

Abstract
Protein translation is an essential cellular process initiated by the association of a methionyl–tRNA with the translation initiation factor eIF2. The Met-tRNA/eIF2 complex then associates with the small ribosomal subunit, other translation factors and mRNA, which together comprise the translational initiation complex. This process is regulated by the phosphorylation status of the α subunit of eIF2 (eIF2α); phosphorylated eIF2α attenuates protein translation. Here, we report a consanguineous family with severe microcephaly, short stature, hypoplastic brainstem and cord, delayed myelination and intellectual disability in two siblings. Whole-exome sequencing identified a homozygous missense mutation, c.1972G>A; p.Arg658Cys, in protein phosphatase 1, regulatory subunit 15b (PPP1R15B), a protein which functions with the PPP1C phosphatase to maintain dephosphorylated eIF2α in unstressed cells. The p.R658C PPP1R15B mutation is located within the PPP1C binding site. We show that patient cells have greatly diminished levels of PPP1R15B–PPP1C interaction, which results in increased eIF2α phosphorylation and resistance to cellular stress. Finally, we find that patient cells have elevated levels of PPP1R15B mRNA and protein, suggesting activation of a compensatory program aimed at restoring cellular homeostasis which is ineffective due to PPP1R15B alteration. PPP1R15B now joins the expanding list of translation-associated proteins which when mutated cause rare genetic diseases.
Introduction
Protein translation is the final step in the genetic expression program, wherein mRNA transcripts are decoded to produce proteins. Translation begins with the association of a methionyl-tRNA with GTP-bound eukaryotic translation initiation factor 2 (a trimer consisting of eIF2α, eIF2β and eIF2γ subunits) (1,2). The methionyl-tRNA/eIF2-GTP complex (termed ternary complex) then binds a 40S ribosomal subunit which, with the help of a host of other translation initiation factors, assembles on the 5′ end of an mRNA which it scans to find the appropriate start codon and begin polypeptide synthesis (1,2). In stress conditions, eukaryotic cells conserve resources by attenuating protein translation. One such mechanism involves a number of stress-induced kinases that phosphorylate eIF2α at serine 51 (p-eIF2α), blocking the production of eIF2-GTP (1,2). Once the stress is resolved, PPP1R15A/GADD34 is induced to restore translation by dephosphorylating eIF2α (3). Unstressed cells also contain basal levels of p-eIF2α, and this is countered by constitutively expressed PPP1R15B/CreP (4). PPP1R15A and PPP1R15B function by recruiting the phosphatase PPP1C to eIF2 (3–6).
For years, it was assumed that translational dysfunction early in development was not compatible with life; however, a growing number of rare disease mutations have been identified in translational constituents, countering this assumption (7). Examples of translation machinery genes associated with disease include the translation factor eIF2B [leukoencephalopathy with vanishing white matter (VWM)] (8,9), 40S ribosomal subunits RPS19 and RPS24 (Diamond–Blackfan anemia) (10,11), ribosome biogenesis genes SBDS and RMPR (Shwachman–Diamond and cartilage-hair hypoplasia, respectively) (12,13) and tRNA maturation and synthesis genes TRNT1, GARS, YARS, KARS and AARS (SIFD syndrome, Charcot–Marie–Tooth disease) (14–18). Given the large number of proteins involved in translation, and the relatively new advent of whole-exome and genome sequencing, it is clear we are just beginning to realize the spectrum of disease-causing mutations impacting this essential biological process.
Here, we report two children from a consanguineous family with a novel autosomal recessive disorder characterized by microcephaly, short stature, hypoplastic brainstem and cord, delayed myelination and intellectual disability. Whole-exome sequencing revealed a homozygous missense mutation in the PPP1R15B gene, and studies in patient cells revealed greatly decreased PPP1R15B–PPP1C interactions, which resulted in increased basal levels of p-eIF2α and resistance to cellular stress, and elevation of PPP1R15B mRNA and protein, suggesting activation of an ineffective compensatory response. Our findings add PPP1R15B to the list of translation pathway components which when mutated cause rare genetic diseases.
Results
Patient description
The female proband was born to second cousin parents following a pregnancy with exposure to maternal smoking and H1N1. Intrauterine growth retardation was detected prenatally at 5 months of gestation. She was born at 36 weeks 1 day of gestational age, weighing 1.64 kg (−3.1 SD) and measuring 38.1 cm (−6.1 SD) in length with a head circumference of 28.5 cm (−5.0 SD). There were no neonatal complications. She was assessed by the Medical Genetics Service at 12 months of chronological age and noted to have significant developmental delay, distinctive facial features and severe symmetric growth retardation (Fig. 1A): head circumference of 37.3 cm (−6 SD), length 58.6 cm (−5.0 SD) and weight of 4.63 kg (−7.3 SD). TSH was increased: 9.53 mmol/l (normal: 0.5–5.5 mmol/l). Despite her hypothyroidism being adequately controlled at 22 months of age, she still had severe symmetrical growth retardation: head circumference of 40.8 cm (−5 to −6 SD), length of 68.9 cm (−4.5 SD) and weight of 7.33 kg (−5.3 SD, 50th percentile for 5–1/2-month-old girl). At her last clinical assessment at 5 years and 2 months of age, she continued to show significant delays in growth and development: head circumference of 43.1 cm (−5 to −6 SD), height of 87.5 cm (−5.0 SD) and weight of 10.95 kg (−5.2 SD). She was interactive, friendly and socially appropriate. She had ∼25 words which she could put together in short sentences, though speech was dysarthric. She had been formally evaluated by a speech pathologist at the age of 5 years, at which time her language skills were approximately equivalent to a 3 year old. Cranial nerve exam was normal with normal extra-ocular movements, saccades and pursuit. There was no oculomotor apraxia. She had decreased axial tone with spasticity in the lower extremities more than upper extremities. Her distal strength was weak, plantars were downgoing, and reflexes brisk (3+). Sensation appeared intact. Her feet were everted. She was able to run, go up and down stairs and jump, though she had a wide-based ataxic gait, with truncal instability accentuated by turning. Gait was deteriorating by report from her parents. She had a low amplitude, high frequency, kinetic tremor in both hands and mild dysmetria. Facial features were distinctive (Fig. 1A).
