these cells, suggesting that pre-insulin is being effectively processed in these cells (Fig. 4C). Moreover, such bi-hormonal cells lacked MafA expression indicating a rather immature transient nature for these cells (Fig. 4C).
Figure 4.
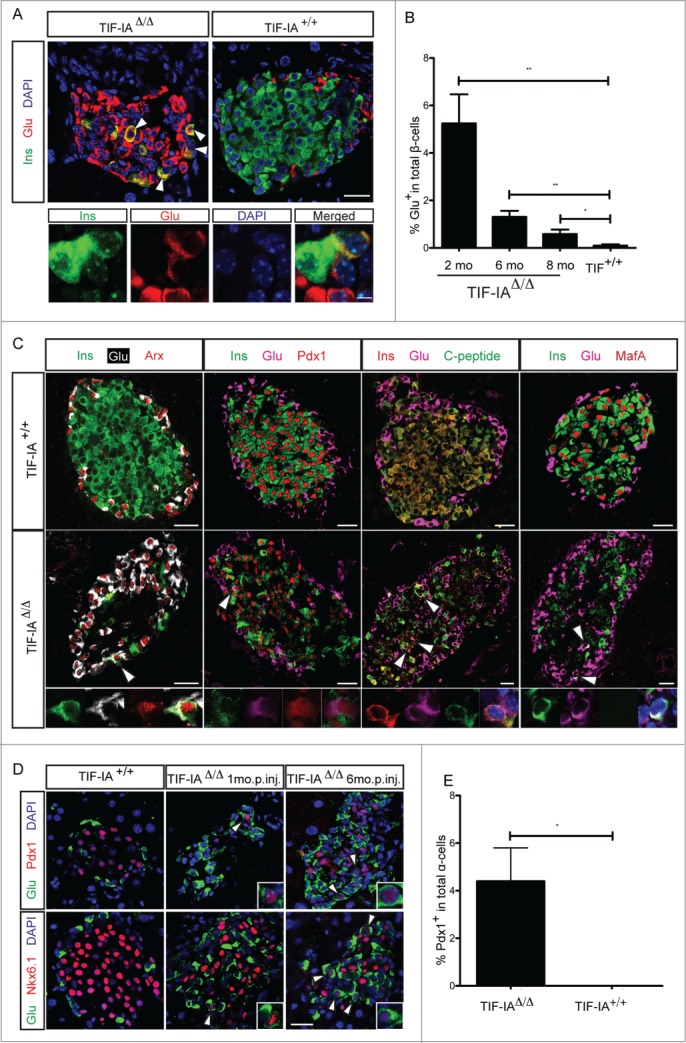
α-to β-cell transdifferentiation is induced in TIF-IAΔ/Δ model. (A) Immunostaining of insulin (green) and glucagon (red) reveals the presence of cells co-expressing insulin and glucagon inside the islets of TIF-IAΔ/Δ mice. (B) The relative abundance of these bihormonal cells is decreased over time. (C) Bihormonal cells express a combination of α- and β-cell markers. Arrows point to the bihormonal cells. (D and E) Expression of Pdx1 and Nkx6.1 is induced in glucagon expressing α-cells. m.p.inj.: months post injection. Scale bar: 25 μm, 5 μm for 4A lower images. Values are presented by mean ± SEM. P-value <0.05: *, P-value <0.01: **. N >3 animals for each experimental condition.
Additionally, we also noticed glucagon-labeled α-cells that expressed typical β-cell transcription markers, such as Pdx1 or Nkx6.1 (Fig. 4D and E). These two transcription factors, along with some others, are essential for development and function of pancreatic β-cells. Therefore, the induction of their expression in α-cells further suggests the acquisition of a β-cell phenotype. All together, our results indicate that the massive and protracted β-cell depletion observed in the TIF-IAΔ/Δ model appears to activate, albeit at a low frequency, some α-cells to acquire β-like cell characteristics. However, a contribution of α-cells to β-cell regeneration will await advanced lineage-tracing experiments.
