

Abstract
There is a connection between nutrient inputs, energy-sensing pathways, lifespan variation and aging. Despite the role of metabolic enzymes in energy homeostasis and their metabolites as nutrient signals, little is known about how their gene expression impacts lifespan. In this report, we use P-element mutagenesis in Drosophila to study the effect on lifespan of reductions in expression of seven central metabolic enzymes, and contrast the effects on normal diet and dietary restriction. The major observation is that for five of seven genes, the reduction of gene expression extends lifespan on one or both diets. Two genes are involved in redox balance, and we observe that lower activity genotypes significantly extend lifespan. The hexokinases also show extension of lifespan with reduced gene activity. Since both affect the ATP/ADP ratio, this connects with the role of AMP-activated protein kinase as an energy sensor in regulating lifespan and mediating caloric restriction. These genes possess significant expression variation in natural populations, and our experimental genotypes span this level of natural activity variation. Our studies link the readout of energy state with the perturbation of the genes of central metabolism and demonstrate their effect on lifespan.
Keywords: aging, redox balance, energy homeostasis, caloric restriction
1. Introduction
In their lifetime, all organisms experience environments that change both temporally and spatially in their nutrient availability and energy content. The optimal utilization and storage of available energy is a physiological challenge that shapes variation in life-history phenotypes, and in principle sets the trade-off between lifespan and reproduction [1]. Failing to allocate energy optimally to the changing availability of nutrients would be expected to have significant fitness costs. As a consequence, nutrient sensing and response networks are strongly conserved pathways [2]. Since there are optimal physiological responses that reset internal energy balance for different environments, we should expect genetic variation associated with these responses.
As potential modifiers of cell energy state through their action on metabolite levels, the genes of central metabolism are potential sources for this genetic variation. The response to changing nutrient input results in intercellular signals that drive a cascade of downstream gene transcription shifts that facilitate energy utilization and storage [3]. The proximal signal is generally derived from specific metabolite levels as they change under shifting nutrient load [4]. There is considerable precedent for this general mechanism; for example, the well-known action of glucose on insulin secretion in vertebrates [5], and the signals associated with the secretion and action of adipokinetic hormone (AKH) in insects [6]. The effect of energy balance on longevity in yeast is well known [7,8]. Since metabolite concentrations act as proximal signals and also appear to correlate with the associated gene expression levels of the component steps in metabolism [9–11], this relationship clearly implicates the natural genetic variation in expression (or activity) of the central metabolic genes as potential sources of genetic variation in setting metabolite levels, and thus play a role in sensing and setting responses. Moreover, we expect specific enzymes will emerge as the targets of natural selection where genetic expression or activity level will act as an ‘energy-stat’. Different genotypes will bracket and set the nutrient levels involved in triggering downstream responses that affect traits associated with fitness, such as lifespan variation and fecundity.
The well-established observation that nutritional or dietary restriction (DR) extends lifespan in many species [12,13] is mechanistically related to energy-state signalling [14]. In Drosophila, many studies have shown DR to extend lifespan [15–19], and using genetic manipulation, many of the signalling pathways have been identified that extend lifespan and thus effectively mimic DR [2,20]. Experimental work has shown connections between energy-signalling steps and other energy correlated phenotypes such as starvation resistance, nutrient storage and stress resistance that have connections to DR [21–24].
In both plants and animals, it is becoming apparent that many metabolic genes and their enzyme products are associated with roles other than simple processing of metabolites [25–29]. Based on evidence from RNAi reduction screens of central metabolic genes in Caenorhabditis elegans, it is estimated that as much as 25% of the genes screened implicate metabolism in longevity extension [30,31]. RNAi knockdown of expression of several genes in the mitochondrial respiratory chain has been shown to extend lifespan in Drosophila [32]. In yeast, the overexpression of several of the genes involved in cofactor shuttles actually extends lifespan [7]. Despite its importance as a model in lifespan studies, and the connection between metabolism and lifespan seen in other models, no studies in Drosophila have examined the mutational perturbation of central metabolic gene activity and their effects on longevity.
Natural populations of Drosophila melanogaster vary in both average lifespan and response of lifespan to dietary challenge [19,33]. In D. melanogaster, the genes of the central metabolic pathway harbour considerable sequence and expression variation [34–36], which often shows change with latitude and season [37–39]. In this regard, unlike the other experimental models used in aging studies, Drosophila offers the unique opportunity to associate aging and signalling pathways with their population genetics [40,41].