Figure 1.

Clinical features of this novel syndrome. (A) Female proband at 4 years 4 months of age. Distinctive facial features included a tall narrow face with bitemporal narrowing, full cheeks and micrognathia. She had hypotelorism, blue sclerae, a right-sided epicanthal fold and synophrys. Her nose was short and upturned, with a short philtrum, and she had a small mouth with downturned corners. Ears were large. Hair was fine and somewhat sparse, and shoulders were rounded and sloping. (B) MRI images of the female proband performed at 3 years (left) and 5 years (middle and right) of age. Left image displays axial T2WI, which showed delayed myelination characterized by the presence of a very thin low Signal Intensity (SI) in the Posterior Limb of the Internal Capsule (PLIC) and Anterior Limb of the Internal Capsule (ALIC) (arrow), as well as high T2WI SI in the subcortical white matter. By this age, in normal individuals, the whole of the white matter should be of low T2WI, including thick low T2WI SI of the ALIC and PLIC. Middle axial T2WI shows interval progression of the myelination pattern but persistent thin low SI of the PLIC and ALIC (arrow). Right sagittal T2WI shows a thin corpus callosum (small arrow), decreased volume of the brainstem (arrow head) and very thin cord. The size of the cerebellum in relation to the size of the cerebrum is smaller, and the ventricular system appears mildly prominent, denoting low white matter volume. (C) Male sibling at 1 year 9 months of age resembles his sister in appearance. Distinctive facial features included a round face, full cheeks and micrognathia. He had hypotelorism, down-slanting palpebral fissures and bilateral epicanthal folds. His nose was longer than his sister’s, and he had a long philtrum with a small mouth and a thin upper vermillion border. Ears were large. Hair was fine and sparse, and shoulders were rounded and sloping. (D) MRI images of the male sibling and 15 months (left) and 3 years (middle and right) of age. Left axial T2WI shows delayed myelination characterized high T2WI SI in the ALIC (black arrow) and genu of the Corpus Callosum (CC) (arrow head), structures that should have low T2WI SI by this age in normal individuals. The PLIC is very thin as well. The cerebral sulci appear mildly prominent but the sulci are preserved. Middle axial T2WI shows persistent high T2WI SI of the ALIC (arrow), and thinning of the CC. In comparison with MRI done at 15 months of age, myelination had progressed but remains delayed. Right sagittal T1WI shows diffusely decreased volume of the brainstem (long arrow) and cord (arrowhead), as well as thinning of the corpus callosum (short arrow). The size of the cerebellum in relation to the size of the cerebrum is smaller, and the ventricular system appears mildly prominent, denoting low white matter volume.
Investigations for this patient included a normal echocardiogram, normal sweat chloride, normal chromosomal breakage studies with mitomycin C and DEB and a microarray on a 180 k oligonucleotide platform which did not reveal any microdeletions/microduplications. CBC was unremarkable: normal hemoglobin, MCV of 85.5 (normal range: 71–85), and reticulocyte count at 29.6 (normal range: 22–92). A skeletal survey showed significantly delayed bone age but no features to suggest a skeletal dysplasia. Brain MRI myelination corresponded to 18–21 months when she was 35 months of age (Fig. 1B) and review of these images identified a small brainstem. A repeat brain MRI at 5 years 3 months of age showed myelination was still delayed but had progressed since the last study (Fig. 1B). However, the brainstem was small and there was decreased cerebellar volume, particularly in the superior vermis. The volume of the cervical cord was also diminished. Additional findings included small hyperintense T2/FLAIR signal intensity in subcortical white matter and a thin corpus callosum. There was no cortical dysplasia.
The male sibling was born following a pregnancy with exposure to maternal smoking and potential alcohol consumption (approximately every 2 weeks, quantity unknown). He was born at term, weighing 2.255 kg (−2.0 SD) and measuring 46 cm (−1.5 SD) in length with a head circumference of 28.5 cm (−4.7 SD). There were no neonatal complications. He was first assessed by the Medical Genetics service at 15 months of age when he presented with developmental delay and growth failure: head circumference of 37 cm (−6 to −7 SD), length of 69 cm (−3.7 SD) and weight of 5.77 kg (−6.4 SD). He could roll over, but not sit independently, and was not able to eat solids or purees. He could mimic sounds, but did not have any clear words. At 1 year 9 months of age, severe developmental and growth delays persisted: head circumference of 39.6 cm (−7 SD), length of 73.4 cm (−3.6 SD) and weight of 6.94 kg (−5.6 SD). He had distinctive facial features (Fig. 1C) which, while similar to his sister, were more striking. At 2 years 10 months of age, significant developmental and growth delay persisted and he drooled excessively: head circumference of 41.5 cm (−6 to −7 SD), height of 81.5 cm (−3.6 SD) and weight of 8.5 kg (−5.3 SD). He could take four steps without assistance, walk with a walker and climb on the sofa. He had 4–5 words and would point to things, but did not use sign language.