Adult precursor cells are not involved in β-cell regeneration
To further assess whether precursor cells might contribute to β-cell regeneration in TIF-IAΔ/Δ model, we took advantage of the YFP reporter system enabling to distinguish between pre-existing islets and newly formed ones. Due to the protracted nature of the β-cell death induced by TIF-IA deletion, we did detect some YFP+ cells up to 3 months after tamoxifen injection (Fig. 1C).Three months after tamoxifen injection, most islets comprised few YFP positive cells. One possibility is that some precursor cells might exist inside the islet, which can replenish the islets by proliferation and then differentiation into β-cells. To test this hypothesis, we used the sequential incorporation of 2 different thymidine analogs to detect more than one round of cell division in β-cells of TIF-IAΔ/Δ and TIF-IA+/+ mice. Both groups were first treated with CldU and then IdU in drinking water for four days, each with one-day washout interval. Tissue sections were stained specifically against CldU and IdU. β-Cells were exclusively labeled with either CldU or IdU, and no double-marked cells were detected in TIF-IAΔ/Δ and TIF-IA+/+ (Fig. 5A and B). This result clearly suggests that proliferation rate is equally distributed among all the β-cells in normal tissue homeostasis, and even in regenerating β-cells following severe β-cell loss.
Figure 5.

The new β-cells do not arise from a rapidly proliferating progenitor cell type. (A and B) double thymidine analog labeling shows even distribution of proliferation in the β-cell population. (C and D) The frequency of insulin positive cells inside the duct structures, marked by Sox9 expression, does not change in response to β-cell loss in TIF-IAΔ/Δ mice. E: Ngn3 expression is not induced at several time points after tamoxifen injection. Scale bar: 25 μm. Values are presented by mean ± SEM. P-value <0.001: *** , P-value <0.0001: ****. N > 3 animals for each experimental condition.
Additionally, we analyzed the association of insulin-positive cells with duct epithelium using a duct specific marker, Sox9. Insulin positive cells inside the duct structures were rare and their frequency did not change in the TIF-IAΔ/Δ compared to wild type littermates (Fig. 5C and D). We also asked whether Neurogenin3 is induced in TIF-IAΔ/Δ pancreata. Ngn3 positive cells were not detected in the TIF-IAΔ/Δ and TIF-IA+/+ pancreata (Fig. 5E), neither Ngn3 mRNA level was changed at any time point following β-cell loss (data not shown). Based on these observations, we conclude that adult precursors are less likely to contribute to β-cell regeneration in response to the protracted β-cell loss condition induced in the TIF-IAΔ/Δ model.
Bihormonal insulin+ glucagon+ cells are also detected in older mice
Next, we investigated how aging affects the induction of different regenerating pathways. β-cell ablation was induced by injecting with tamoxifen 3 mo old mice. The pattern of β-cell loss and subsequent increase in β-cell numbers was comparable to their younger counterparts (Fig. 6A and B). The changes in blood glucose levels were also consistent with β-cell number at different time points (Fig. 6C). However, β-cell regeneration and, consequently, recovery rate were slower in older mice. Indeed, β-cell proliferation rate was lower in 3 mo old injected TIF-IAΔ/Δ animals compared to one-month old injected TIF-IAΔ/Δ mice analyzed at similar stages following ablation initiation, though it was always higher than in the wild type littermates with similar fold-changes compared to the basal level at any given age (Fig. 6D). These results indicate that the basal β-cell proliferation rate, although reduced in older mice, increased in the presence of β-cell loss, supporting the activation of an adaptive proliferative response.
Figure 6.

Declined regenerative rate in TIF-IAΔ/Δ mice with older onset of β-cell loss. (A and B) Immunostaining and quantification of insulin (green) and glucagon (red) expressing cells in the pancreata of TIF-IA+/+ and TIF-IAΔ/Δ mice at different time points post injection (A). β-cell number is increased gradually in mice injected at three-months, but at a slower rate compared tot he younger mice (B). (C) Reduction of blood glucose level takes place at a slower manner in older mice. (D) β-cell proliferation rate is increased in mice with the older onset ablation. (E) The relative abundance of bihormonal cells is similar in one- and 3 mo old injected mice. In both cases, their frequencies decrease by time following initial ablation. (F) Adaptive β-cell proliferation and α- to β-cell transdifferentiation contribute to β-cell regeneration in TIF-IAΔ/Δ model. Bihormonal cells are shown in violet. mo.: months post injection. Scale bar: 25 μm. Values are presented by mean ± SEM. P-value <0.05: *, P-value <0.01 : **, P-value <0.001: ***. N >3 animals for each experimental condition.
