

Abstract
Dysfunctions in mitochondrial complex I (NADH:ubiquinone oxidoreductase) are both genetically and clinically highly diverse and a major cause of human mitochondrial diseases. The genetic determinants of individual clinical cases are increasingly being described, but how these genetic defects affect complex I on the molecular and cellular level, and have different clinical consequences in different individuals, is little understood. Furthermore, without molecular-level information innocent genetic variants may be misassigned as pathogenic. Here, we have used a yeast model system (Yarrowia lipolytica) to study the molecular consequences of 16 single amino acid substitutions, classified as pathogenic, in the NDUFV1 subunit of complex I. NDUFV1 binds the flavin cofactor that oxidizes NADH and is the site of complex I-mediated reactive oxygen species production. Seven mutations caused loss of complex I expression, suggesting they are detrimental but precluding further study. In two variants complex I was fully assembled but did not contain any flavin, and four mutations led to functionally compromised enzymes. Our study provides a molecular rationale for assignment of all these variants as pathogenic. However, three variants provided complex I that was functionally equivalent to the wild-type enzyme, challenging their assignment as pathogenic. By combining structural, bioinformatic and functional data, a simple scoring system for the initial evaluation of future NDUFV1 variants is proposed. Overall, our results broaden understanding of how mutations in this centrally important core subunit of complex I affect its function and provide a basis for understanding the role of NDUFV1 mutations in mitochondrial dysfunction.
Introduction
Mitochondrial complex I (NADH:ubiquinone oxidoreductase) catalyzes oxidation of NADH in the mitochondrial matrix, reduction of ubiquinone in the inner mitochondrial membrane and transfer of protons across the membrane (1). It thus regenerates NAD+ in the matrix, to sustain crucial metabolic processes including the tricarboxylic acid cycle and β-oxidation of fatty acids, supplies electrons to respiratory complex III, and contributes to the proton motive force that drives ATP synthesis and transport processes. Complex I is also a significant source of reactive oxygen species generation in mitochondria and thus contributes to cellular oxidative stress (2). Mammalian complex I consists of 45 subunits (1,3,4). Fourteen of them are the conserved core subunits that are sufficient for catalysis; detailed structural information on them is available from complex I from the bacterium Thermus thermophilus (5). The remaining 31 are supernumerary subunits that have been accumulated through evolution; the locations of some of them have been assigned in the structure of the mammalian complex I from Bos taurus (4).
Dysfunctions in complex I, which account for around a third of all early-onset mitochondrial disorders, are both genetically and clinically highly diverse (6,7). Genetically, they are caused by mutations in both the mitochondrial and nuclear genomes, in both the subunits of the mature enzyme and the assembly factors required for its biogenesis. Clinically, the most common presentations include Leigh syndrome, leuko-encephalopathy and other early-onset neurodegenerative disorders, as well as lactic acidosis, cardiomyopathy and exercise intolerance (6,7). Although, on the molecular level, different genetic defects must result in specific and distinguishable defects (defective assembly or increased degradation, increased reactive oxygen species production or impaired catalysis or regulation), the in vivo consequences of individual mutations vary significantly between tissues and produce different clinical profiles in different patients. The complex relationships between a specific molecular defect and its cellular and clinical consequences remain poorly understood.
Studies to define the molecular-level defects arising from mutations in complex I subunits, and to confirm the diagnosis of pathogenic variants (required for genetic counseling and patient support), require functional and structural studies of the isolated enzyme and so are significantly hampered by the small, experimentally limiting amounts of material available from cultured human cells and biopsy samples. In addition, in many cases (see Supplementary Material, Table S1), the complex I defects observed in tissue biopsy analyses are not expressed in cultured fibroblasts, precluding functional investigations. Consequently, reproducing the mutations in a clean and well-characterized model system, which is both readily amenable to genetic manipulation and provides large amounts of material for structural and functional studies, is an attractive option. Using this strategy, several mutations in nuclear-encoded core subunits that cause Leigh syndrome have been analyzed in the yeast Yarrowia lipolytica and found to decrease stability and activity and/or alter ubiquinone reduction (8,9). Decreased levels of complex I resulted from mutations tested in the fungus Neurospora crassa (10) and mutations in the mitochondrial-encoded subunits that cause Leber's hereditary optic neuropathy and mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) have been studied in the bacteria Paracoccus denitrificans and Escherichia coli and suggested to affect enzyme assembly, stability, ubiquinone reduction and proton translocation (11–14). Although bacterial systems allow mutagenesis of those subunits that are mitochondrial encoded in eukaryotes, the yeast enzymes are much closer to the human enzyme in both their subunit compositions and protein sequences (15).
Here, we use the yeast Y. lipolytica to elucidate the molecular consequences of the set of pathological mutations identified in mitochondrial disease patients in the NDUFV1 subunit of complex I, a nuclear-encoded subunit that contains the highly conserved flavin mononucleotide (FMN) and NADH-binding sites. NDUFV1 was chosen because it has a relatively high number of pathological variants (6,7), because we have extensive mechanistic knowledge of the two physiologically relevant reactions (NADH oxidation and reactive oxygen species production) that are catalyzed by the flavin in it (16–20), and to investigate the origin of the beneficial effects of riboflavin supplementation reported in some clinical cases of complex I dysfunction (21,22). We use Y. lipolytica to evaluate the effects of each mutation, revealing a set of highly variable effects that provide varying support for their assignment as pathogenic. Furthermore, by mapping each variant onto the structure of NDUFV1, and considering the phylogenetic conservation, we identify mutation hot-spots and consider the extent to which the disruptive potential of an NDUFV1 mutation can be evaluated without extensive experimentation. Thus, we broaden understanding of how mutations around the flavin site affect complex I, evaluate Y. lipolytica and other model systems for their suitability for studying human complex I mutations, and develop approaches for evaluating the pathogenic potential of NDUFV1 variants identified clinically in the future.
Results
Mutations identified as pathological in human NDUFV1
Nineteen single amino acid substitutions that have been classified as pathological in the human NDUFV1 subunit of complex I were identified by searching the literature and in the Human Gene Mutation Database (23) (see Table 1 and Supplementary Material, Table S1). Two further mutations that introduce stop codons to produce premature terminations [R59X (30) and Q344X (25)] are not discussed here. Aligning the sequence of NDUFV1 with NUBM, its Y. lipolytica homolog, revealed that 16 of the mutations affect conserved residues (see Fig. 1 and Table 2), within an overall sequence identity of 67% (85% similarity). Therefore, these 16 mutations can be studied in Y. lipolytica. The three non-conserved mutations are S56P, K111E and A211V. Figure 1 also shows the high sequence identity (98%) between the human NDUFV1 subunit and its homolog in Bos taurus, the 51-kDa subunit, so the human mutations can be mapped to the B. taurus structure (see Table 2 and Fig. 2). Note that we use the nomenclature and numbering for the human subunit throughout for consistency, regardless of the species referred to. In Figure 2, the subunit has been separated into its four constituent domains for clarity: an N-terminal domain that ends in a glycine-rich loop, followed by a Rossmann-fold domain, an ubiquitin-like domain and a C-terminal four-helix bundle that ligates the iron–sulfur (FeS) cluster known as cluster N3 (34).
Table 1.