At 3 years 8 months of age, he was able to walk but fell approximately 8 times per day. He could run, but not jump or climb stairs. Physiotherapy had evaluated his fine motor skills as equivalent to a 9–12-month-old. Significant delay in growth continued: head circumference of 42.3 cm (−6 to −7 SD), height of 86.2 cm (−3.5 SD) and weight of 9.71 kg (−5.0 SD). Facial features were distinctive with some resemblance to his sister. He had a pectus excavatum with strikingly visible veins and spine showed loss of lumbar lordosis. He had tapering fingers and long feet. Neurological examination revealed a friendly child. Cranial nerves were normal. Appendicular and axial tone were low throughout. Distal strength appeared decreased in upper and lower extremities compared with proximal strength. He had an ulnar grasp. Plantars were upgoing bilaterally. Gait was ataxic with everted feet. There was some truncal ataxia. No tremor was obvious and there was no dysmetria when reaching for objects. Brain MRI done at 3 years 8 months of age showed significantly delayed myelination pattern equivalent to 2 years of age (Fig. 1D). A prior MRI performed at an outside facility when the patient was 13 months of age was reviewed and showed a myelination pattern corresponding to ∼6 months of age (Fig. 1D). Therefore, the patient's myelination had progressed between the two studies, but not at a normal rate. Similar to his sister, the MRI also showed a small brainstem and decreased cerebellar volume, a thin corpus callosum and diminished volume of the cervical cord. CBC showed a normal red blood cell count, but a slightly low hemoglobin of 102 (normal range 105–135), in the context of a normal MCV. A reticulocyte count had previously been measured and was normal. A microarray had previously shown a maternally inherited duplication of uncertain significance, though his mother is of normal stature. This microarray also showed three blocks of loss of heterozygosity of >10 Mb, one of which includes the PP1R15B gene. Both siblings were enrolled in the Care4Rare Canada research study due to their family history and lack of a molecular diagnosis.
Whole-exome sequencing identified homozygous missense mutation in PPP1R15B
Given the family history of consanguinity, we predicted this rare disease was caused by a homozygous mutation. Exome sequencing was performed on DNA from both affected children to search for shared homozygous rare variants. Average coverage for the exomes was 152× and 170× for each child, respectively, and 97% of CCDS exons in both exomes were covered at >10×. After excluding common variants (≥5% minor allele frequency represented in the NHLBI exome server or the 1000 genome phase 1 data set, or ∼2000 internal exomes), two shared homozygous missense variants were identified in the genes PPP1R15B and AQR. The AQR variant was located in the 5′ UTR and had been reported with a heterozygous frequency of 0.3% in the 1000 Genomes Project (rs200116349), whereas the PPP1R15B variant, c.1972G>A;p.R658C (NM_032833.4), had not been annotated in any databases to date. We also searched for shared multiple heterozygous variants in the same gene and identified two rare variants in KRTAP9-9. These variants affect non-conserved residues and were predicted to be non-damaging; upon further investigation, it was found these variants are located on the same allele in cis. Thus, there were no shared compound heterozygous mutations to possibly explain this phenotype identified using WES.
Based on WES data analysis, the PPP1R15B variant was the most promising candidate. PPP1R15B position 1972/residue 658 is highly conserved (GERP 4.19), and the p.R658C variant was predicted to be ‘probably damaging’ by SIFT (19) and Polyphen-2 (20) (SIFT 0, Polyphen-2 1.0). In the ExAC database, only seven individuals have been documented with a PPP1R15B loss-of-function variant, none of which are homozygous and residue 658 has only one documented heterozygous variant in one individual, p.R658H. PPP1R15B functions by association with PPP1C through a binding site comprising residues 513–713 (4); the familial variant is located within this critical domain. PCR followed by Sanger sequencing confirmed the variant in both siblings (Fig. 2A). An evaluation of the literature revealed that Ppp1r15bnull mice have similar phenotypes to our patients, they are small and fail to thrive; these mice also have defects in erythropoiesis and die shortly after birth (21). The Ppp1r15bnull phenotype is rescued by a concomitant mutation, S51A, which blocks phosphorylation of eIF2αS51, implicating the PPP1C–PPP1R15B partnership to dephosphorylate eIF2α as responsible for the phenotypic presentation in this mouse model (21). Taken together, these findings indicated that the homozygous missense variant in PPP1R15B is likely the cause of this novel autosomal recessive condition and prompted us to further investigate the functional impact.
Figure 2.