The long-term goal of our studies is to integrate geographical and seasonal variation in metabolic genes with life-history phenotypes, such as longevity and its fitness correlates [42]. In this report, we use matched sets of P-element excision alleles in D. melanogaster to create genotypes that possess modest reductions in gene expression and subsequently examine the impact of these perturbations on lifespan under normal and restricted diets. We study seven metabolic genes: Idh, Mdh2, Hex-C, Hex-A, Gpdh, Gdh and Men. These enzymes involve possible signalling via glucose, ATP/ADP, NAD/NADH and NADP/NADPH ratios, citrate, pyruvate, malate and glutamate, and they also involve genes with primary expression restricted to the cytoplasm or the mitochondria. We observe a range of effects on lifespan, from none at all to very significant increases in lifespan that can depend on diet.
2. Material and methods
(a). Lifespan design
Under any diet, each genotype was represented by 20 vials; each experiment (genotype and diet) consisted of a total of 80 vials. Each vial has 20 five-day-old virgin females, for a total of 1600 flies phenotyped for each enzyme examined. Flies were transferred to fresh media every 3–4 days and surviving individuals counted at 1–3-day intervals. To make all results comparable with other published studies, food treatments were matched with the dietary formulae used by Min et al. [43]. Normal or high-nutrient food (normal diet; DN) consisted of 16 g yeast (SAF Dry Autolyzed), 16 g sucrose and 5.2 g cornmeal. Low-nutrient (DR) food consisted of 4 g yeast (SAF Dry Autolyzed), 4 g sucrose and 5.2 g cornmeal per 100 ml. Vials were not seeded with live yeast. All lines and lifespan studies were maintained at 25°C, and all genotype and dietary experiments for each gene were carried out concurrently.
All experiments were carried out using paired P-element excision lines. All genetic backgrounds were replaced as described in Merritt et al. [44] and therefore each paired set differs only in the gene of interest [44–46]. The P-element insertions used to generate the excision allele pairs were: Idh, IdhEP3729; Mdh2, Mdh2EY01940; Gdh, GdhKG00965; Men, MenBG02790; Hex-A (20817) [EP(X)352]; Hex-C, CG8079KG09154. Hex-A, Idh and Gpdh alleles have been described previously [44,46,47]. In each gene, we have an excision allele with impaired activity and an excision allele with normal activity. Enzyme activities were assayed as in earlier work [44,46–49] and are shown in units of change in optical density per minute (ΔOD). The Men9D allele is a perfect excision. The IdhΔ1 allele had three single base substitutions in the first 100 nucleotides of the transcript and is an Idh knockout [47]. The Men7SC2 allele possessed a small 14-nucleotide retention of the 3′ end of the GT element at the insertion site, and acquired a 788-nucleotide deletion 120 nucleotides up-stream of the insertion site. The Gdh9.3 allele is a perfect excision. The Gdh24.1 allele is an imperfect excision that retains a large portion of the original P-element. The Mdh220B1 excision allele was homozygous lethal and possessed a large 986-nucleotide deletion that removed part of exon 1.
The survival data were analysed using a Cox proportional-hazards model. Individual flies were counted for survival. These observations were used in a Cox proportional-hazards regression against the covariates of genotype and replicate vial for each diet separately. Conversely, diet effects were tested for each genotype separately using the covariates of diet and vial. The relative risk associated with each of the covariates is evaluated statistically as a log-likelihood test with corresponding χ2 and probability. These were carried out in JMP v. 11.2.0 (SAS Institute Inc.).
(b). Hexokinase activities
Determining the relative activity reductions associated with Hex-A and Hex-C genotypes is complex since whole fly hexokinase assays combine the activities for both HEXA and HEXC enzymes. The Hex-A gene is primarily expressed in the nervous tissue and muscle. The Hex-A74 allele possesses an eight-nucleotide deletion that results in a truncated protein and should be a complete knockout of HEXA. Hex-C is largely fat body expressed [50], and the Hex-C35 allele possesses a large deletion that removes all of the 3′UTR and much of the exon and should be a knockout. We estimated the activities associated with the combination of the genotypes for alleles Hex-A79 and Hex-A74, and Hex-C4 and Hex-C35 (figure 1), all in 6326 backgrounds. The genotypes were produced by the four crosses (1) Hex-A74/6326;6326 females X 6326/Y; Hex-C4/6326 males, (2) Hex-A79/6326;6326 females X 6326/Y; Hex-C4/6326 males, (3) Hex-A79/6326;6326 females X 6326/Y; Hex-C35/6326 males and (4) Hex-A74/6326;6326 females X 6326/Y; Hex-C35/6326 males. The two-way analysis-of-variance (ANOVA) showed statistically significant reduction in activity associated with each allele (table 1). We estimated that, compared with the presumed full-activity Hex-A79/6326 and Hex-C4/6326 genotypes, the Hex-A74/6326 genotype reduced overall whole-body hexokinase activity by 27%, and the Hex-C35/6326 genotype reduces it by 8% (table 2).
Figure 1.