Clinical cases with mutations identified in NDUFV1 and comparison with effects in Y. lipolytica
| Mutation(s) | Zygosity | Clinical reporta | References | Effects in Y. lipolytica |
|---|---|---|---|---|
| S56P+T423M | Compound heterozygous | Leuko-encephalopathy (alive at 2.5 years) | (7) | Not conserved+ complex I absent |
| R88G+R199P | Compound heterozygous | Fatal infantile lactic acidosis (died 4 months) | (24) | Complex I absent+ normal |
| K111E+R386H | Compound heterozygous | Not known | (25) | Not conserved+ impaired |
| A117T+E246K | Compound heterozygous | Fatal infantile lactic acidosis (died 3 days) | (26) | Complex I absent+ complex I absent |
| R147W mtDNA | Heterozygous+mtDNA | Not known | (25) | Normal +not studied |
| Y204C+C206G | Compound heterozygous | Leigh syndrome (alive at 7 years) | (27,28) | Complex I absent+ no flavin |
| A211V+R257Q | Compound heterozygous | Leuko-encephalopathy (alive at 16 months) | (29) | Not conserved+ complex I absent |
| E214K | Compound heterozygous; second variant not expressed owing to mutation in donor splice-site | Leigh syndrome (died 3 years) |
(28) | Impaired |
| P252R+R386H | Compound heterozygous | Not known | (25) | Complex I absent+ impaired |
| A341V | Homozygous | Leuko-encephalopathy (alive at 10 years) | (30) | Normal |
| E377K | Homozygous | Fatal infantile lactic acidosis (died 4 months) | (25) | No flavin |
| R386C | Homozygous | Leuko-encephalopathy (alive at 32 months) | (24,31) | Impaired |
| R386H | Homozygous | Leigh syndrome (died 5–8 months) | (32) | Impaired |
| T423M+R59X | Compound heterozygous; R59X variant not expressed | Myoclonic epilepsy (died 18 months) | (30) | Complex I absent |
| A432P | Compound heterozygous; second variant not expressed owing to two base deletion | Leigh syndrome (died 1.5 years) | (28) | Impaired |
aA detailed summary of clinical data is presented in Supplementary Material, Table S1.
Figure 1.

Alignment of the NDUFV1 sequences for Homo sapiens, Bos taurus and Y. lipolytica. The accession codes are P49821 (Hs), P25708 (Bt) and Q9UUU2 (Yl), and the sequences shown include the mitochondrial target peptides (gray) that are removed to form the mature proteins. The sequences are colored as in Figure 2 to show the four domains of the protein: blue, N-terminal domain; magenta, Rossmann-fold domain; green, ubiquitin-like domain; red, 4Fe-4S four-helix bundle domain. Variant/mutated residues are shaded in cyan, and the ligands of the 4Fe-4S cluster are shaded in yellow. Out-line boxes are blue for residues within 5 Å of the flavin, orange for additional residues within 5 Å of the bound nucleotide and green for additional residues within 5 Å of the FeS cluster [calculated using the nucleotide-bound structure of the hydrophilic domain of T. thermophilus complex I (33)].
Table 2.
Characterization of the 19 mis-sense mutations identified clinically in NDUFV1
| H. sapiens variant | 4UQ8. pdb (B. taurus)a | Y. lipolytica mutation | Effect in Y. lipolytica | Structure | Distance from flavinb | Distance from FeSb | Conservationc | Score |
|---|---|---|---|---|---|---|---|---|
| S56P | S36 | – | – | N-terminus | 23.7 | 38.3 | None | 0—none |
| R88G | R68 | R87G | Complex I absent | N-terminus | 4.5 | 21.0 | All | 16—strongly impaired |
| K111E | K91 | – | – | Rossmann fold | 16.7 | 25.1 | Metazoans | 6—mild |
| A117T | A97 | A119T | Complex I absent | Rossmann fold | 7.0 | 19.8 | Metazoans | 12—impaired |
| R147W | R127 | R149W | Normal | Rossmann fold | 16.7 | 31.9 | Chordates | 4—mild |
| R199P | R179 | R201P | Normal | Rossmann fold | 14.5 | 21.1 | Eukaryotes | 10—mild |
| Y204C | Y184 | Y206C | Complex I absent | Rossmann fold | 5.9 | 8.9 | All | 20—strongly impaired |
| C206G | C186 | C208G | No flavin | Rossmann fold | 7.0 | 9.2 | All | 20—strongly impaired |
| A211V | A191 | – | – | Rossmann fold | 8.5 | 8.5 | Metazoans | 16—strongly impaired |
| E214K | E194 | E216K | Impaired | Rossmann fold | 12.5 | 12.0 | Eukaryotes | 14—impaired |
| E246K | E226 | E248K | Complex I absent | Rossmann fold | 7.2 | 23.8 | All | 14—impaired |
| P252R | P232 | P254R | Complex I absent | Rossmann fold | 13.3 | 31.5 | Eukaryotes | 10—mild |
| R257Q | R237 | R259Q | Complex I absent | Rossmann fold | 19.9 | 38.5 | Eukaryotes | 8—mild |
| A341V | A321 | A343V | Normal | Ubiquitin-like | 17.5 | 26.7 | Chordates | 4—none |
| E377K | E357 | E379K | No flavin | FeS domain | 8.3 | 6.0 | All | 20—strongly impaired |
| R386C | R366 | R388C | Impaired | FeS domain | 14.0 | 6.3 | All | 18—strongly impaired |
| R386H | R366 | R388H | Impaired | FeS domain | 14.0 | 6.3 | All | 18—strongly impaired |
| T423M | T403 | T425M | Complex I absent | FeS domain | 10.0 | 5.1 | Eukaryotes | 16—strongly impaired |
| A432P | A412 | A434P | Impaired | FeS domain | 10.1 | 8.9 | Eukaryotes | 16—strongly impaired |
aResidues in the structure file are numbered from the beginning of the mature protein, so the mitochondrial target sequences are not included.
b(Center-to-center) distance from the Cα of the variant residue to the nearest atom in the cofactor.
cOverview of the level of conservation within chordates, metazoans, eukaryotes and all species. The category listed is the most general level in which the residue is conserved.
Figure 2.
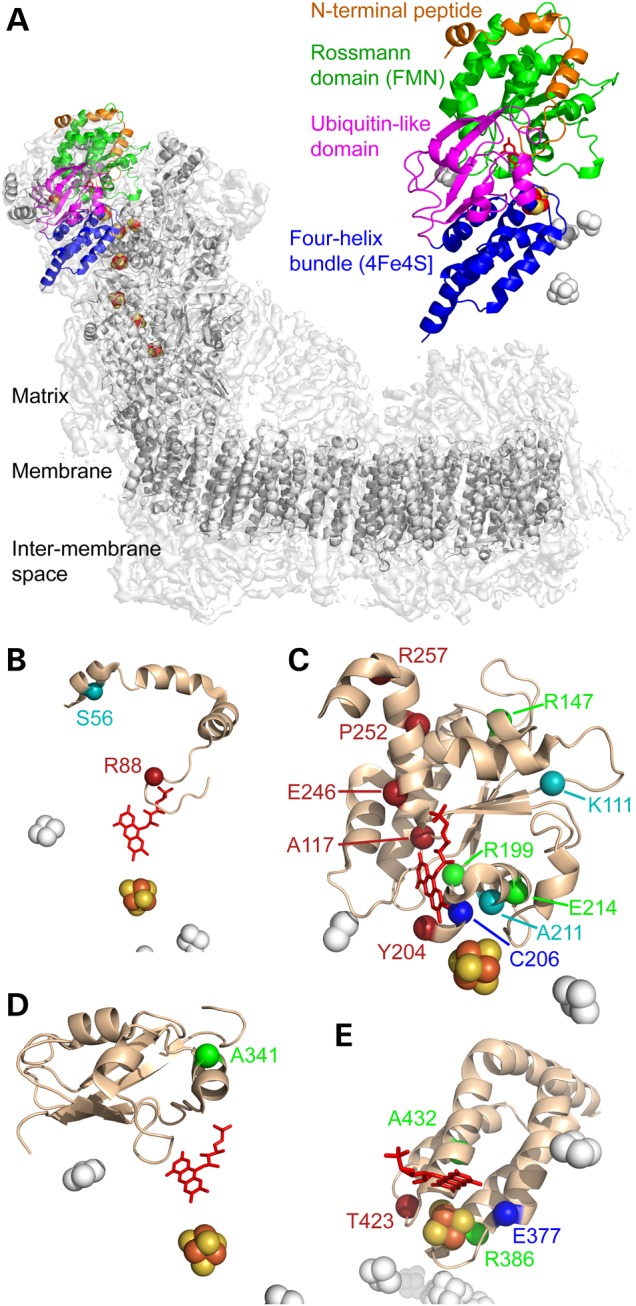
The 19 clinically identified mutations in the structure of B. taurus NDUFV1. (A) Location of NDUFV1 (the 51-kDa subunit) in the structure of complex I. The density for the whole enzyme is in light gray, with the core subunit models in dark gray and NDUFV1 in color. NDUFV1 is shown expanded on the right to illustrate its four domains. (B–E). The four domains of NDUFV1: the N-terminal peptide (B), FMN-binding Rossmann domain (C), ubiquitin-like domain (D) and FeS-binding four-helix bundle (E), with mutated residues labeled. Cyan, residues not conserved in Y. lipolytica; red, residues for which mutations produced no complex I in Y. lipolytica; blue, residues for which mutations produced complex I without any flavin in Y. lipolytica; green, residues for which mutations produced Y. lipolytica complex I with wild-type or variant properties. Figure created using 4UQ8.pdb (4).