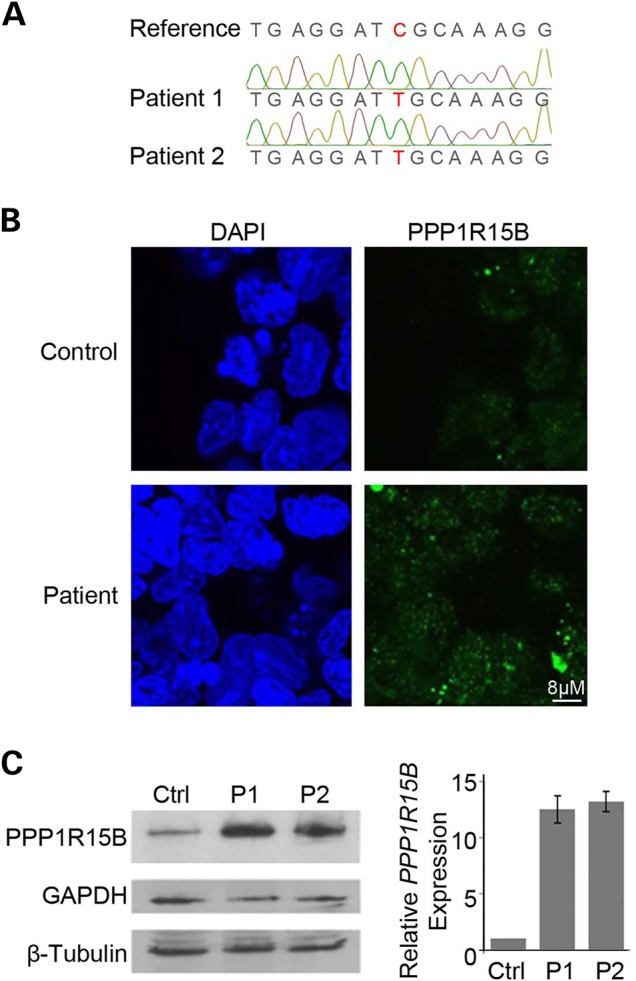
PPP1R15B is mutated, upregulated and correctly localized in patient cells. (A) Sanger sequencing demonstrating confirmation of the c.1972G>A;p.R658C PPP1R15B variant (NM_032833.4) complimentary strand shown. (B) Immunodetection of PPP1R15B in control and patient lymphoblast cell lines demonstrated no overt changes in PPP1R15B localization. (C) Western blot (left) and real-time PCR analysis (right) on extracts from control and patient cells showing elevated levels of PPP1R15B transcript and protein.
PPP1R15B is more abundant and properly localized in patient cells
We began evaluating impact of the PPP1R15B R658C mutation by assessing PPP1R15B localization in control and patient lymphoblast cells. Immunofluorescence staining demonstrated that PPP1R15B was localized throughout the cell as punctate foci in both control and patient cells. These foci appeared similar in shape and size, but brighter in patient cells (Fig. 2B). This suggested patient cells had increased PPP1R15B abundance with correct cellular localization. Western blot analysis on extracts from patient and control cells confirmed elevated levels of PPP1R15B in patient cells (Fig. 2C). To determine whether the increase in PPP1R15B originated at a transcript level, we performed real-time PCR and found a similar increase in transcript abundance (Fig. 2C). We conclude that PPP1R15B is elevated but properly localized in patient cells. This suggests patient cells have activated a pathway-driven response to compensate for defective PPP1R15B or that the patient mutation increases transcript stability. This increase of the affected gene product is intriguing and, to our knowledge, not commonly observed with recessive conditions
Patient mutations in eIF2α abolish interactions with PPP1c leading to increased levels of eIF2α phosphorylation and stress resistance
PPP1R15B partners with PPP1C to maintain levels of global protein translation. The PPP1R15B mutation observed in the siblings is located within the PPP1R15B–PPP1C binding site, suggesting it may affect this interaction. Reciprocal co-immunoprecipitation experiments showed a strong interaction of PPP1R15B with PPP1C in control lymphoblast cells and very little interaction with non-specific IgG, indicating the specificity of the immunoprecipitation reactions (Fig. 3A). We found that PPP1R15B–PPP1C binding was virtually abolished in patient cells, indicating that the p.R658C protein change significantly influences this interaction (Fig. 3A).
Figure 3.

The p.R658C PPP1R15B mutation substantially interferes with the PPP1C interaction, leading to an increase in p-eIF2α and protection from cellular stress. (A) PPP1R15B and PPP1C were immunoprecipitated from control and patient lymphoblast extracts, and western blot analysis performed for the reciprocal protein. PPP1R15B forms a strong interaction in control cells, which is lost with the cells harboring the p.R658.C PPP1R15B mutation. Control reactions were done with IgG. (B) Western blot analysis of extracts from control and patient lymphoblasts demonstrates elevated levels of p-eIF2α. Total eIF2α and PPP1R15A were unchanged. (C) Real-time RT–PCR analysis of PPP1R15A mRNA expression in control and patient cells relative to controls, corrected to GAPDH. Graphed data represent the mean relative expression level, and error bars depict standard deviation from three technical replicates. Expression of PPP1R15A is unaltered. (C) Cell viability of patient and control lymphoblast cultures following exposure to oxidative (H2O2) or ER (tunicamyacin) stress for 48 h as measured by trypan blue dye exclusion. Patient cells display increased viability following 5 µm H202, 10 µm H202 and tunicamyacin treatments. One hundred cells were counted for each experiment; graphed data represent the mean of three technical replicates and error bars depict standard deviation.
The PPP1R15B–PPP1C complex functions to dephosphorylate eIF2α. As the patient cells have lost this interaction, we predicted this would have adverse effects on p-eIF2α control. Western blot analysis confirmed highly elevated basal levels of p-eIF2α in patient cells compared with controls, whereas levels of total eIF2α were unaltered (Fig. 3B). In mice, a lack of PPP1R15B and associated increased p-eIF2α has been shown to trigger compensatory expression of PPP1R15A (21). Western blot and real-time PCR analysis showed that this does not occur in patient cells (Fig. 3B and C). This discrepancy may be due to the fact that Ppp1r15bnull mice have a complete lack of protein whereas our patients have only a point mutation, although the mechanics of PPP1R15B compensation remain to be elucidated. Overall, it is clear that the c.1972G>A;p.R658C PPP1R15B mutation in these patients leads to defective PPP1R15B function.