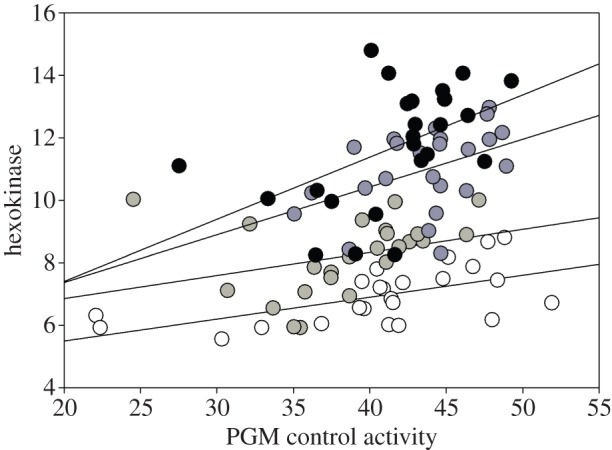
Hexokinase activity of progeny genotypes. The genotypes were produced by the four crosses: (1) Hex-A74/6326;6326 females X 6326/Y; Hex-C4/6326 males, (2) Hex-A79/6326;6326 females X 6326/Y; Hex-C4/6326 males, (3) Hex-A79/6326;6326 females X 6326/Y; Hex-C35/6326 males and (4) Hex-A74/6326;6326 females X 6326/Y; Hex-C35/6326 males. Black = Hex-A79/6326; Hex-C4/6326. Dark grey = Hex-A79/6326;6326/Hex-C35. Light grey = Hex-A74/6326; Hex-C4/6326; white = Hex-A74/6326; Hex-C35/6326. (Online version in colour.)
Table 1.
Two-way ANOVA of hexokinase activities of the Hex-A and Hex-C genotypes.
| genotype | estimate | s.e. | t-ratio | p |
|---|---|---|---|---|
| Hex-A74 | −1.716 | 0.133 | −12.82 | <0.0001 |
| Hex-C35 | −0.609 | 0.133 | −4.64 | <0.0001 |
| Hex-A74X Hex-C35 | −0.141 | 0.129 | −1.09 | 0.2782 |
| PGM control activity | 0.1055 | 0.0241 | 4.38 | <0.0001 |
Table 2.
Mean hexokinase activities of the Hex-A and Hex-C genotypes.
| genotype | hex activity | s.e. |
|---|---|---|
| Hex-A79/6326; Hex-C4/6326 | 11.65 | 0.258 |
| Hex-A79/6326; Hex-C35/6326 | 10.71 | 0.265 |
| Hex-A74/6326; Hex-C4/6326 | 8.49 | 0.265 |
| Hex-A74/6326; Hex-C35/6326 | 7.00 | 0.258 |
However, the tissue-specific reduction of hexokinase activity associated with each gene and not whole-body activity is the relevant phenotype and its estimation depends on the relative contribution of each tissue to whole-body HEX activity, as well as the activities of the Hex-A and Hex-C alleles on the X and second 6326 chromosomes. The relative contribution of the two enzymes estimated from the isoelectric focusing data [50] is a notably larger contribution of the Hex-A gene to whole-body HEX activity. This would appear to explain the smaller reduction for the Hex-C35/6326 genotype. If, as the data suggest, that HEX-C is about 25% of whole-body activity, then it is predicted that the 8% reduction associated with Hex-C35/6326 translates into a 45% HEX reduction in Hex-C tissues. The overall analysis supports the expectation that the tissue-associated activities have reduced the knockout heterozygotes by about 50% of normal activity.
3. Results
Idh (isocitrate dehydrogenase (cytosolic)). The results are shown in figure 2a. The IdhΔ1/6326 genotype has a 50% activity reduction in IDH activity (figure 2a, inset) [47]. While there is an overall highly significant DR effect in both genotypes (IdhΔ1/6326, χ2 = 45.50, p < 0.0001; IdhΔ4/6326, χ2 = 65.53, p < 0.0001), there are no significant Idh genotype effects on lifespan on either diet.
Figure 2.

The survival of Idh and Men genotypes of female D. melanogaster under normal and DR. (a) Idh genotypes IdhΔ1/6326 (grey) and Δ4/6326 (black) under normal (DN, circles) and DR (squares). Relative Idh genotype activities are shown in inset. (b) Men genotypes Men7SC2/6326 (grey) and 9D/6326 (black) under normal (DN, circles) and DR (squares). Relative Men genotype activities are shown in inset.
Men (malic enzyme (cytosolic)). The Men7SC2/6326 genotype has 52.3% of the activity of the Men9D/6326 genotype (F1,28 = 37.6, p < 0.0001). The results are shown in figure 2b. There is a highly significant diet effect, for the high-activity Men genotype (Men9D/6326, χ2 = 288.28, p < 0.0001), but not the low-activity Men7SC2/6326 genotype (less than 1 day, χ2 = 1.15, p < 0.2837). Under DR, the Men genotypes had significantly different lifespans (χ2 = 11.19, p < 0.0008), where the normal activity genotype showed a large and highly significant extension of lifespan compared with the reduced activity genotype (more than 12 days; χ2 = 461.41, p < 0.0001).