Seven mutations result in loss of complex I expression
The 16 mutants for study were expressed in NDUFV1 in Y. lipolytica complex I, by expressing NUBM (NDUFV1) on the pUB26 plasmid in a GB10 Δnubm deletion strain of Y. lipolytica (35). The strain also expresses a matrix-targeted NDH1 (alternative dehydrogenase), to support cell growth even when complex I is inactive (36). None of the variants displayed abnormal growth, although variants in which intact complex I could not be observed (see below) grew up to twice as slowly as the wild-type and other variants.
Mitochondrial membranes were prepared from each variant and investigated using blue-native (BN)-PAGE (by visualizing the band from intact complex I and by detecting complex I with an in-gel assay for NADH oxidation), and by measuring the rate of NADH:ferricyanide (FeCN) oxidoreduction by the complex I flavin site in solution assays (see Fig. 3). Intact complex I could be detected in only nine cases and so enzyme assembly or stability is disrupted in the seven variants (R88G, A117T, Y204C, E246K, P252R, R257Q and T423M) that did not contain detectable complex I. The seven mutations are all located in functionally important regions of the enzyme and are well conserved between species (see Fig. 2 and Table 2). R88 is present on the glycine-rich loop that forms part of the flavin-binding cavity. E246, P252 and R257 are on the final helix of the Rossmann fold, with E246 close to the flavin and adjacent to R88. A117 is on a loop that also forms part of the flavin-binding site, between a strand and a helix of the Rossmann fold. Y204 is within the Rossmann fold, close to both the flavin isoalloxazine ring system and 4Fe-4S cluster N3, and T423 is adjacent to the cluster, on the cluster-coordinating loop that carries Cys425, close to the 75-kDa subunit.
Figure 3.
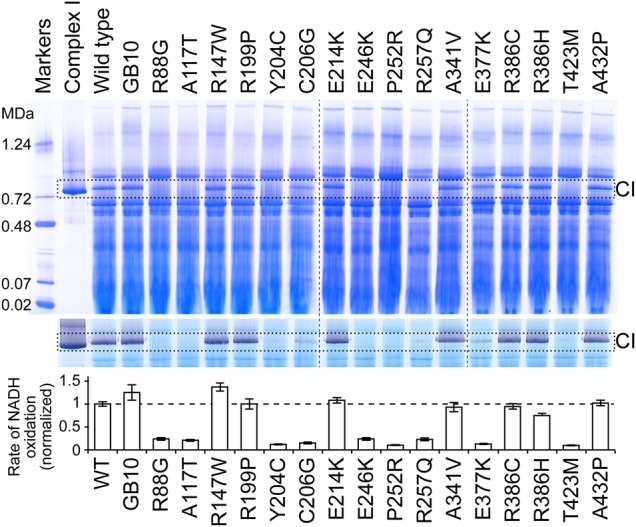
Analyses of 16 NDUFV1 variants in complex I in mitochondrial membranes from Y. lipolytica. BN-PAGE analyses of Y. lipolytica mitochondrial membranes from the 16 pathological variants are presented at the top, alongside membranes isolated from the parent GB10 strain and the wild-type variant (the wild-type subunit expressed in the GB10 Δnubm strain from the pUB26 plasmid, to match the expression of the variants). Isolated Y. lipolytica complex I is included for reference, and dashed vertical lines indicate where different gels have been combined. Membranes were solubilized using DDM at 2:1 protein:detergent ratio, 40 µg of solubilized proteins were loaded per lane and proteins were visualized using Coomassie Blue. Middle: NADH oxidation by complex I was detected as a purple color using 0.5 mg ml−1 NBT and 120 µm NADH. Bottom: NADH oxidation was measured using 100 µm NADH and 1 mm FeCN in the presence of piericidin A and normalized to the wild-type value.
Two mutations result in flavin-deficient complex I
In two cases, C206G and E377K, BN-PAGE revealed the presence of intact complex I, but in-gel activity assays (NADH oxidation coupled to non-physiological reduction of a purple-blue dye) revealed no evidence that the complexes are able to oxidize NADH (see Fig. 3). To investigate whether the variants are inactive because they lack flavin, or because of the different amino acid side chain, standard protocols were used to purify the variant complexes. As expected, neither in-gel assays on the isolated complexes nor solution assays for NADH oxidation coupled to the reduction of either FeCN (that reacts at the flavin site), or decylubiquinone (DQ, an ubiquinone-10 analog), revealed any catalytic activity (see Fig. 4). SDS–PAGE analyses did not identify any changes to the subunit compositions; the bands corresponding to NDUFV1 were clearly visible and confirmed using mass spectrometry. Subsequently, flavin analyses (see Fig. 4) revealed that the two variants are inactive because their levels of flavin are negligible. Both mutations are close to the flavin site (E377 is at the end of one of the four helices of the cluster-coordinating bundle, close to both the flavin and the FeS cluster, and C206 is within the Rossmann fold, see Fig. 2 and Table 2), and they are located on each side of Y204 (the Y204C mutation resulted in lack of complex I).
Figure 4.
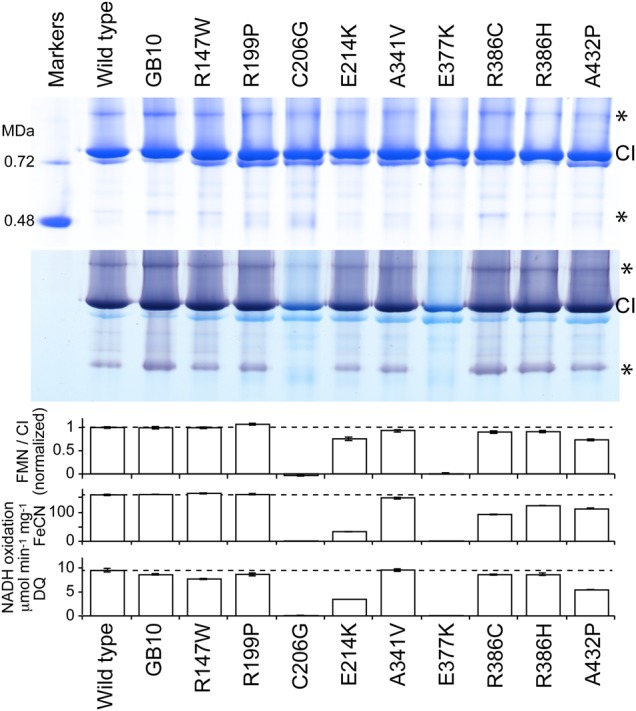
Analyses of nine NDUFV1 variants of complex I isolated from Y. lipolytica. Top: BN-PAGE analyses of Y. lipolytica complex I from the nine variants, alongside enzyme from the parent GB10 strain and the wild-type variant. Eight micrograms of protein was loaded per lane and visualized using Coomassie Blue. Asterisks indicate catalytically-active complex I dimers and subcomplexes. Middle: NADH oxidation by complex I was detected as a purple color using 0.5 mg ml−1 NBT and 120 µm NADH. Bottom: the flavin content of the purified enzymes, relative to the WT variant, equivalent to the number of flavins bound per complex I; the rate of NADH oxidation measured using 100 µm NADH and 1 mm FeCN; the rate of NADH oxidation measured using 100 µm NADH and 100 µm DQ.