Interestingly, the amount of bihormonal α-cells that express β-cell markers was similar to that observed in younger animals. Quantification of bihormonal cells at different time points following β-cell ablation showed a similar fraction of β-cells expressing glucagon at 2- and 6 mo post injection (Fig. 6E). We concluded that although the basal and adaptive proliferation rate of β-cells is reduced by age, massive β-cell depletion in the TIF-IA model appears to still activate, albeit at low frequency, some α-cells to acquire β-cell like cell characteristics.
Discussion
In this study, we used TIF-IA conditional knockout mice to establish a novel model of progressive β-cell ablation allowing the monitoring of β-cell regeneration in the endocrine pancreas. Following 95% ablation, adaptive proliferation of pre-existing β-cells mainly contributes to the recovery of the β-cell mass (Fig. 6F). It appears that, in contrast to the previously described extreme β-cell ablation model,19 the low number of non-ablated β-cells in the TIF-IAΔ/Δ model is sufficient to drive the adaptive proliferation, and contribute significantly to β-cell regeneration.
Using TIF-IAΔ/Δ mice as a regeneration model offers several advantages compared to previously studied models: first, the deletion of TIF-IA induces a protracted endogenous cell death in insulin producing cells that results in a gradual ablation of the β-cell pool, which mimics type 1 diabetes disease progression.23-25 Second, the efficient recombination promoted by tamoxifen induction leads to ablation of most of β-cells, but, at the same time, keeps a number of residual insulin-producing cells intact. This aspect is of particular relevance in light of the finding that such residual β-cells might have a critical function in response to therapeutic approaches.23 Third, since the elevation of blood glucose is detected as early as 10 d upon tamoxifen induction, the regeneration procedure is initiated while islets are still not completely depleted of insulin producing β-cells (Fig. 1D and 2A). The gradual loss and presence of the healthy residual populations of β-cells help to sustain animal survival without exogenous insulin administration, which has been used for animal survival in another reported genetic model of extreme β-cell loss,19 the effect of such treatment on regeneration remaining to be characterized. For instance, in the diphtheria toxin-inducible system,19 all β-cells are lost within 15 days after induction. In such conditions, mice have to be treated with insulin to survive the extreme β-cells loss. This will indeed influence glucose metabolism, which is a key factor in the stimulation of β-cells regeneration.33 Exogenous insulin administration is suggested to reduce β-cell proliferation in mice lacking normal β-cells by decreasing the glucose metabolism rate and individual β-cells workload.33 This may explain why β-cell proliferation never increased neither in the remaining β-cells population immediately after ablation nor after partial increase in β-cell mass.19 In the absence of exogenous insulin treatment, newly formed β-cells could increase their proliferation rate and gain control of regeneration in later stages, as we have seen in our TIF-IAΔ/Δ model.