Hex-A (hexokinase-A). The results for Hex-A are summarized in figure 3a. Both genotypes responded to DR significantly (Hex79/6326, χ2 = 35.08, p < 0.0001; Hex74/6326, χ2 = 6.924, p < 0.0085, less than 1 day). There was a significant genotype difference under DN diet (χ2 = 67.92, p < 0.0001), with the lower-activity Hex-A74/6326 genotype living 10% longer (more than 4 days). There was no significant difference between genotypes under DR (χ2 = 0.0036, p < 0.952)
Figure 3.

The survival of Hex-A and Hex-C genotypes of female D. melanogaster under normal and DR. (a) Hex-A genotypes 74/6326 (grey) and 79/6326 (black) under normal (DN, circles) and DR (squares). (b) Hex-C genotypes 35/6326 (grey) and 4/6326 (black) under normal (DN, circles) and DR (squares). Relative activities are discussed in text.
Hex-C (hexokinase-C). Both diet and Hex-C genotype had effects on lifespan (figure 3b). There was a DR effect for both the normal full-activity Hex-C4/6326 genotype (χ2 = 116.5, p < 0.0001) and Hex-C35 (χ2 = 48.1, p < 0.0001) genotypes. There were significant extensions of lifespan (more than 6 days) for the low Hex-C35/6326 genotype compared with the normal activity Hex-C4/6326 genotype under both diets, although the DR genotype effect was small (DN, χ2 = 50.58, p < 0.0001; DR, χ2 = 12.41, p < 0.0004).
Gdh (glutamate dehydrogenase (mitochondrial) (figure 4a)). The Gdh24.1/6326 genotype had 71% of the activity of the normal Gdh9.3/6326 genotype (F1,87 = 364.2, p < 0.0001). Both genotypes showed an effect of DR on lifespan (Gdh24.1/6326; χ2 = 145.70, p < 0.0001; Gdh9.3/6326, χ2 = 43.48, p < 0.0001). The normal and reduced Gdh genotypes had identical lifespans under DN diet (χ2 = 0.461, p < 0.4970). Under DR, the Gdh24.1/6326 genotype shows a significantly longer lifespan (more than 14 days; χ2 = 89.37, p < 0.0001).
Figure 4.

The survival of Gdh and Mdh2 genotypes of female D. melanogaster under normal and DR. (a) Gdh genotypes 24.1/6326 (grey) and 9.3/6326 (black) under normal (DN, circles) and DR (squares). Relative Gdh genotype activities are shown in inset. (b) Mdh2 genotypes 20B1/6326 (grey) and 21W1/6326 (black) under normal (DN, circles) and DR (squares). Relative Mdh2 genotype activities are shown in inset.
Mdh2 (malate dehydrogenase (mitochondrial) (figure 4b)). About 85% of the malate dehydrogenase activity in the whole body is MDH2 (mitochondrial) in origin [51]. Mdh-220b1/6326 had 56% of the activity of the normal Mdh21w1/6326 genotype (F1,27 = 99.75, p < 0.0001). For both Mdh2 genotypes, there was a large DR effect (Mdh21w1/6326, χ2 = 138.84, p < 0.0001; Mdh-220b1/6326, χ2 = 259.62, p < 0.0001). The low-activity Mdh220B1/6326 genotype possessed a significantly longer lifespan than the higher-activity genotype under DR (χ2 = 12.68, p < 0.0004), but not under DN (χ2 = 0.48, p < 0.4894)
Gpdh (glycerol-3-phosphate dehydrogenase (figure 5)). The Gpdh9.2 and Gpdh10.3 alleles were intercrossed to produce the test genotypes. The Gpdh9.2 allele is a full knockout and the Gpdh9.2/Gpdh10.3 genotype had 59.6% normal activity (F1,69 = 339.5, p < 0.0001). The Gpdh genotypes individually showed no effect of DR (Gpdh10.3/Gpdh10.3, χ2 = 2.22, p < 0.1359; Gpdh9.2/Gpdh10.3, χ2 = 0.49, p < 0.4831). There was a large and highly significant difference in the effect of Gpdh genotype on lifespan seen at both diets (DN, χ2 = 28.39, p < 0.0001; DR, χ2 = 40.37, p < 0.0001), with the lower-activity Gpdh9.2/Gpdh10.3genotype possessing the significantly longer average lifespan under both diets (5.5 days or 20% extension in lifespan).
Figure 5.