The following experiments were designed to test whether the flavin contents could be increased by supplementation. First, catalytic activity measurements with FeCN and DQ were carried out with FMN added to the assay solution, to determine whether the FMN is absent simply because it is too weakly bound. For both variants, the activities increased rapidly at low FMN concentrations and reached their maximal values at ∼5 µm. However, the maximal rates from C206G and E377K were only 10–15% and ∼2% of the wild-type rates, respectively, suggesting that (as the FMN:complex I ratio is very high) either a significant proportion of enzyme is unable to accept FMN, or FMN binds to each molecule but the enzymes formed have very low activity. To further examine whether C206G could stably incorporate FMN, 100 µm FMN was added to a stock of 10 mg ml−1 complex I and incubated at 32°C for 2 min. The mixture was then diluted 2000-fold and the FeCN activity assayed and compared with the rate from enzyme incubated without FMN. The high-concentration incubation increased the rate to around two-thirds of the level attained by adding 5 µm FMN to the assay buffer, indicating that any ‘reconstituted’ FMN is not tightly bound and slowly dissociates. Finally, extra riboflavin was added to the Y. lipolytica cell growth medium to increase its concentration 10-fold (from ∼10 to 100 µg ml−1); wild-type cell growth was unaffected and no improvement was observed in the activities of either flavin-deficient variant measured in membranes. Subsequently, we investigated the FeS clusters in the two flavin-free variants using EPR spectroscopy (37); cluster N3 was not observed in either case (the rest of the FeS cluster spectra were normal). Our results suggest that the mutations obstruct flavin incorporation during assembly, causing misfolding of the flavin-free NDUFV1 protein.
Mutations with normal complex I activities
Three variants (R147W, R199P and A341V) closely matched the WT/GB10 variants in all the experiments presented so far, suggesting that they have no effect on the assembly, flavin content or catalytic activity of Y. lipolytica complex I (see Fig. 4). [The WT strain has NDUFV1 encoded on an expression plasmid (like the mutants), whereas in the GB10 strain NDUFV1 is present in the genome.] Additional experiments were thus performed for each variant, to challenge this conclusion (see Fig. 5).
Figure 5.

Characteristics of the seven NDUFV1 variants of complex I isolated from Y. lipolytica that retain catalytic activities. Top: the dissociation temperature of the flavin from complex I, measured using the ThermoFMN assay, relative to the WT value of 51.9 ± 0.2°C. Middle: flavin-site catalytic activity measurements using 100 µm NADH and 1 mm APAD+, 3.5 mm HAR and 0.25 mm paraquat, respectively. Bottom: reactive oxygen species production measured using the Amplex Red assay for H2O2 production at two NADH concentrations.
First, the propensity of the flavin to dissociate was measured using the ‘ThermoFMN’ assay, in which loss of flavin from the complex is monitored by fluorescence as the temperature is increased (38,39). For R147W, R199P and A341V, the dissociation temperatures were not significantly different from the values for the WT/GB10 complexes. Second, a further set of flavin-site assays, the NADH:APAD+, NADH:HAR and NADH:paraquat assays, were carried out. APAD+ (3-acetylpyridine adenine dinucleotide) is an NAD+ analog (17); HAR (hexaammineruthenium (III) or [Ru(NH3)6]3+) is an artificial electron acceptor that reacts at the flavin by a different mechanism to FeCN (19); and paraquat is a redox-cycling molecule that is reduced by the flavin then reoxidized by O2 (19,20). None of the values were appreciably different from the WT/GB10 values, except that the rate of HAR reduction by A341V is negligible. The physiological relevance of this intriguing result is unclear, and we are currently unable to explain it. Finally, the rate of H2O2 production by NADH was tested at two different NADH concentrations using the Amplex Red assay (16) but, again, no significant differences were observed. Thus, our data do not provide a basis for assigning functional complex I defects to any of these three variants.
Consistent with their lack of deleterious effect, none of the three residues are close to the flavin, form part of the NADH-binding site or interact with the bound nucleotide or are close to the FeS cluster. Figure 2 and Table 2 show that R147 is located toward the end of one of the helices of the Rossmann fold and conserved only in the majority of chordates; R199 is in a loop connecting two strands of the Rossmann fold (at the interface with the 24-kDa subunit) and conserved in the majority of eukaryotes; A341 is in the ubiquitin-like domain (on the external surface of the subunit) and generally conserved only in chordates.
Mutations resulting in intermediate complex I activities
Four NDUFV1 variants (E214K, R386H, R386C and A432P) produce Y. lipolytica complex I with intermediate behavior. First, they all have decreased FMN content, from ∼0.75 FMN/complex I for E214K and A432P to ∼0.9 for R386H and R386C (see Fig. 4); addition of FMN to assays of E214K and A432P did not increase their activities. The low FMN contents explain the decreased activities of R386H and R386C in NADH:DQ assays (Fig. 4) as the flavin-normalized rates are the same as for WT (within 5%), but for A432P and particularly E214K, the normalized values are still low. Because NADH oxidation is much faster than DQ reduction and so not rate limiting, this result suggests that subpopulations of the FMN-bound enzymes are inactive. For all four variants, the flavin-normalized rates for the NADH:FeCN reaction are slower than those for WT, with the greatest effect for E214K and the mildest for A432P. In the ThermoFMN assay, the FMN in E214K dissociates at significantly lower temperature (see Fig. 5), so it is severely destabilized in the complex; R386H, R386C and A432P are slightly destabilized. In the NADH:APAD+, NADH:HAR and NADH:paraquat assays, the values observed are decreased moderately with respect to the WT values, and in most cases, the rates of H2O2 production were normal. The exception is that H2O2 production in the highest NADH concentration is increased significantly for E214K (see Fig. 5). Interestingly, these four mutations that cause a range of detrimental effects on the flavin-site stability and/or reactivity are all in well-conserved residues located in the vicinity of the FeS cluster (see Fig. 2 and Table 2). E214 is located on a short helix of the Rossmann fold, R386 at the bottom of one of the four helices that ligate the cluster, and A432 (Pro is a common variant in prokaryotes) is also on one of the helices that ligate the cluster.
Discussion
Comparison of results with clinical data
Six of the seven mutations that prevented formation of intact complex I in Y. lipolytica were identified clinically as compound heterozygotes, and T423M as an apparent homozygote (Table 1) (30). T423M showed decreased activity in both muscle tissue and cultured fibroblasts (see Supplementary Material, Table S2) (7,30), whereas no detectable activity was observed here. This result highlights a deficiency in the Y. lipolytica model system, because, although lack of complex I likely indicates the severity of a mutation, it precludes further experimental investigation. R88G, Y204C and P252R were heterozygous with variants that supported complex I expression in Y. lipolytica, and R257Q with a non-conserved variant, hindering comparison of clinical (see Supplementary Material, Table S2) and Y. lipolytica activities. A117T and E246K were observed in the same compound heterozygote (26) and in one of the most severe clinical cases (see Table 1 and Supplementary Material, Table S1), but no biochemical data were presented (see Supplementary Material, Table S2). Differences between the effects of these mutations in patients (in which complex I must be present at some level) and in Y. lipolytica likely reflect differences between the assembly and/or quality-control pathways, perhaps enhanced by plasmid expression of the Y. lipolytica subunit.