ChIP- and RNA-sequencing analyses have shown that several genes involved in the regulation of the β-cell identity are bivalently marked by activating and repressing histone modifications in human α-cells.34 These observations confer an innate epigenetic plasticity to α-cells, which allows their reprograming into β-like cells in extreme conditions. This might point to the action of supportive mechanisms of α-cells for β-cell mass maintenance, thereby allowing β-cell regeneration even in extreme conditions, when β-cells are completely depleted. It is essential to understand how a massive β-cell loss can lead to the epigenetic modifications essential for this identity switch. Toward this goal, it is worth mentioning the suggestion that the cytokine(s) and chemokine(s) released from injured β-cells, such as stromal cell factor-1 (SDF-1) and interleukin-6 (IL-6), could mediate α-cell hyperplasia and α-to-β-cell transdifferentiation.35-37
Additionally, we observed a regeneration delay in the mice with older onset of β-cell ablation; however, their regenerative capacity is not altered. While, basal β-cell proliferation rates decline with age, upon challenge, their proliferation rate still rises with a similar fold-change relative to the basal level, which is consistent with earlier reports.32,38 Our findings sustain the notion that the declined proliferation capacity reduces β-cell regeneration in older mice.32,39,40 α−to-β-cell conversion was demonstrated to mainly contribute to β-cell regeneration in condition of extreme β-cell loss19 and in a PDL (pancreatic duct ligation) model combined with alloxan-induced β-cell ablation.20 Moreover, it was shown that adult α-cells can be reprogramed into β-cell upon ectopic overexpression of Pax4 with the similar efficiency at different ages, suggesting that aging does not restrict α-cells plasticity.21,41
The detection of α-cells displaying some β-cell markers, such as insulin, Pdx1, and Nkx6.1, but lacking MafA, suggests that these cells may represent α-cells transitioning toward a β-cell identity. However, a definitive proof of the contribution of these cells to the regenerated β-cell mass will await lineage tracing experiments. It is interesting to notice that the appearance and the low abundance of such cells is age independent in TIF-IAΔ/Δ model. As TIF-IA is expressed ubiquitously in different cell types, we could not incorporate a Cre-LoxP based lineage tracing system into our model. In the future, other types of “hit and run” recombination systems could be used to determine the extent of the contribution from the two cellular sources, that is pre-existing β-cell and α-cells, to the regenerated β-cell mass. In addition, it is conceivable that due to the diabetic condition induced by TIF-IA KO, the observed bihormonal cells may emanate from a dedifferentiation of β-cells into α-cells, as reported by Talchai et al., 2012.42
The contribution of adult progenitor cells to endocrine lineages is still under debate. We could not find any evidence supporting their involvement in β-cell regeneration in TIF-IAΔ/Δ model. Adult progenitors have been shown to play a role in β-cell regeneration, when the pancreas tissue is surgically damaged in pancreatic duct ligation and pancreatectomy models,43 and in Pax4-misexpressing mice. Following Pax4 forced expression in α-cells of transgenic mice, duct-lining cells were shown to contribute to β-cell regeneration.21,22 This mechanism was not observed in this specific β-cell ablation model.
In conclusion, our study indicates that TIF-IAΔ/Δ model can be used in a great range of studies addressing different aspects of β-cell regeneration in type I diabetes.
Research Design and Methods
Ethics statement
All animal works have been conducted according to the German animal welfare law (LAVES Niedersachsen).
Animals
TIF-IAfl/fl;RIPCreERT2;Rosa26YFP/YFP mice were generated by crossing TIF-IAfl/fl line28 with RIPCreERT2 line3 and Rosa26YFP/YFP reporter line.44 One- to 3-month old male mice were used for experiments.
Tamoxifen (Sigma-Aldrich) was injected to mice for 7 d (2 mg/day per 20 gr body weight).
For BrdU labeling experiment, 5-bromo-2′-deoxyuridine (Sigma-Aldrich) was administrated in drinking water (0.8 mg/ml), and changed every 5 d.
For double thymidine analogs experiments, CldU and IdU (Sigma-Aldrich) were given in drinking water each for 4 d (1 mg/ml) with one-day washout interval.
Blood glucose and insulin measurements
Blood glucose levels were measured with AccuChek glucose monitoring system using tail tip blood.
Insulin ELISA was performed using Mercodia Ultrasensitive Mouse Insulin ELISA according to according to manufacturer's instruction.
Tissue preparation and immunohistochemistry
Pancreata were isolated and immediately fixed in 4% paraformaldehyde for 2 hours at 4°C. Fixed tissues were then washed 4 time, 30 minutes each, and incubated overnight in 30% (w/v) sucrose solution in PBS. Tissues were cryopreserved in Jung tissue freezing medium (Leica Microystems, Nussloch, Germany). 8 μm cryosections were air-dried, washed in PBS and then blocked in 10% FCS solutions. For nuclear stainings, tissue sections were permeabilized using 0.5% Triton-X100 treatment for 30 minutes before blocking. Primary antibodies were incubated overnight at 4°C. On the second day, sections were washed with PBS and secondary antibodies were incubated for one hour at room temperature. DNA counterstaining and mounting was done using DAPI (Vectashield).