The survival of Gpdh genotypes of female D. melanogaster under normal and DR. Gpdh genotypes 9.2/10.3 (grey) and 10.3/10.3 (black) under normal (DN, circles) and DR (squares). Relative Gpdh genotype activities are shown in inset.
4. Discussion
Energy homeostasis is intimately connected to the signals of nutrient sensing, and it is expected that energy balance ultimately sets the life-history trade-offs that establish longevity. In D. melanogaster, we observe that the genetic perturbation of central metabolic genes has significant effects on lifespan in our experimental setting. Moreover, the general observation is that low-activity genotypes show extension of lifespan. Perhaps not surprisingly, some genotype effects are also diet dependent. Some genes (Idh, Mdh2, Hex-C) show an increase in lifespan under DR, but no effect of genotype, while others show a strong (Gdh, Men) genotype dependence in their response to DR. Finally, Gpdh shows a strong dependence on genotype activity, yet no effect of DR. These differences are expected because our observations represent seven enzymes that act on different metabolites and cofactors, and are limited to mitochondrial or cytosolic function.
In our work, we cannot strictly compare experimental outcomes across genes. First, while we are studying single gene effects within identical genetic backgrounds, the backgrounds differ across the seven gene sets. The P-element progenitor lines differ in the type of element used in the excision series (e.g. KG versus EP), and while the replacement backgrounds possess some chromosomes in common (often the 6326 chromosomes), they generally differ in others. Second, while all of our genotype comparisons involve reductions of activity of 50% or less, we should not expect the same level of flux control across enzymes [42]. Thus, cytosolic IDH may possess little flux control over NADPH/NADP levels, while MEN may exercise greater control over these metabolites, especially at reduced nutrient levels. It should be pointed out that in the Raleigh population the cytosolic Idh gene bears little molecular polymorphism and the few SNPs seen show little cis-based expression effect or clinal change [38].
It should also be emphasized that unlike many gene-targeted lifespan studies, we are not using full knockout genotypes, or genotypes where the relative functional reduction is unknown, and we have precise estimates of genotype activity. Moreover, these activity differences are representative of the range of much of the cis-associated SNP expression variation seen in natural populations [34,38]. The observation that metabolic genes, when perturbed modestly in activity, have an effect on lifespan is certainly relevant to discussions of the maintenance of genetic variation in these genes in natural populations, especially as nutrient levels vary geographically and seasonally [38,39].
Gpdh, Gdh and Hex-A were highlighted in our study of clinal SNP expression variation in the pathway [38]. These genes, among others, showed significant changes in gene transcript expression with latitude. Our observations here add a fitness component to the causes of genetic expression variation of metabolic genes in natural populations of Drosophila. Also, the expectation that the gene-specific extension of lifespan can depend on dietary level adds complexity, since we expect nutritional background to shift locally and seasonally in this species. The effect of reduced activity is incrementally small in terms of daily survival, but when integrated over the average lifespan of a fly this can be very significant. This relationship is in contrast to flight metabolism, where similar activity changes have no effects on flight performance [46].
Our enzymes were targeted because they act on different metabolites and cofactors, and are limited to either mitochondrial or cytosolic function. Both IDH and MEN are cytosolic enzymes and NADPH dependent, and, along with the pentose shunt enzymes, provide a significant contribution to the NADPH/NADP pool [52]. Both glutamate (GDH) and malate dehydrogenase (MDH2) are limited to mitochondrial function and, like GPDH, are dependent on NAD/NADH, and will impact that redox balance and its effect on signalling and aging [53]. The two hexokinases, HEX-A and HEX-C, potentially affect the ADP/ATP ratio and have different tissue expressions. They could vary the ADP/ATP content, and in that fashion set energy-state response via regulating AMPK signalling [54–56]. This AMP-activated kinase is a sensor that has effects in Drosophila [57,58], which provides a link between lifespan and caloric restriction [59].
Our observations for the Gpdh and Gdh genes implicate mitochondrial function and the redox balance with lifespan extension. GPDH is part of the essential mitochondrial phosphoglycerol shuttle in insects and is often considered a point of ROS production [60]. The NAD/NADH redox balance is emerging as an important element of lifespan extension in yeast [8,61] and is often considered a direct readout of metabolic state. Moreover, the lifespan extension associated with these genes might also act through the Sir-like enzymes, which are NAD-dependent histone deacetylases that silence chromatin and thus control transcription in a fashion directly coupled to energy-state imbalance [62]. This relationship is important because starvation in Drosophila has been clearly shown to significantly raise the NAD/NADH ratio [63], although the role of Sir2 in lifespan extension in Drosophila has been questioned [45]. GDH is also limited to mitochondrial function, and potentially affects the redox balance and NAD/NADH ratio. It connects glutamate, a key energy-state signalling molecule, to metabolic control, and sits at the important crossroads of carbohydrate and amino acid metabolism [64,65]. As well, glutamate is at the hub of connectivity in the large central metabolic network [66]. Both of these enzymes show significant extension of lifespan with only 40% reductions in whole-body activity. However, all mitochondrial or NAD-dependent genes are not similar in affecting lifespan; comparable activity changes in mitochondrial and NAD-dependent MDH (Mdh2) have little effect on lifespan in either dietary condition.