Our results indicate that E377K and C206G are pathogenic because they lack flavin and cannot oxidize NADH. E377K was identified clinically as a homozygous variant that was 11% active relative to the control value in muscle (7,25) and C206G as a compound heterozygote with Y204C that was 32% active relative to the lowest control value (7,27) (see Supplementary Material, Table S2). The Y204C variant did not express Y. lipolytica complex I, making the effect of C206G difficult to deconvolute. However, for these two variants, the levels of activity observed in both systems are consistent.
Three mutations that had no discernable effect on Y. lipolytica complex I, R147W, R199P and A341V, were heterozygous plus a mutation in mitochondrial-encoded ND5 (25), compound heterozygous with R88G (24) and homozygous (30,40), respectively (Table 1). The mutation in ND5 was assigned as pathological (25), consistent with our assignment of R147W as non-pathogenic. R199P was identified in a Leigh syndrome patient as compound heterozygous with R88G (a variant that did not assemble Y. lipolytica complex I), and no biochemical data were reported (see Supplementary Material, Table S2) (24). Although R199 is at the interface between NDUFV1 and NDUFV2, no effect was observed in Y. lipolytica, so the detrimental nature of R199P may be particular to human complex I, or owing to an effect not identified in our study, such as increased susceptibility to proteolysis. However, an independent, unidentified effect, such as a mutation in a different complex I protein, may also contribute: two severe mutations in two different complex I-related proteins would combine to produce an enzyme with only 25% activity and confound the genetic analysis. Indeed, Marin and coworkers also questioned the pathogenicity of R199P on the basis of mixed predictions from SIFT and PolyPhen (24) (see Supplementary Material, Table S2). For A341V, biochemical and clinical data from two studies argue that it is a recessive, pathogenic variant because only the homozygous patient exhibited severe symptoms whereas heterozygous family members are unaffected (see Supplementary Material, Table S2) (30,40). In our study, A341V behaved normally in all respects, except for its unexplained behavior in the HAR activity assay, an effect lacking obvious physiological relevance. It is possible that this observation may provide a future key to understanding the molecular effect of this mutation, or, because only a small number of genes were sequenced in these two cases (see Supplementary Material, Table S2), the A341V variant may have been misidentified as pathological. We note that, despite an ever increasing number of known mitochondrial disease genes, a significant proportion of mitochondrial disease patients lack molecular diagnoses, even after investigation by exome sequencing (41,42).
Of the four variants with intermediate activity in Y. lipolytica complex I, none are controversial. All of E214K, R386C, R386H and A432P exhibited evidence of decreased complex I activity in either tissue or cultured fibroblasts (Supplementary Material, Table S2) and also in Y. lipolytica, supporting their assignments as pathogenic. Notably, only one variant, E214K, displayed increased reactive oxygen species production (see Fig. 5). Thus, even for mutations around the flavin site, the increased reactive oxygen species production that is a common feature of complex I dysfunctions (6,43) is not (generally) a primary effect of the molecular defect, but a secondary effect mediated by the increased NADH levels that arise from inhibition of catalysis (16,44).
Structure–function relationships for mutations identified as pathological in NDUFV1
Table 2 summarizes structural data on the variant residues. Only one variant in a conserved residue has been identified in the N-terminal region of NDUFV1 that wraps around the Rossmann-fold domain and then ends in a glycine-rich loop that forms part of the flavin-binding site. Mutation R88G, within this final loop, caused lack of assembled complex I, most likely because it disrupts the interactions of the Arg side chain. Similarly, only one pathological mutation has been identified in the ubiquitin-like domain (that has no known functional role), and it had little effect on the properties of Y. lipolytica complex I. As may be expected, most of the pathogenic mutations cluster in the flavin/NADH-binding Rossmann-fold domain and the FeS-binding four-helix bundle. Three mutations that caused a complete lack of assembled complex I in Y. lipolytica are located on the final helix (residues 244–265) of the Rossmann-fold domain, making this a hotspot for detrimental mutations. This helix is enclosed and likely stabilized by the N-terminal domain, indicating its central structural role. Two further mutations in the Rossmann-fold domain that abolish Y. lipolytica complex I are adjacent to the flavin, A117T and Y204C. Y204 is on a highly conserved loop/helix element immediately underneath the flavin that positions the Tyr side chain adjacent to one of the isoalloxazine rings. Close to the Tyr, mutation of C206 caused loss of flavin from the complex, and within the same region of the protein, but at increased distance from the flavin are A211 (non-conserved) and E214 (intermediate behavior). The remaining three variant residues in the Rossmann-fold domain, K91, R127 and R179, are all distant from both the flavin and the FeS cluster (see Table 2), K91 is not conserved and the Y. lipolytica variants of the two Arg residues are indistinguishable from wild type. Thus, for these residues, increasing distance from the flavin correlates with decreasing effect on Y. lipolytica complex I. Table 2 reveals that a second set of disruptive mutations, in the FeS domain, are located within 10 Å of the FeS cluster, providing a rational explanation for their effects in both patients and Y. lipolytica.
Evaluating the disruptive potential of residues using structure and sequence conservation
Table 2 presents the distances from each variant residue to the FMN and the [4Fe-4S] center [measured using the structure of NDUFV1 from B. taurus complex I (4)], alongside the phylogenetic conservation of each residue. The levels of conservation reported are from the manual inspection of a multiple sequence alignment of 200 NDUFV1 proteins from all regions of the phylogenetic tree; the levels reflect majority (>95%) conservation within the given classes of chordata, metazoa, eukaryota and all species. Table 2 highlights clear correlations between the effect of the mutations in Y. lipolytica and the structure/conservation information, forming the basis for a basic scoring system to evaluate the ‘disruptive potential’ of each mutation (see Table 2). For the two distances, the scores were 0 points for >20 Å, 2 for 15–20 Å, 4 for 10–15 Å, 6 for 5–10 Å and 8 for <5 Å. For conservation, the scores were 0 points for none, 2 for chordata, 4 for metazoa, 6 for eukaryota and 8 for all species. The scores for each variant were summed, and the predicted effects classified as none for <5 points, mild for 6–10 points, impaired for 11–15 points and strongly impaired for >16 points.
Two variants are predicted to have no effect: S56P (not conserved) and A341V, consistent with our data. Five variants are predicted to have mild effects: K111E (not conserved), R147W and R199P (no effect in Y. lipolytica), and P252R and R257Q (no complex I in Y. lipolytica). The scoring system underestimates the effects of P252R and R257Q because it does not account for their location on the structurally important final helix of the Rossmann fold. Three mutations are predicted as impaired: A117T and E246K (no complex I in Y. lipolytica) and E214K (impaired enzyme). Finally, eight variants are predicted to be strongly impaired: R88G, Y204C and T423M (did not form complex I), C206G and E377K (complex I containing no flavin) and R386H, R386C and A432P (impaired enzyme). The predicted and observed effects are thus consistent in almost all cases, providing a method for initial evaluation of future variants; to aid these evaluations Supplementary Material, Figure S1 presents the distances of all the structurally defined residues in NDUFV1 to the flavin and FeS cluster. For the three residues that could not be tested in Y. lipolytica, S56P and K111E are not predicted to be pathogenic, and only the assignment of A211V as pathogenic is supported. The low scores of the R147W, R199P and A341V variants underscore the need to question the assignment of these mutations as pathogenic. Finally, SIFT (45) and PolyPhen-2 (46) analyses are commonly used to evaluate the disruptive potential of identified variants (see Supplementary Material, Table S2). Here, SIFT scored all the variants ‘Damaging’ except for K111E and R147W, which were scored ‘Tolerated’, whereas PolyPhen-2 scored S56P as ‘Benign’, K111E, A117T and A341V as ‘Possibly Damaging’, and all the others as ‘Probably Damaging’. Thus, SIFT and PolyPhen-2 disagree about S56P and R147W, and together they are more likely to predict a variant to be damaging than either our scoring system or our experimental method. Both SIFT and PolyPhen-2 are based on sequence information and do not take account of structure-function data pertaining specifically to the protein under consideration.