The following antibodies were used. Guinea-pig anti-insulin and rat anti-Ki67 (Dako), mouse anti-insulin (Sigma), rabbit anti-glucagon, chicken anti-GFP, and rabbit anti-Glut2 (Abcam), rabbit anti-MafA (BETHYL Laboratories), rabbit anti-C-peptide (Cell signaling), mouse anti-BrdU (Roche-applied-science), rabbit anti-Arx and rabbit anti-Sox9 (Millipore), rabbit anti-Pdx1, rabbit anti-Nkx6.1 (kindly provided by C.Wright), and guinea-pig anti-ngn3 (kindly provided by M.Sander). Secondary antibodies were obtained from Invitrogen and Jackson Immunoresearch Laboratories. Images were taken using a Leica SP5 confocal laser-scanning microscope.
For BrdU staining, sections were treated in 2N HCl for 30 minutes at 37°C and then washed in PBS prior to blocking step.
The staining of CldU/IdU labeled tissue was done according to the established protocol5 with minor modifications. Briefly, sections were treated with 1.5 N HCl, blocked with 10% FCS, and incubated overnight with mouse anti-BrdU (BD Biosciences, Franklin Lakes, NJ) and guinea-pig anti-insulin (Dako). The next day, sections were washed with high salt PBST (0.5 M NaCl, 36 mM Tris-HCl pH8.0, 0.5% Tween20) and again incubated overnight with rat anti-BrdU antisera (BU1/75; Accurate Chemical, Westbury, NY). Secondary antibodies were incubated for one hour at room temperature.
TUNEL assay was performed using Apoptag apoptosis detection kit (Millipore) according to manufacturer's protocol.
Quantifications and Statistics
For each experiments, at least 40 islets from 4 equally separated sections were counted per animal. The values are presented as mean ± SEM from at least 3 animals at the same experimental condition. Statistical significance between the mean values was analyzed using ANOVA, Tukey's Multiple Comparison Test, Kruskal-Wallis test, Dunn's Multiple Comparison Test, and Student t-test. P-values less than 0.05 were considered as statistically significant.
Supplementary Material
Acknowledgments
This work was supported by the Max-Planck Society, the Juvenile Diabetes Research foundation (17-2011-16, 2-2010-567, 26-2008-639, 17-2013-426), the INSERM AVENIR program, the INSERM, the European Research Council (StG-2011-281265) the FMR (DRC20091217179), the ANR/BMBF (2009 GENO 105 01/01KU0906), the ‘‘Investments for the Future’’ LABEX SIGNALIFE (ANR-11-LABX-0028-01), Club Isatis, Mr and Mrs Dorato, the Fondation Générale de Santé, and the Foundation Schlumberger pour l’Education et la Recherche.
References
- 1. Ziv O, Glaser B, Dor Y. The plastic pancreas. Dev Cell 2013; 26:3-7; PMID:23867225; http://dx.doi.org/ 10.1016/j.devcel.2013.06.013 [DOI] [PubMed] [Google Scholar]
- 2. Bernard-Kargar C, Ktorza A. Endocrine pancreas plasticity under physiological and pathological conditions. Diabetes 2001; 50 Suppl 1:S30-5; PMID:11272194; http://dx.doi.org/ 10.2337/diabetes.50.2007.S30 [DOI] [PubMed] [Google Scholar]
- 3. Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004; 429:41-6; PMID:15129273; http://dx.doi.org/ 10.1038/nature02520 [DOI] [PubMed] [Google Scholar]
- 4. Georgia S, Bhushan A. Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J Clin invest 2004; 114:963-8; PMID:15467835; http://dx.doi.org/ 10.1172/JCI200422098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Teta M, Rankin MM, Long SY, Stein GM, Kushner JA. Growth and regeneration of adult beta cells does not involve specialized progenitors. Dev Cell 2007; 12:817-26; PMID:17488631; http://dx.doi.org/ 10.1016/j.devcel.2007.04.011 [DOI] [PubMed] [Google Scholar]
- 6. Cano DA, Rulifson IC, Heiser PW, Swigart LB, Pelengaris S, German M, Evan GI, Bluestone JA, Hebrok M. Regulated beta-cell regeneration in the adult mouse pancreas. Diabetes 2008; 57:958-66; PMID:18083786; http://dx.doi.org/ 10.2337/db07-0913 [DOI] [PubMed] [Google Scholar]