Several in vitro studies have shown that elevated levels of p-eIF2α provide resistance to cellular stress. To test whether increased p-eIF2α levels in patient cells correlate with increased stress resistance, we treated them with 2.5, 5.0 or 10.0 µm hydrogen peroxide (induces oxidative stress) or 2 µg/ml tunicamyacin (induces endoplasmic reticulum stress) for 48 h. Treatments and duration were chosen based on previous literature (21,22). Cells were stained with tryphan blue and counted using a hemocytometer; 100 cells were counted from each population in triplicate. We found that patient cells exhibited an approximately 20% increase in survival when treated with 5 and 10 µm hydrogen peroxide, as well as 2 µg/ml tuncamyacin (Fig. 3D). Both patient and control cell lines were unaffected by 2.5 µm hydrogen peroxide, as evidenced by the lack of cellular death with this treatment (Fig. 3D).
Finally, given the persistent p-eIF2α and stress resistance capacity, we wondered whether the patient cells had altered rates of cellular growth and protein translation. To assess cellular growth, cells were plated at 100 000 cells per well in triplicate and counted daily with a flow cytometer for 6 days. As expected, control cells grew well for the first 4 days and then plateaued as nutrients were depleted from the media (Supplementary Material, Fig. S1). The growth curve for patient cells showed a trend of slowed initial growth which continued despite nutrient depletion (Supplementary Material, Fig. S1). It is noted that some variability is seen between technical replicates, likely due to the artificial system used; however, the trends observed were consistent and data from Days 5 and 6 further support the aforementioned stress resistance capacity. Protein synthesis was measured by 35S-methionine/cysteine incorporation assay, which revealed no overt change in translation, likely reflecting a compensatory adaptation to persistent p-eIF2α or selective effect on specific transcripts. Together these results show that the PPP1R15B mutation abolishes PPP1R15B–PPP1C interactions, resulting in elevated levels of p-eIF2α, decreased cell growth rates and an associated resistance to cellular stress.
Discussion
We have identified a novel autosomal recessive disease characterized by microcephaly, short stature, delayed myelination and intellectual disability in two siblings. Both children carry a homozygous missense mutation in the gene encoding the translational regulator PPP1R15B, in the PPP1C binding site. Molecular profiling of patient cells showed that the PPP1R15B mutation adversely affects protein function, providing substantial support for the causality of this novel variant.
Mice have been engineered, which lack global PPP1R15A and/or PPP1R15B (21,23). Ppp1r15anull mice are indistinguishable from wild-type littermates (23). Ppp1r15bnull mice exhibit severe growth retardation, impaired erythropoiesis and die by postnatal day one (21). Combined Ppp1r15a/Ppp1r15bnull mice are early embryonic lethal (21). Notably, the phenotypes in Ppp1r15a/Ppp1r15bnull and Ppp1r15bnull mice are rescued by the introduction of a concomitant mutation in eIF2α, which prevents phosphorylation (S51A), suggesting that misregulated phosphorylation is predominantly responsible for the phenotypes observed (21). Our patients show similar defects in eIF2α phosphorylation and growth deficiency. Interestingly, our patients do not display an erythropoietic phenotype, though it is noted a bone marrow analysis has not been conducted to rule out subtle effects at this level. This variance may be due to differences in expression patterns, molecular pathways for erythropoiesis or compensatory factors between mice and humans. Notably, PPP1R15B has a documented role in membrane transport, which is independent of PPP1C (24). Given the mouse data and location of our patient mutation, it is unlikely this aspect of PPP1R15B function is a major contributor to the disease presentation in the family reported here.
There is an expanding list of factors involved in protein translation that when mutated cause rare genetic diseases. These include proteins involved in translation initiation, ribosomal components, tRNAs and amino-acyl tRNA synthases (7). Given the critical importance of translation, one might expect that each of these mutations would result in severe multi-systemic disorders. However, the diseases documented thus far are varied in terms of tissues affected and severity. One potential explanation for this is that many of these proteins have additional functions beyond translation and that disease mutations primarily affect alternate functions. For example, mutations in eIF2B, a binding partner for eIF2α in translation initiation, cause leukoencephalopathy with VWM, a childhood onset autosomal recessive neurodegenerative disorder (8,9). Studies on VWM patient cells have shown that eIF2B activity is only decreased by 20–30% (25) and that this decrease does not affect rates of protein synthesis (26). It was proposed that eIF2B activity does not limit translation and that an alternative yet undiscovered eIF2B function is predominantly responsible for the VWM phenotype. Another explanation may exist in tissue-specific expression patterns and/or susceptibility of some cell types. For example, mutations in PERK, an endoplasmic reticulum stress-induced eIF2α kinase, cause a form of infantile-onset diabetes mellitus known as Wolcott–Rallison syndrome (27). The predominantly pancreatic phenotype has been attributed to high expression levels of PERK in secretory cells of the pancreas (28). In the absence of PERK function, it is believed pancreatic β-cells produce an overabundance of insulin which leads to stress-induced apoptosis (28). Alternate cell types have significantly less PERK expression indicating decreased dependence on this kinase. Finally, while no disease examples have been documented thus far, it is plausible that reserve systems able to compensate for defects in the translation pathway are triggered in specific cell types, sparing them from deleterious effects. Clearly mutations in the translation pathway can have diverse effects; our report now expands this list to include severe microcephaly, short stature, hypoplastic brainstem and cord, delayed myelination and intellectual disability.
In summary, we have used whole-exome sequencing to identify a homozygous missense mutation in PPP1R15B in two children with a novel syndrome characterized by severe microcephaly, short stature, hypoplastic brainstem and cord, delayed myelination and intellectual disability. While independent data from further families are required to confirm the link between this phenotype and PPP1R15B variants, the molecular data strongly support this connection. Our findings expand the list of rare diseases caused by dysregulation of translational machinery, while providing insight into the role of PPP1R15B and the translation initiation pathway in human development.