Riboflavin supplementation as a route to increasing complex I activity
Two Y. lipolytica variants, C206G and E377K, contained essentially no flavin, and two variants, E214K and A432P, contained decreased levels. Riboflavin supplementation has been tested in Y204C–C206G patients (see Supplementary Material, Table S1), but while Benit and coworkers noted a positive effects (28), Laugel and coworkers noted no additional positive effect following treatment by idebenone (27), providing no coherent picture. In our study, we found no improvement from supplementing the growth medium for Y. lipolytica C206G and E377K with riboflavin, and only limited improvement upon attempts to reconstitute the flavin into the purified enzyme. The mechanism of flavin insertion into complex I during assembly is not known, and the mutations may disrupt either (or both) the enzyme affinity for flavin or insertion of flavin into the site. In any case, our results provide little support for a direct, beneficial effect of dietary supplementation by riboflavin on complex I activity in these cases. In contrast, there is a clear rationale for the beneficial effects of riboflavin supplementation in patients with mutations in a riboflavin transporter (22,47), and it is possible that mutations in ACAD9 (an assembly factor of complex I that is also involved in fatty acid oxidation) that respond to riboflavin supplementation may be detrimental because they decrease affinity for the FAD cofactor (48).
Conclusions and perspectives
The Y. lipolytica complex I system has provided many insights into the molecular effects of variants identified as pathogenic in the NDUFV1 subunit. However, two limitations of the model system quickly became clear. First, 3 of the 19 pathological variants identified clinically could not be studied in Y. lipolytica because the residues are not conserved. Second, seven variants created in Y. lipolytica did not provide any enzyme for detailed study, probably reflecting differences in assembly, stability or degradation pathways between the mammalian and yeast systems. The lack of Y. lipolytica complex I may be taken to indicate a detrimental phenotype, but it is clear that the same effect cannot be present in the patient to the same extent. Although these two limitations refer to around half the cohort of variants, we note that similar studies using material isolated from mammalian systems, either from rodent models (49) or cultured human cells, which may be considered more representative of the clinical situation, suffer from significant technical challenges (notably, in the scale of material they provide) that preclude their routine application for structure-function studies at present. For variants for which Y. lipolytica complex I is assembled, fundamental insights, which may be transferred to the human variants, have been obtained. The results of our experimental study have also been used to test a simple scoring system for the initial evaluation of NDUFV1 variants identified in the future. Together, our results and evaluation challenge the assignment of 5 of the 19 variants (S56P, K111E, R147W, R199P and A341V) as pathogenic, suggesting the need for further investigation in these cases.
Materials and Methods
Growth of Y. lipolytica cells and preparation of mitochondrial membranes and complex I
Y. lipolytica complex I was prepared as described previously (50–52). Briefly, cells were grown at 27°C in 2% yeast extract, 4% peptone and 4% glucose, initially at pH 5.5, and harvested in late-exponential phase. Typically, 50 g of cells were obtained per liter of medium. All the following steps were at 4°C. Cells were disrupted by two passes through a Dyno®-Mill bead mill (Willy A. Bachofen UK), cell debris removed, then mitochondrial membranes collected by ultracentrifugation (140 000g) and washed twice. Proteins were solubilized at 20 mg ml−1 in 2.9% (w/v) DDM (dodecyl-β-D-maltoside, Glycon Biochemicals) by stirring for 30 min, and centrifuged, then the solubilized material was supplemented with 50 mm imidazole and NaCl, loaded on to a Nickel-Sepharose 6 Fast Flow column (GE Healthcare) and eluted in 140 mm imidazole. Complex I fractions were pooled, concentrated and eluted from a Superose 6 size-exclusion column in 20 mm Na-MOPS (pH 7.45), 150 mm NaCl, 10% glycerol and 0.05% w/v DDM, concentrated and stored at −80°C.
Preparation of Y. lipolytica NUBM (NDUFV1) complex I variants
The Y. lipolytica NUBM deletion strain, GB10 Δnubm, and the pUB26 plasmid were from Professor U. Brandt (Nijmegen Centre for Mitochondrial Disorders). The full-length NUBM gene was amplified from genomic DNA extracted from the parent GB10 strain with ∼700-bp flanking regions, inserted into the pUB26 plasmid between the ClaI and NheI restriction sites to create the wild-type NUBM expression plasmid and verified by sequencing. Site-directed mutagenesis was carried out by PCR using KOD Xtreme Hot Start DNA Polymerase (Novagen) with non-overlapping primers. The linear products were 5′-phosphorylated, blunt-end-ligated and transformed into E. coli strain 10-β (Invitrogen) for sequencing. Plasmid transformations were performed as described previously (53). Briefly, Δnubm cells were grown at 27°C in 2xYPD overnight, collected by centrifugation and resuspended in buffer containing 45% (w/v) poly(ethylene glycol) 4000, 0.1 m lithium acetate (pH 6.0), 0.1 m DTT and 0.25 µg µl−1 single-stranded carrier DNA. Approximately 100 ng of plasmid DNA was added to 120 µl, and the cells incubated at 40°C for 1 h then plated onto YPD containing 50 µg ml−1 Hygromycin B and incubated for 3 days. Single transformant colonies were selected, grown in 2xYPD with Hygromycin B and cell stocks frozen in 50% (v/v) glycerol.
Analytical techniques
Kinetic measurements were carried out at 32°C in 96-well microtiter plates using a SpectraMax Plus spectrophotometer as described previously (19,54). Assay buffers contained 10 mm Tris–HCl (pH 7.5), 100 µm NADH and 1 mm K3[Fe(CN)6] (ferricyanide, FeCN), 1 mm 3-acetylpyridine adenine dinucleotide (APAD+), 3.5 mm [Ru(NH3)6]Cl3 (hexaammineruthenium III, HAR), 0.25 mm paraquat or 100 µm decylubiquinone (DQ) as required. Activities were monitored at 340–380 nm (ε = 4.81 mm−1 cm−1 or 5.84 mm−1 cm−1 for NADH:APAD+) except for the NADH:FeCN reaction at 420–450 nm (ε = 1.03 mm−1 cm−1). For isolated complex, 0.05% (w/v) asolectin and 0.05% (w/v) CHAPS were added to NADH:DQ oxidoreduction assays. When required, piericidin A was added to 2 µm to inhibit ubiquinone reduction. H2O2 production was measured using 10 µm Amplex Red and 2 U ml−1 horseradish peroxidase (HRP, Sigma) at 557–620 nm (ε = 51.6 mm−1 cm−1) as described previously (16). Flavin dissociation and flavin content (relative to WT) were assessed using a method described previously (38,39): specifically, an ABI 7900HT real-time PCR machine was used to monitor the flavin fluorescence; the temperature was held at 20°C for 2 min and then increased by 1.5° C every 30 s. The data were fit to sigmoidal curves, with the total intensity change proportional to the total amount of complex I-bound flavin, and the steepest section of the sigmoid at the characteristic dissociation temperature.
SDS–PAGE was carried out using 10–20% acrylamide gels (Invitrogen) and visualized with 0.2% Coomassie Blue R250. For BN-PAGE, membranes were solubilized by incubation on ice for 30 min in DDM (1.5 mg DDM per milligram protein), centrifuged, applied to 3–12% Bis–Tris native-PAGE gels (Invitrogen) and run according to the manufacturer's instructions. Proteins were visualized using colloidal Coomassie R250, or the gels were destained with MilliQ water for in-gel complex I assays performed using 120 µm NADH and 0.5 mg ml−1 nitro blue tetrazolium (NBT) (55).
Supplementary Material
Funding
This work was supported by the Medical Research Council (grant number U105663141 to J.H.). E.A. was supported by the Amgen Scholars programme. Funding to pay the Open Access publication charges for this article was provided by The Medical Research Council.