- 7. Parsons JA, Brelje TC, Sorenson RL. Adaptation of islets of Langerhans to pregnancy: increased islet cell proliferation and insulin secretion correlates with the onset of placental lactogen secretion. Endocrinology 1992; 130:1459-66; PMID:1537300 [DOI] [PubMed] [Google Scholar]
- 8. Rieck S, Kaestner KH. Expansion of beta-cell mass in response to pregnancy. Trends Endocrin Met: TEM 2010; 21:151-8; PMID:20015659; http://dx.doi.org/ 10.1016/j.tem.2009.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nir T, Melton DA, Dor Y. Recovery from diabetes in mice by beta cell regeneration. J Clin Invest 2007; 117:2553-61; PMID:17786244; http://dx.doi.org/ 10.1172/JCI32959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xu X, D'Hoker J, Stange G, Bonne S, De Leu N, Xiao X, Van de Casteele M, Mellitzer G, Ling Z, Pipeleers D, et al. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell 2008; 132:197-207; PMID:18243096; http://dx.doi.org/ 10.1016/j.cell.2007.12.015 [DOI] [PubMed] [Google Scholar]
- 11. Inada A, Nienaber C, Katsuta H, Fujitani Y, Levine J, Morita R, Sharma A, Bonner-Weir S. Carbonic anhydrase II-positive pancreatic cells are progenitors for both endocrine and exocrine pancreas after birth. Proc Nat Acad Sci U S A 2008; 105:19915-9; PMID:19052237; http://dx.doi.org/ 10.1073/pnas.0805803105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bonner-Weir S, Baxter LA, Schuppin GT, Smith FE. A second pathway for regeneration of adult exocrine and endocrine pancreas. A possible recapitulation of embryonic development. Diabetes 1993; 42:1715-20; PMID:8243817; http://dx.doi.org/ 10.2337/diab.42.12.1715 [DOI] [PubMed] [Google Scholar]
- 13. Chen S, Shimoda M, Chen J, Matsumoto S, Grayburn PA. Transient overexpression of cyclin D2/CDK4/GLP1 genes induces proliferation and differentiation of adult pancreatic progenitors and mediates islet regeneration. Cell Cycle 2012; 11:695-705; PMID:22373529; http://dx.doi.org/ 10.4161/cc.11.4.19120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen S, Bastarrachea RA, Roberts BJ, Voruganti VS, Frost PA, Nava-Gonzalez EJ, Arriaga-Cazares HE, Chen J, Huang P, DeFronzo RA, et al. Successful beta cells islet regeneration in streptozotocin-induced diabetic baboons using ultrasound-targeted microbubble gene therapy with cyclinD2/CDK4/GLP1. Cell Cycle 2014; 13:1145-51; PMID:24553120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xiao X, Chen Z, Shiota C, Prasadan K, Guo P, El-Gohary Y, Paredes J, Welsh C, Wiersch J, Gittes GK. No evidence for beta cell neogenesis in murine adult pancreas. J Clin Invest 2013; 123:2207-17; PMID:23619362; http://dx.doi.org/ 10.1172/JCI66323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kopp JL, Dubois CL, Schaffer AE, Hao E, Shih HP, Seymour PA, Ma J, Sander M. Sox9+ ductal cells are multipotent progenitors throughout development but do not produce new endocrine cells in the normal or injured adult pancreas. Development 2011; 138:653-65; PMID:21266405; http://dx.doi.org/ 10.1242/dev.056499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kopinke D, Brailsford M, Shea JE, Leavitt R, Scaife CL, Murtaugh LC. Lineage tracing reveals the dynamic contribution of Hes1+ cells to the developing and adult pancreas. Development 2011; 138:431-41; PMID:21205788; http://dx.doi.org/ 10.1242/dev.053843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Solar M, Cardalda C, Houbracken I, Martin M, Maestro MA, De Medts N, Xu X, Grau V, Heimberg H, Bouwens L, et al. Pancreatic exocrine duct cells give rise to insulin-producing beta cells during embryogenesis but not after birth. Dev Cell 2009; 17:849-60; PMID:20059954; http://dx.doi.org/ 10.1016/j.devcel.2009.11.003 [DOI] [PubMed] [Google Scholar]