The dependence of our results on diet is important. Over the past two decades, DR has been shown to impose a trade-off where it extends lifespan and reduces reproduction fecundity [1]. It has gained prominence because of its association with aging research in general [12], but the phenomenon of DR has obvious relevance to studies of life-history evolution because it will be associated with plastic responses to nutritional challenges in nature, and the potential maintenance of genetic variation. Studies on model organisms have led to the discovery of many genes where mutational perturbation extends lifespan and thus mimics DR restriction [12]. This is most notable in the parallel effects of disruption of genes specifically associated with energy-sensing pathways and signalling of dietary state and DR [12]. Despite its general occurrence in D. melanogaster [19], we see that an effect of DR on lifespan is not seen in some of our experimental lines. Two (Gpdh, Hex-A) of the seven genes show no DR effect in general. For Gdh, DR is seen just for the low-activity genotype. This different response to DR is suggested by studies where line-by-diet interactions are noted [67], but is shown more definitively in studies in mice [68] and yeast, where it becomes clear that genetic background affects the response to DR [69,70]. In Schleit et al. [70], 166 single, non-essential genes were made deficient in yeast, and a large proportion of genes showed loss of DR extension capability, as well as an enhanced DR response. Clearly, DR response can be modified genetically and it would not be surprising if genetic variation in natural populations were to reflect this observation.
Does the failure of some expression modified genotypes to respond to DR imply a mechanistic connection to the signalling associated with DR? An interaction between genotype and diet would suggest that the lifespan responses to DR may be coupled to metabolic signals associated with these enzymes or pathways. For example, the full-activity Men genotype shows a significant DR lifespan response, yet the Men low-activity genotype appears resistant to lifespan extension under DR. This may suggest that the NADPH/NADP ratio in the case of MEN is a signal associated with DR. However, the NADPH-dependent IDH shows no genotypic effect on DR, which may contradict this suggestion or simply be because IDH possesses low control over cofactor pool levels. Conversely, the Gdh normal activity genotype shows no DR response, while a genotype reduction in Gdh activity strongly enhances a DR response. Perhaps the reduction in amino acids that is associated with DR in Drosophila [71,72] is enhanced by reduction of Gdh activity, since glutamate and GDH sit at the crossover of carbohydrate and amino acid metabolism in the mitochondria [65]. However, this interaction must be interpreted with caution, since tests of genotype dependence of DR should be tested by using a range of diet changes [73].
Where might the mechanism of action reside that extends lifespan or is associated with a genotype-dependent response to DR for these metabolic genes? Discussions of energy-signalling pathways typically start with the statement that nutrient levels are first ‘sensed’ and then the pathway of interest is addressed (e.g. the insulin receptor insulin/TOR pathway in Drosophila [74]). Presumably, this initial sensing must emanate from direct immediate readouts of the cell's metabolic state (i.e. metabolites). In Drosophila, dietary sugars induce significant metabolite changes [4]. These metabolite levels changes trigger the secretion of neuropeptides from specialized neurosecretory cells [6]. This model of sensing is similar to the regulation of glucagon in mammalian pancreatic cells, and a similar case has been made by Kim & Rulifson [75] for the sensing and regulation of AKH by the corpora cardiac cells in Drosophila. It is possible that either these cell-specific or just systemic metabolite levels initiate the signalling process. Genetic variation will regulate these metabolite levels in conjunction with nutrient inputs. In this sense, lifespan extension by some metabolic genes is top-down.
Over the past two decades, a large number of genes in several model species have been observed to extend lifespan when mutated. In Drosophila, most studies have emphasized the signalling cascades emanating from neurosecretory cells [6]. However, the most proximal steps of central metabolism must set the signalling environments because their metabolite levels respond immediately to nutrient inputs. Here, we place the metabolic pathway in the discussion of energy signalling and look at the impact of genetic perturbation of some key genes on lifespan. The outcome is that there are numerous examples where reductions in activity extend lifespan. Perhaps not surprisingly, this extension depends on the nutrient environment. We propose that this setting of lifespan response to gene expression variation also provides a selective context for naturally segregating metabolic gene variation, and moreover may contribute to unravelling the patterns of genetic variation observed in natural populations.
Acknowledgements
Thomas Merritt donated the Idh excision allele series.
Data accessibility
The raw data for all the lifespan experiments is deposited in Dryad under http://dx.doi.org/10.5061/dryad.r620q.