Supplementary Material
Acknowledgements
We thank Shamima Rahman (UCL) and Massimo Zeviani (MRC) for advice on complex I-linked mitochondrial disorders and Alan J. Robinson and Cassandra L. Smith (MBU) for the NDUFV1 sequence alignment.
Conflict of Interest statement. None declared.
References
- 1.Hirst J. (2013) Mitochondrial complex I. Annu. Rev. Biochem., 82, 551–575. [DOI] [PubMed] [Google Scholar]
- 2.Murphy M.P. (2009) How mitochondria produce reactive oxygen species. Biochem. J., 417, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carroll J., Fearnley I.M., Skehel J.M., Shannon R.J., Hirst J., Walker J.E. (2006) Bovine complex I is a complex of 45 different subunits. J. Biol. Chem., 281, 32724–32727. [DOI] [PubMed] [Google Scholar]
- 4.Vinothkumar K.R., Zhu J., Hirst J. (2014) Architecture of mammalian complex I. Nature, 515, 80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baradaran R., Berrisford J.M., Minhas G.S., Sazanov L.A. (2013) Crystal structure of the entire respiratory complex I. Nature, 494, 443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fassone E., Rahman S. (2012) Complex I deficiency: clinical features, biochemistry and molecular genetics. J. Med. Genet., 49, 578–590. [DOI] [PubMed] [Google Scholar]
- 7.Koene S., Rodenburg R.J., van der Knaap M.S., Willemsen M.A.A.P., Sperl W., Laugel V., Ostergaard E., Tarnopolsky M., Martin M.A., Nesbitt V. et al. (2012) Natural disease course and genotype-phenotype correlations in complex I deficiency caused by nuclear gene defects: what we learned from 130 cases. J. Inherit. Metab. Dis, 35, 737–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahlers P.M., Garofano A., Kerscher S.J., Brandt U. (2000) Application of the obligate aerobic yeast Yarrowia lipolytica as a eucaryotic model to analyse Leigh syndrome mutations in the complex I core subunits PSST and TYKY. Biochim. Biophys. Acta, 1459, 258–265. [DOI] [PubMed] [Google Scholar]
- 9.Kerscher S., Grgic L., Garofano A., Brandt U. (2004) Application of the yeast Yarrowia lipolytica as a model to analyse human pathogenic mutations in mitochondrial complex I (NADH:ubiquinone oxidoreductase). Biochim. Biophys. Acta, 1659, 197–205. [DOI] [PubMed] [Google Scholar]
- 10.Duarte M., Schulte U., Ushakova A.V., Videira A. (2005) Neurospora strains harboring mitochondrial disease-associated mutations in iron-sulfur subunits of complex I. Genetics, 171, 91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zickermann V., Barquera B., Wikström M., Finel M. (1998) Analysis of the pathogenic human mitochondrial mutation ND1/3460, and mutations of strictly conserved residues in its vicinity, using the bacterium Paracoccus denitrificans. Biochemistry, 37, 11792–11796. [DOI] [PubMed] [Google Scholar]
- 12.Kervinen M., Hinttala R., Helander H.M., Kurki S., Uusimaa J., Finel M., Majamaa K., Hassinen I.E. (2006) The MELAS mutations 3946 and 3949 perturb the critical structure in a conserved loop of the ND1 subunit of mitochondrial complex I. Hum. Mol. Genet., 15, 2543–2552. [DOI] [PubMed] [Google Scholar]
- 13.Pätsi J., Kervinen M., Finel M., Hassinen I.E. (2008) Leber hereditary optic neuropathy mutations in the ND6 subunit of mitochondrial complex I affect ubiquinone reduction kinetics in a bacterial model of the enzyme. Biochem. J., 409, 129–137. [DOI] [PubMed] [Google Scholar]
- 14.Pätsi J., Maliniemi P., Pakanen S., Hinttala R., Uusimaa J., Majamaa K., Nyström T., Kervinen M., Hassinen I.E. (2012) LHON/MELAS overlap mutation in ND1 subunit of mitochondrial complex I affects ubiquinone binding as revealed by modeling in Escherichia coli NDH-1. Biochim. Biophys. Acta, 1817, 312–318. [DOI] [PubMed] [Google Scholar]
- 15.Gabaldón T., Rainey D., Huynen M.A. (2005) Tracing the evolution of a large protein complex in the eukaryotes, NADH:ubiquinone oxidoreductase (complex I). J. Mol. Biol., 348, 857–870. [DOI] [PubMed] [Google Scholar]
- 16.Kussmaul L., Hirst J. (2006) The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Natl Acad. Sci. USA, 103, 7607–7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yakovlev G., Hirst J. (2007) Transhydrogenation reactions catalyzed by mitochondrial NADH-ubiquinone oxidoreductase (complex I). Biochemistry, 46, 14250–14258. [DOI] [PubMed] [Google Scholar]
- 18.King M.S., Sharpley M.S., Hirst J. (2009) Reduction of hydrophilic ubiquinones by the flavin in mitochondrial NADH:ubiquinone oxidoreductase (complex I) and production of reactive oxygen species. Biochemistry, 48, 2053–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Birrell J.A., Yakovlev G., Hirst J. (2009) Reactions of the flavin mononucleotide in complex I: a combined mechanism describes NADH oxidation coupled to the reduction of APAD+, ferricyanide or molecular oxygen. Biochemistry, 48, 12005–12013. [DOI] [PubMed] [Google Scholar]
- 20.Birrell J.A., King M.S., Hirst J. (2011) A ternary mechanism for NADH oxidation by positively charged electron acceptors, catalyzed at the flavin site in respiratory complex I. FEBS Lett., 585, 2318–2322. [DOI] [PubMed] [Google Scholar]
- 21.Pfeffer G., Horvath R., Klopstock T., Mootha V.K., Suomalainen A., Koene S., Hirano M., Zeviani M., Bindoff L.A., Yu-Wai-Man P. et al. (2013) New treatments for mitochondrial disease - no time to drop our standards. Nat. Rev. Neurol., 9, 474–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horvath R. (2012) Update on clinical aspects and treatment of selected vitamin-responsive disorders II (riboflavin and CoQ10). J. Inherit. Metab. Dis., 35, 679–687. [DOI] [PubMed] [Google Scholar]
- 23.Stenson P.D., Mort M., Ball E.V., Shaw K., Phillips A.D., Cooper D.N. (2014) The human gene mutation database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum. Genet., 133, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marin S.E., Mesterman R., Robinson B., Rodenburg R.J., Smeitink J., Tarnopolsky M.A. (2013) Leigh syndrome associated with mitochondrial complex I deficiency due to novel mutations In NDUFV1 and NDUFS2. Gene, 516, 162–167. [DOI] [PubMed] [Google Scholar]
- 25.Calvo S.E., Tucker E.J., Compton A.G., Kirby D.M., Crawford G., Burtt N.P., Rivas M., Guiducci C., Bruno D.L., Goldberger O.A. et al. (2010) High-throughput, pooled sequencing identifies mutations in NUBPL and FOXRED1 in human complex I deficiency. Nat. Genet., 42, 851–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dinwiddie D.L., Smith L.D., Miller N.A., Atherton A.M., Farrow E.G., Strenk M.E., Soden S.E., Saunders C.J., Kingsmore S.F. (2013) Diagnosis of mitochondrial disorders by concomitant next-generation sequencing of the exome and mitochondrial genome. Genomics, 102, 148–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laugel V., This-Bernd V., Cormier-Daire V., Speeg-Schatz C., de Saint-Martin A., Fischbach M. (2007) Early-onset ophthalmoplegia in Leigh-like syndrome due to NDUFV1 mutations. Pediatr. Neurol., 36, 54–57. [DOI] [PubMed] [Google Scholar]