- 19. Thorel F, Nepote V, Avril I, Kohno K, Desgraz R, Chera S, Herrera PL. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature 2010; 464:1149-54; PMID:20364121; http://dx.doi.org/ 10.1038/nature08894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chung CH, Hao E, Piran R, Keinan E, Levine F. Pancreatic beta-cell neogenesis by direct conversion from mature alpha-cells. Stem Cells 2010; 28:1630-8; PMID:20653050; http://dx.doi.org/ 10.1002/stem.482 [DOI] [PubMed] [Google Scholar]
- 21. Al-Hasani K, Pfeifer A, Courtney M, Ben-Othman N, Gjernes E, Vieira A, Druelle N, Avolio F, Ravassard P, Leuckx G, et al. Adult duct-lining cells can reprogram into beta-like cells able to counter repeated cycles of toxin-induced diabetes. Dev Cell 2013; 26:86-100; PMID:23810513; http://dx.doi.org/ 10.1016/j.devcel.2013.05.018 [DOI] [PubMed] [Google Scholar]
- 22. Collombat P, Xu X, Ravassard P, Sosa-Pineda B, Dussaud S, Billestrup N, Madsen OD, Serup P, Heimberg H, Mansouri A. The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into alpha and subsequently beta cells. Cell 2009; 138:449-62; PMID:19665969; http://dx.doi.org/ 10.1016/j.cell.2009.05.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Akirav E, Kushner JA, Herold KC. Beta-cell mass and type 1 diabetes: going, going, gone? Diabetes 2008; 57:2883-8; PMID:18971435; http://dx.doi.org/ 10.2337/db07-1817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cnop M, Welsh N, Jonas JC, Jorns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes 2005; 54 Suppl 2:S97-107; http://dx.doi.org/ 10.2337/diabetes.54.suppl_2.S97 [DOI] [PubMed] [Google Scholar]
- 25. Eisenbarth GS. Type I diabetes mellitus. A chronic autoimmune disease. New Engl J Med 1986; 314:1360-8; PMID:3517648; http://dx.doi.org/ 10.1056/NEJM198605223142106 [DOI] [PubMed] [Google Scholar]
- 26. Bodem J, Dobreva G, Hoffmann-Rohrer U, Iben S, Zentgraf H, Delius H, Vingron M, Grummt I. TIF-IA, the factor mediating growth-dependent control of ribosomal RNA synthesis, is the mammalian homolog of yeast Rrn3p. EMBO Reports 2000; 1:171-5; PMID:11265758; http://dx.doi.org/ 10.1093/embo-reports/kvd032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schnapp A, Pfleiderer C, Rosenbauer H, Grummt I. A growth-dependent transcription initiation factor (TIF-IA) interacting with RNA polymerase I regulates mouse ribosomal RNA synthesis. EMBO J 1990; 9:2857-63; PMID:2390974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yuan X, Zhou Y, Casanova E, Chai M, Kiss E, Grone HJ, Schutz G, Grummt I. Genetic inactivation of the transcription factor TIF-IA leads to nucleolar disruption, cell cycle arrest, and p53-mediated apoptosis. Mol Cell 2005; 19:77-87; PMID:15989966; http://dx.doi.org/ 10.1016/j.molcel.2005.05.023 [DOI] [PubMed] [Google Scholar]
- 29. Kiryk A, Sowodniok K, Kreiner G, Rodriguez-Parkitna J, Sonmez A, Gorkiewicz T, Bierhoff H, Wawrzyniak M, Janusz AK, Liss B, et al. Impaired rRNA synthesis triggers homeostatic responses in hippocampal neurons. Front Cell Neurosci 2013; 7:207; PMID:24273493; http://dx.doi.org/ 10.3389/fncel.2013.00207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Parlato R, Kreiner G, Erdmann G, Rieker C, Stotz S, Savenkova E, Berger S, Grummt I, Schutz G. Activation of an endogenous suicide response after perturbation of rRNA synthesis leads to neurodegeneration in mice. J Neurosci : the official J Soc Neurosci 2008; 28:12759-64; PMID:19036968; http://dx.doi.org/ 10.1523/JNEUROSCI.2439-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Collombat P, Xu X, Heimberg H, Mansouri A. Pancreatic beta-cells: from generation to regeneration. Semin Cell Dev Biol 2010; 21:838-44; PMID:20688184; http://dx.doi.org/ 10.1016/j.semcdb.2010.