Authors' contributions
M.E.T., B.B. and R.H. carried out the lifespan experiments. M.E.T. and W.F.E. designed and conceptualized the project. W.F.E. and E.B. drafted the document. W.F.E and E.L. carried out the data analysis. S.K. and E.B. characterized the sequence changes. M.E.T., B.B., M.A. and K.K. recovered the mutant alleles and measured activities.
Competing interests
We declare we have no competing interests.
Funding
This study was supported by National Institutes of Health grant no. (GM090094) to W.F.E. and John True.
References
- 1.Flatt T. 2011. Survival costs of reproduction in Drosophila. Exp. Gerontol. 46, 369–375. ( 10.1016/j.exger.2010.10.008) [DOI] [PubMed] [Google Scholar]
- 2.Owusu-Ansah E, Perrimon N. 2014. Modeling metabolic homeostasis and nutrient sensing in Drosophila: implications for aging and metabolic diseases. Dis. Models Mech. 7, 343–350. ( 10.1242/dmm.012989) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gershman B, Puig O, Hang L, Peitzsch RM, Tatar M, Garofalo RS. 2007. High-resolution dynamics of the transcriptional response to nutrition in Drosophila: a key role for dFOXO. Phys. Genomics 29, 24–34. ( 10.1152/physiolgenomics.00061.2006) [DOI] [PubMed] [Google Scholar]
- 4.Colinet H, Larvor V, Bical R, Renault D. 2013. Dietary sugars affect cold tolerance of Drosophila melanogaster. Metabolomics 9, 608–622. ( 10.1007/s11306-012-0471-z) [DOI] [Google Scholar]
- 5.Polakof S, Mommsen TP, Soengas JL. 2011. Glucosensing and glucose homeostasis: from fish to mammals. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 160, 123–149. ( 10.1016/j.cbpb.2011.07.006) [DOI] [PubMed] [Google Scholar]
- 6.Toivonen JM, Partridge L. 2008. Endocrine regulation of aging and reproduction in Drosophila. Mol. Cell Endocrinol. 299, 39–50. ( 10.1016/j.mce.2008.07.005) [DOI] [PubMed] [Google Scholar]
- 7.Easlon E, Tsang F, Skinner C, Wang C, Lin SJ. 2008. The malate-aspartate NADH shuttle components are novel metabolic longevity regulators required for calorie restriction-mediated life span extension in yeast. Genes Dev. 22, 931–944. ( 10.1101/gad.1648308) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin SJ, Ford E, Haigis M, Liszt G, Guarente L. 2004. Calorie restriction extends yeast life span by lowering the level of NADH. Genes Dev. 18, 12–16. ( 10.1101/gad.1164804) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fendt SM, Buescher JM, Rudroff F, Picotti P, Zamboni N, Sauer U. 2010. Tradeoff between enzyme and metabolite efficiency maintains metabolic homeostasis upon perturbations in enzyme capacity. Mol. Syst. Biol. 6, 356 ( 10.1038/msb.2010.11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan EKF, Rowe HC, Hansen BG, Kliebenstein DJ. 2010. The complex genetic architecture of the metabolome. PLoS Genet. 6, e1001198 ( 10.1371/journal.pgen.1001198) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bradley PH, Brauer MJ, Rabinowitz JD, Troyanskaya OG. 2009. Coordinated concentration changes of transcripts and metabolites in Saccharomyces cerevisiae. PLoS Comput. Biol. 5, e1000270 ( 10.1371/journal.pcbi.1000270) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fontana L, Partridge L, Longo VD. 2010. Extending healthy life span—from yeast to humans. Science 328, 321–326. ( 10.1126/science.1172539) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Piper MDW, Bartke A. 2008. Diet and aging. Cell Metab. 8, 99–104. ( 10.1016/j.cmet.2008.06.012) [DOI] [PubMed] [Google Scholar]
- 14.Houtkooper RH, Williams RW, Auwerx J. 2010. Metabolic networks of longevity. Cell 142, 9–14. ( 10.1016/j.cell.2010.06.029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chippindale AK, Leroi AM, Kim SB, Rose MR. 1993. Phenotypic plasticity and selection in Drosophila life-history evolution. 1. Nutrition and the cost of reproduction. J. Evol. Biol. 6, 171–193. ( 10.1046/j.1420-9101.1993.6020171.x) [DOI] [Google Scholar]
- 16.Clancy DJ, Gems D, Hafen E, Leevers SJ, Partridge L. 2002. Dietary restriction in long-lived dwarf flies. Science 296, 319 ( 10.1126/science.1069366) [DOI] [PubMed] [Google Scholar]