- 28.Bénit P., Chretien D., Kadhom N., de Lonlay-Debeney P., Cormier-Daire V., Cabral A., Peudenier S., Rustin P., Munnich A., Rotig A. (2001) Large-scale deletion and point mutations of the nuclear NDUFV1 and NDUFS1 genes in mitochondrial complex I deficiency. Am. J. Hum. Genet., 68, 1344–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zafeiriou D.I., Rodenburg R.J.T., Scheffer H., van den Heuvel L.P., Pouwels P.J.W., Ververi A., Athanasiadou-Piperopoulou F., van der Knaap M.S. (2008) MR spectroscopy and serial magnetic resonance imaging in a patient with mitochondrial cystic leukoencephalopathy due to complex I deficiency and NDUFV1 mutations and mild clinical course. Neuropediatrics, 39, 172–175. [DOI] [PubMed] [Google Scholar]
- 30.Schuelke M., Smeitink J., Mariman E., Loeffen J., Plecko B., Trijbels F., Stöckler-Ipsiroglu S., van den Heuvel L. (1999) Mutant NDUFV1 subunit of mitochondrial complex I causes leukodystrophy and myoclonic epilepsy. Nat. Genet., 21, 260–261. [DOI] [PubMed] [Google Scholar]
- 31.Breningstall G.N., Shoffner J., Patterson R.J. (2008) Siblings with leukoencephalopathy. Semin. Pediatr. Neurol., 15, 212–215. [DOI] [PubMed] [Google Scholar]
- 32.Vilain C., Rens C., Aeby A., Balériaux D., Van Bogaert P., Remiche G., Smet J., Van Coster R., Abramowicz M., Pirson I. (2012) A novel NDUFV1 gene mutation in complex I deficiency in consanguineous siblings with brainstem lesions and Leigh syndrome. Clin. Genet., 82, 264–270. [DOI] [PubMed] [Google Scholar]
- 33.Berrisford J.M., Sazanov L.A. (2009) Structural basis for the mechanism of respiratory complex I. J. Biol. Chem., 284, 29773–29783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sazanov L.A., Hinchliffe P. (2006) Structure of the hydrophilic domain of respiratory complex I from Thermus thermophilus. Science, 311, 1430–1436. [DOI] [PubMed] [Google Scholar]
- 35.Kerscher S., Dröse S., Zwicker K., Zickermann V., Brandt U. (2002) Yarrowia lipolytica, a yeast genetic system to study mitochondrial complex I. Biochim. Biophys. Acta, 1555, 83–91. [DOI] [PubMed] [Google Scholar]
- 36.Kerscher S.J., Okun J.G., Brandt U. (1999) A single external enzyme confers alternative NADH:ubiqinone oxidoreductase activity in Yarrowia lipolytica. J. Cell Sci., 112, 2347–2354. [DOI] [PubMed] [Google Scholar]
- 37.Hirst J. (2010) Towards the molecular mechanism of respiratory complex I. Biochem. J., 425, 327–339. [DOI] [PubMed] [Google Scholar]
- 38.Forneris F., Orru R., Bonivento D., Chiarelli L.R., Mattevi A. (2009) ThermoFAD, a Thermofluor-adapted flavin ad hoc detection system for protein folding and ligand binding. FEBS J., 276, 2833–2840. [DOI] [PubMed] [Google Scholar]
- 39.Birrell J.A., Morina K., Bridges H.R., Friedrich T., Hirst J. (2013) Investigating the function of [2Fe-2S] cluster N1a, the off-pathway cluster in complex I, by manipulating its reduction potential. Biochem. J., 456, 139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bugiani M., Invernizzi F., Alberio S., Briem E., Lamantea E., Carrara F., Moroni I., Farina L., Spada M., Donati M.A. et al. (2004) Clinical and molecular findings in children with complex I deficiency. Biochim. Biophys. Acta, 1659, 136–147. [DOI] [PubMed] [Google Scholar]
- 41.Mayr J.A., Haack T.B., Freisinger P., Karall D., Makowski C., Koch J., Feichtinger R.G., Zimmermann F.A., Rolinski B., Ahting U. et al. (2015) Spectrum of combined respiratory chain defects. J. Inherit. Metab. Dis., 38, 629–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lieber D.S., Calvo S.E., Shanahan K., Slate N.G., Liu S., Hershman S.G., Gold N.B., Chapman B.A., Thorburn D.R., Berry G.T. et al. (2013) Targeted exome sequencing of suspected mitochondrial disorders. Neurology, 80, 1762–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Verkaart S., Koopman W.J.H., van Emst-de Vries S.E., Nijtmans L.G., van den Heuvel L.W.P.J., Smeitink J.A.M., Willems P.H.G.M. (2007) Superoxide production is inversely related to complex I activity in inherited complex I deficiency. Biochim. Biophys. Acta, 1772, 373–381. [DOI] [PubMed] [Google Scholar]
- 44.Pryde K.R., Hirst J. (2011) Superoxide is produced by the reduced flavin in mitochondrial complex I: a single, unified mechanism that applies during both forward and reverse electron transfer. J. Biol. Chem., 286, 18056–18065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kumar P., Henikoff S., Ng P.C. (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc., 4, 1073–1082. [DOI] [PubMed] [Google Scholar]
- 46.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. (2010) A method and server for predicting damaging missense mutations. Nat. Methods, 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Foley A.R., Menezes M.P., Pandraud A., Gonzalez M.A., Al-Odaib A., Abrams A.J., Sugano K., Yonezawa A., Manzur A.Y., Burns J. et al. (2014) Treatable childhood neuronopathy caused by mutations in riboflavin transporter RFVT2. Brain, 137, 44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schiff M., Haberberger B., Xia C., Mohsen A.-W., Goetzman E.S., Wang Y., Uppala R., Zhang Y., Karunanidhi A., Prabhu D. et al. (2015) Complex I assembly function and fatty acid oxidation enzyme activity of ACAD9 both contribute to disease severity in ACAD9 deficiency. Hum. Mol. Genet., 24, 3238–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin C.S., Sharpley M.S., Fan W., Waymire K.G., Sadun A.A., Carelli V., Ross-Cisneros F.N., Baciu P., Sung E., McManus M.J. et al. (2012) Mouse mtDNA mutant model of Leber hereditary optic neuropathy. Proc. Natl Acad. Sci. USA, 2012, 20065–20070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kashani-Poor N., Kerscher S., Zickermann V., Brandt U. (2001) Efficient large scale purification of his-tagged proton translocating NADH:ubiquinone oxidoreductase (complex I) from the strictly aerobic yeast Yarrowia lipolytica. Biochim. Biophys. Acta, 1504, 363–370. [DOI] [PubMed] [Google Scholar]
- 51.Dröse S., Galkin A., Brandt U. (2005) Proton pumping by complex I (NADH:ubiquinone oxidoreductase) from Yarrowia lipolytica reconstituted into proteoliposomes. Biochim. Biophys. Acta, 1710, 87–95. [DOI] [PubMed] [Google Scholar]
- 52.Bridges H.R., Grgic L., Harbour M.E., Hirst J. (2009) The respiratory complexes I from the mitochondria of two Pichia species. Biochem. J., 422, 151–159. [DOI] [PubMed] [Google Scholar]
- 53.Chen D.-C., Beckerich J.-M., Gaillardin C. (1997) One-step transformation of the dimorphic yeast Yarrowia lipolytica. Appl. Microbiol. Biotechnol., 48, 232–235. [DOI] [PubMed] [Google Scholar]
- 54.Sharpley M.S., Shannon R.J., Draghi F., Hirst J. (2006) Interactions between phospholipids and NADH:ubiquinone oxidoreductase (complex I) from bovine mitochondria. Biochemistry, 45, 241–248. [DOI] [PubMed] [Google Scholar]
- 55.Wittig I., Karas M., Schägger H. (2007) High resolution clear native electrophoresis for in-gel functional assays and fluorescence studies of membrane protein complexes. Mol. Cell Proteomics, 6, 1215–1225. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.