Materials and Methods
Patients
A family with two children with extreme microcephaly, short stature and developmental delay presented to the Medical Genetics service for evaluation. Parents were second cousins. The family was enrolled in the Care4Rare Canada research study due to the absence of a molecular diagnosis. Approval of the study design was obtained from the institutional research ethics board (Children's Hospital of Eastern Ontario), and informed consent was obtained.
Whole-exome sequencing, rare variant identification and variant validation
Exonic DNA was selected using the Agilent SureSelect 50 Mb (V5) All Exon Kit, then sequenced on an Illumina HiSeq 2000. Read alignment, variant calling and annotation were done as outlined for previous FORGE and Care4Rare Canada projects (29) with a pipeline based on Burrows–Wheeler Aligner (30), Picard (31), ANNOVAR (32) and custom annotation scripts. Variants were disregarded if they were present at ≥5% in the 1000 genome phase 1 data set (April 2012 release), the 6500 exomes of the National Institute of Health Heart, Lung and Blood Institute, GO Exome Sequencing Project (Seattle, WA, USA, data downloaded March 10, 2012) and seen in >30 samples from our in-house database (∼2000 exomes previously sequenced at the McGill University and Genome Quebec Innovation Centre). PCR amplification followed by bi-directional Sanger sequencing was used to validate mutations identified by WES and to evaluate segregation of variants in the family. Primers: Fwd AGCATAAAGGGTTATTGACCTC and Rev CAAAGGACAGCTGCCAAG.
Real-time PCR analysis
Immortalized patient lymphobast cell lines were established from blood samples at The Centre for Applied Genomics (Toronto, Canada). Total RNA was obtained from patient and control lymphoblast cell lines with the RNeasy Mini kit (QIAGEN) and reverse-transcribed into complementary DNA (cDNA) with iScript kit (BioRad Laboratories) according to manufacturer's instructions. Control reactions without reverse transcriptase were prepared in parallel. cDNA was amplified with gene-specific primers and iQ SYBR Green mastermix under the following conditions: 35 cycles of 95°C for 10 s, 55°C for 20 s, 72°C for 30 s and a final melting curve generated in increments of 0.5°C per plate read on a CFX96 Touch Real-time PCR Detection System (BioRad Laboratories). Gene expression was quantified using the standard Ct method with CFX software (BioRad Laboratories), and all data corrected against GAPDH as an internal control. Primers: GADD34_Fwd AGGAAGAGGAAGCTGCTGAG, GADD34_Rev AATGGACAGTGACCTTCTCG; PPP1R15B_Fwd GTTGGGGAGCCAAGAAAGTG, PPP1R15B_Rev TGGTCCTTTGCGATCCTCAT.
Western blot analysis
Western blot analysis was conducted to assess protein levels in patient-derived lymphoblast cells. Cells were lysed in radioimmunoprecipitation assay buffer containing 10 mg/ml each of aprotinin, phenylmethanesulfonyl fluoride and leupeptin (all from Sigma) for 20 min at 4°C, followed by centrifugation at 13 000×g for 15 min and retrieval of supernatants. Total protein concentrations were determined by Bradford protein assay (BioRad Laboratories). Protein samples were resolved by 11% SDS–PAGE, transferred onto nitrocellulose membrane and incubated in blocking solution (Tris-buffered saline (TBS), 5% non-fat milk, 0.05% Tween-20) for 1 h at room temperature followed by overnight incubation with primary antibody at 4°C (PPP1R15B, Proteintech 14634-1-AP; GADD34, Santa Cruz Biotechnology, Inc. sc-8327; GAPDH, ImmunoChemical, Inc. 200-901-BJ4; β-Tubulin and PPP1c, Abcam ab6046 and ab16387; p-eIF2α and total eIF2α, Cell Signalling 9721 and 9722). Membranes were washed with TBS and 0.5% Tween-20 three times followed by incubation with secondary antibody (HRP-conjugated anti-rabbit or anti-mouse; Cell Signalling) for 1 h at room temperature. Blots were visualized by autoradiography using the Clarity Western ECL substrate (BioRad Laboratories).
Co-immunoprecipitation
Whole protein lysates were extracted as described earlier. 200 µg was diluted to 500 µl with 0.02% Tween-20 in phosphate-buffered saline (PBS) and incubated overnight with PPP1C (Abcam) or PPP1R15B (Proteintech) antibodies rotating at 4°C. Normal rabbit IgG (Santa Cruz Biotechnology, Inc.) was used as a negative control. Samples were then incubated with Dynabeads protein G (Invitrogen) for 1 h at 4°C. Immunoprecipitates were washed three times with 1 ml 0.04% Tween-20 in PBS, eluted and resolved on 11% SDS–PAGE. Western blot analysis was conducted as described earlier. Input samples represent 5% of total input.
Immunofluorescence and confocal microscopy
For immunofluorescence detection, cells were fixed in 4% paraformaldehyde, permeated in PBS with 0.3% TX-100 and incubated with PP1R15B antibody (1:500, Proteintech) for 1 h at room temperature. Cells were washed three times in PBS with 0.3% TX-100 and then incubated with goat α-rabbit Alexa 488 (1:1000, Millipore), washed again, stained with DAPI and mounted with Vectashield H-1000 (Vector Laboratories). Images were acquired using the Olympus FV1000 confocal microscope and FV10-ASW 2.1 image acquisition software (Olympus).
Cell viability assays
Cells were cultured in media supplemented with 0, 2.5, 5.0 or 10.0 µm hydrogen peroxide or 2 µg/ml tunicamyacin for 48 h. Cells were stained with tryphan blue (Life Technologies) and counted using a hemocytometer, 100 cells were counted from each population and the experiment was repeated in 3 technical replicates.