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stolovich-Rain M, Hija A, Grimsby J, Glaser B, Dor Y. Pancreatic beta cells in very old mice retain capacity for compensatory proliferation. J Biol Chem 2012; 287:27407-14; PMID:22740691; http://dx.doi.org/ 10.1074/jbc.M112.350736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Porat S, Weinberg-Corem N, Tornovsky-Babaey S, Schyr-Ben-Haroush R, Hija A, Stolovich-Rain M, Dadon D, Granot Z, Ben-Hur V, White P, et al. Control of pancreatic beta cell regeneration by glucose metabolism. Cell Metab 2011; 13:440-9; PMID:21459328; http://dx.doi.org/ 10.1016/j.cmet.2011.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bramswig NC, Everett LJ, Schug J, Dorrell C, Liu C, Luo Y, Streeter PR, Naji A, Grompe M, Kaestner KH. Epigenomic plasticity enables human pancreatic alpha to beta cell reprogramming. J Clin Invest 2013; 123:1275-84; PMID:23434589; http://dx.doi.org/ 10.1172/JCI66514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ellingsgaard H, Ehses JA, Hammar EB, Van Lommel L, Quintens R, Martens G, Kerr-Conte J, Pattou F, Berney T, Pipeleers D, et al. Interleukin-6 regulates pancreatic alpha-cell mass expansion. Proc Nat Acad Sci U S A 2008; 105:13163-8; PMID:18719127; http://dx.doi.org/ 10.1073/pnas.0801059105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu Z, Stanojevic V, Avadhani S, Yano T, Habener JF. Stromal cell-derived factor-1 (SDF-1)/chemokine (C-X-C motif) receptor 4 (CXCR4) axis activation induces intra-islet glucagon-like peptide-1 (GLP-1) production and enhances beta cell survival. Diabetologia 2011; 54:2067-76; PMID:21567300; http://dx.doi.org/ 10.1007/s00125-011-2181-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yano T, Liu Z, Donovan J, Thomas MK, Habener JF. Stromal cell derived factor-1 (SDF-1)/CXCL12 attenuates diabetes in mice and promotes pancreatic beta-cell survival by activation of the prosurvival kinase Akt. Diabetes 2007; 56:2946-57; PMID:17878289; http://dx.doi.org/ 10.2337/db07-0291 [DOI] [PubMed] [Google Scholar]
- 38. Gunasekaran U, Hudgens CW, Wright BT, Maulis MF, Gannon M. Differential regulation of embryonic and adult beta cell replication. Cell Cycle 2012; 11:2431-42; PMID:22659844; http://dx.doi.org/ 10.4161/cc.20545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rankin MM, Kushner JA. Adaptive beta-cell proliferation is severely restricted with advanced age. Diabetes 2009; 58:1365-72; PMID:19265026; http://dx.doi.org/ 10.2337/db08-1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gunasekaran U, Gannon M. Type 2 diabetes and the aging pancreatic beta cell. Aging 2011; 3:565-75; PMID:21765202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pfeifer A, Courtney M, Ben-Othman N, Al-Hasani K, Gjernes E, Vieira A, Druelle N, Avolio F, Faurite B, Mansouri A, et al. Induction of multiple cycles of pancreatic beta-cell replacement. Cell Cycle 2013; 12:3243-4; PMID:24036539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 2012; 150:1223-34; PMID:22980982; http://dx.doi.org/ 10.1016/j.cell.2012.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pan FC, Bankaitis ED, Boyer D, Xu X, Van de Casteele M, Magnuson MA, Heimberg H, Wright CV. Spatiotemporal patterns of multipotentiality in Ptf1a-expressing cells during pancreas organogenesis and injury-induced facultative restoration. Development 2013; 140:751-64; PMID:23325761; http://dx.doi.org/ 10.1242/dev.090159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biology 2001; 1:4; PMID:11299042; http://dx.doi.org/ 10.1186/1471-213X-1-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.