Cellular growth curves
Cells were plated at 100 000 cells per well in triplicate. Each subsequent day a well was pelleted and resuspended in 1 ml media with CountBright absolute counting beads (Life Technologies) and bead and cell counts measured at the University of Ottawa Flow Cytometry Core Facility on a Beckman Coulter Cyan ADP9. Absolute cell counts were calculated using cell:bead ratio according to manufacturer's instructions.
Protein translation assay
Cells were cultured in DMEM with 10% FBS and 10 µl/ml 35S-methionine/cysteine (EasyTag 35S labeling mix, PerkinElmer) for 25 min at 37°C. Cells were washed, proteins extracted and resolved by SDS–PAGE and then comassie-stained and exposed to X-ray film. Experiment was repeated in two technical replicates.
Supplementary Material
Funding
This work was supported by the Care4Rare Canada Consortium (Enhanced Care for Rare Genetic Diseases in Canada) funded by Genome Canada, the Canadian Institutes of Health Research, the Ontario Genomics Institute, Ontario Research Fund, Genome Quebec and Children's Hospital of Eastern Ontario Foundation. Funding to pay the Open Access publication charges for this article was provided by Care4Rare Canada Consortium which is funded by Genome Canada, Canadian Institute of Health Research, Ontario Genomics Institute, Ontario Research Fund, Genome Quebec and Children's Hospital of Eastern Ontario Foundation.
Supplementary Material
Acknowledgements
The authors would first and foremost like to thank the study participants and their family. We would also like to acknowledge the contributions of the high-throughput sequencing platform of the McGill University and Genome Quebec Innovation Centre, Montreal, Canada and the Care4Rare project coordinators Taila Hartley and Chandree Beaulieu. The project was selected for analysis by the Care4Rare Consortium Gene Discovery Steering Committee consisting of Kym Boycott (lead; University of Ottawa), Alex MacKenzie (co-lead; University of Ottawa), Jacek Majewski (McGill University), Michael Brudno (University of Toronto), Dennis Bulman (University of Ottawa) and David Dyment (University of Ottawa). M.T. received a post-doctoral fellowship from the Réseau de médecine génétique appliquée.
Conflict of Interest statement. None declared.
References
- 1.Hinnebusch A.G., Lorsch J.R. (2012) The mechanism of eukaryotic translation initiation: new insights and challenges. Cold Spring Harb. Persp. Biol., 4, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kapp L.D., Lorsch J.R. (2004) The molecular mechanics of eukaryotic translation. Ann. Rev. Biochem., 73, 657–704. [DOI] [PubMed] [Google Scholar]
- 3.Novoa I., Zeng H., Harding H.P., Ron D. (2001) Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J. Cell Biol., 153, 1011–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jousse C., Oyadomari S., Novoa I., Lu P., Zhang Y., Harding H.P., Ron D. (2003) Inhibition of a constitutive translation initiation factor 2alpha phosphatase, CReP, promotes survival of stressed cells. J. Cell Biol., 163, 767–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Connor J.H., Weiser D.C., Li S., Hallenbeck J.M., Shenolikar S. (2001) Growth arrest and DNA damage-inducible protein GADD34 assembles a novel signaling complex containing protein phosphatase 1 and inhibitor 1. Mol. Cell. Biol., 21, 6841–6850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kojima E., Takeuchi A., Haneda M., Yagi A., Hasegawa T., Yamaki K., Takeda K., Akira S., Shimokata K., Isobe K. (2003) The function of GADD34 is a recovery from a shutoff of protein synthesis induced by ER stress: elucidation by GADD34-deficient mice. FASEB J., 17, 1573–1575. [DOI] [PubMed] [Google Scholar]
- 7.Scheper G.C., van der Knaap M.S., Proud C.G. (2007) Translation matters: protein synthesis defects in inherited disease. Nat. Rev. Genet., 8, 711–723. [DOI] [PubMed] [Google Scholar]
- 8.Fogli A., Dionisi-Vici C., Deodato F., Bartuli A., Boespflug-Tanguy O., Bertini E. (2002) A severe variant of childhood ataxia with central hypomyelination/vanishing white matter leukoencephalopathy related to EIF21B5 mutation. Neurology, 59, 1966–1968. [DOI] [PubMed] [Google Scholar]
- 9.van der Knaap M.S., Leegwater P.A., Konst A.A., Visser A., Naidu S., Oudejans C.B., Schutgens R.B., Pronk J.C. (2002) Mutations in each of the five subunits of translation initiation factor eIF2B can cause leukoencephalopathy with vanishing white matter. Ann. Neurol., 51, 264–270. [DOI] [PubMed] [Google Scholar]
- 10.Draptchinskaia N., Gustavsson P., Andersson B., Pettersson M., Willig T.N., Dianzani I., Ball S., Tchernia G., Klar J., Matsson H. et al. (1999) The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat. Genet., 21, 169–175. [DOI] [PubMed] [Google Scholar]
- 11.Gazda H.T., Kho A.T., Sanoudou D., Zaucha J.M., Kohane I.S., Sieff C.A., Beggs A.H. (2006) Defective ribosomal protein gene expression alters transcription, translation, apoptosis, and oncogenic pathways in Diamond-Blackfan anemia. Stem Cells, 24, 2034–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Menne T.F., Goyenechea B., Sanchez-Puig N., Wong C.C., Tonkin L.M., Ancliff P.J., Brost R.L., Costanzo M., Boone C., Warren A.J. (2007) The Shwachman-Bodian-Diamond syndrome protein mediates translational activation of ribosomes in yeast. Nat. Genet., 39, 486–495. [DOI] [PubMed] [Google Scholar]
- 13.Thiel C.T., Horn D., Zabel B., Ekici A.B., Salinas K., Gebhart E., Ruschendorf F., Sticht H., Spranger J., Muller D. et al. (2005) Severely incapacitating mutations in patients with extreme short stature identify RNA-processing endoribonuclease RMRP as an essential cell growth regulator. Am. J. Hum. Genet., 77, 795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Antonellis A., Ellsworth R.E., Sambuughin N., Puls I., Abel A., Lee-Lin S.Q., Jordanova A., Kremensky I., Christodoulou K., Middleton L.T. et al. (2003) Glycyl tRNA synthetase mutations in Charcot-Marie-Tooth disease type 2D and distal spinal muscular atrophy type V. Am. J. Hum. Genet., 72, 1293–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.James P.A., Cader M.Z., Muntoni F., Childs A.M., Crow Y.J., Talbot K. (2006) Severe childhood SMA and axonal CMT due to anticodon binding domain mutations in the GARS gene. Neurology, 67, 1710–1712. [DOI] [PubMed] [Google Scholar]
- 16.Jordanova A., Irobi J., Thomas F.P., Van Dijck P., Meerschaert K., Dewil M., Dierick I., Jacobs A., De Vriendt E., Guergueltcheva V. et al. (2006) Disrupted function and axonal distribution of mutant tyrosyl-tRNA synthetase in dominant intermediate Charcot-Marie-Tooth neuropathy. Nat. Genet., 38, 197–202. [DOI] [PubMed] [Google Scholar]
- 17.McLaughlin H.M., Sakaguchi R., Liu C., Igarashi T., Pehlivan D., Chu K., Iyer R., Cruz P., Cherukuri P.F., Hansen N.F. et al. (2010) Compound heterozygosity for loss-of-function lysyl-tRNA synthetase mutations in a patient with peripheral neuropathy. Am. J. Hum. Genet., 87, 560–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chakraborty P.K., Schmitz-Abe K., Kennedy E.K., Mamady H., Naas T., Durie D., Campagna D.R., Lau A., Sendamarai A.K., Wiseman D.H. et al. (2014) Mutations in TRNT1 cause congenital sideroblastic anemia with immunodeficiency, fevers, and developmental delay (SIFD). Blood, 124, 2867–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar P., Henikoff S., Ng P.C. (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc., 4, 1073–1081. [DOI] [PubMed] [Google Scholar]
- 20.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. (2010) A method and server for predicting damaging missense mutations. Nat. Methods, 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harding H.P., Zhang Y., Scheuner D., Chen J.J., Kaufman R.J., Ron D. (2009) Ppp1r15 gene knockout reveals an essential role for translation initiation factor 2 alpha (eIF2alpha) dephosphorylation in mammalian development. Proc. Natl Acad. Sci. USA, 106, 1832–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zeng N., Li Y., He L., Xu X., Galicia V., Deng C., Stiles B.L. (2011) Adaptive basal phosphorylation of eIF2alpha is responsible for resistance to cellular stress-induced cell death in Pten-null hepatocytes. Mol. Cancer Res., 9, 1708–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marciniak S.J., Yun C.Y., Oyadomari S., Novoa I., Zhang Y., Jungreis R., Nagata K., Harding H.P., Ron D. (2004) CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Gene. Dev., 18, 3066–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kloft N., Neukirch C., von Hoven G., Bobkiewicz W., Weis S., Boller K., Husmann M. (2012) A subunit of eukaryotic translation initiation factor 2alpha-phosphatase (CreP/PPP1R15B) regulates membrane traffic. J. Biol. Chem., 287, 35299–35317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fogli A., Schiffmann R., Hugendubler L., Combes P., Bertini E., Rodriguez D., Kimball S.R., Boespflug-Tanguy O. (2004) Decreased guanine nucleotide exchange factor activity in eIF2B-mutated patients. Eur. J. Hum. Genet., 12, 561–566. [DOI] [PubMed] [Google Scholar]
- 26.van Kollenburg B., Thomas A.A., Vermeulen G., Bertrand G.A., van Berkel C.G., Pronk J.C., Proud C.G., van der Knaap M.S., Scheper G.C. (2006) Regulation of protein synthesis in lymphoblasts from vanishing white matter patients. Neurobiol. Dis., 21, 496–504. [DOI] [PubMed] [Google Scholar]
- 27.Senee V., Vattem K.M., Delepine M., Rainbow L.A., Haton C., Lecoq A., Shaw N.J., Robert J.J., Rooman R., Diatloff-Zito C. et al. (2004) Wolcott-Rallison Syndrome: clinical, genetic, and functional study of EIF2AK3 mutations and suggestion of genetic heterogeneity. Diabetes, 53, 1876–1883. [DOI] [PubMed] [Google Scholar]
- 28.Zhang P., McGrath B., Li S., Frank A., Zambito F., Reinert J., Gannon M., Ma K., McNaughton K., Cavener D.R. (2002) The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol. Cell Biol., 22, 3864–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Srour M., Schwartzentruber J., Hamdan F.F., Ospina L.H., Patry L., Labuda D., Massicotte C., Dobrzeniecka S., Capo-Chichi J.M., Papillon-Cavanagh S. et al. (2012) Mutations in C5ORF42 cause Joubert syndrome in the French Canadian population. Am. J. Hum. Genet., 90, 693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H., Durbin R. (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M. et al. (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res., 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang K., Li M., Hakonarson H. (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucl. Acids. Res., 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.