- 17.Mair W, Goymer P, Pletcher SD, Partridge L. 2003. Demography of dietary restriction and death in Drosophila. Science 301, 1731–1733. ( 10.1126/science.1086016) [DOI] [PubMed] [Google Scholar]
- 18.Partridge L, Piper MDW, Mair W. 2005. Dietary restriction in Drosophila. Mech. Ageing Dev. 126, 938–950. ( 10.1016/j.mad.2005.03.023) [DOI] [PubMed] [Google Scholar]
- 19.Metaxakis A, Partridge L. 2013. Dietary restriction extends lifespan in wild-derived populations of Drosophila melanogaster. PLoS ONE 8, e74681 ( 10.1371/journal.pone.0074681) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mair W. 2013. Tipping the energy balance toward longevity. Cell Metab. 17, 5–6. ( 10.1016/j.cmet.2012.11.011) [DOI] [PubMed] [Google Scholar]
- 21.Grewal SS. 2009. Insulin/TOR signaling in growth and homeostasis: a view from the fly world. Int. J. Biochem. Cell Biol. 41, 1006–1010. ( 10.1016/j.biocel.2008.10.010) [DOI] [PubMed] [Google Scholar]
- 22.Lee G, Park JH. 2004. Hemolymph sugar homeostasis and starvation-induced hyperactivity affected by genetic manipulations of the adipokinetic hormone-encoding gene in Drosophila melanogaster. Genetics 167, 311–323. ( 10.1534/genetics.167.1.311) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bharucha KN, Tarr P, Zipursky SL. 2008. A glucagon-like endocrine pathway in Drosophila modulates both lipid and carbohydrate homeostasis. J. Exp. Biol. 211, 3103–3110. ( 10.1242/jeb.016451) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baker KD, Thummel CS. 2007. Diabetic larvae and obese flies—emerging studies of metabolism in Drosophila. Cell Metab. 6, 257–266. ( 10.1016/j.cmet.2007.09.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moore B, Zhou L, Rolland F, Hall Q, Cheng WH, Liu YX, Hwang I, Jones T, Sheen J. 2003. Role of the Arabidopsis glucose sensor HXK1 in nutrient, light, and hormonal signaling. Science 300, 332–336. ( 10.1126/science.1080585) [DOI] [PubMed] [Google Scholar]
- 26.Kim JW, Dang CV. 2005. Multifaceted roles of glycolytic enzymes. Trends Biochem. Sci. 30, 142–150. ( 10.1016/j.tibs.2005.01.005) [DOI] [PubMed] [Google Scholar]
- 27.Marden JH. 2013. Nature's inordinate fondness for metabolic enzymes: why metabolic enzyme loci are so frequently targets of selection. Mol. Ecol. 22, 5743–5764. ( 10.1111/mec.12534) [DOI] [PubMed] [Google Scholar]
- 28.Cho YH, Yoo SD. 2011. Signaling role of fructose mediated by FINS1/FBP in Arabidopsis thaliana. PLoS Genet. 7, e1001263 ( 10.1371/journal.pgen.1001263) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. 2009. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080. ( 10.1126/science.1164097) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hansen M, Hsu AL, Dillin A, Kenyon C. 2005. New genes tied to endocrine, metabolic, and dietary regulation of lifespan from a Caenorhabditis elegans genomic RNAi screen. PLoS Genet. 1, 119–128. ( 10.1371/journal.pgen.0010017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hamilton B, Dong Y, Shindo M, Liu W, Odell I, Ruvkun G, Lee SS. 2005. A systematic RNAi screen for longevity genes in C. elegans. Genes Dev. 19, 1544–1555. ( 10.1101/gad.1308205) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Copeland JM, Cho J, Lo J, Hur JH, Bahadorani S, Arabyan T, Rabie J, Soh J, Walker DW. 2009. Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr. Biol. 19, 1591–1598. ( 10.1016/j.cub.2009.08.016) [DOI] [PubMed] [Google Scholar]
- 33.Schmidt PS, Matzkin L, Ippolito M, Eanes WF. 2005. Geographic variation in diapause incidence, life-history traits, and climatic adaptation in Drosophila melanogaster. Evolution 59, 1721–1732. ( 10.1111/j.0014-3820.2005.tb01821.x) [DOI] [PubMed] [Google Scholar]
- 34.Massouras A, et al. 2012. Genomic variation and its impact on gene expression in Drosophila melanogaster. PLoS Genet. 8, e1003055 ( 10.1371/journal.pgen.1003055) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ayroles JF, et al. 2009. Systems genetics of complex traits in Drosophila melanogaster. Nat. Genet. 41, 299–307. ( 10.1038/ng.332) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Flowers J, Sezgin E, Kumagai S, Duvernell D, Matzkin L, Schmidt P, Eanes W. 2007. Adaptive evolution of metabolic pathways in Drosophila. Mol. Biol. Evol. 24, 1347–1354. ( 10.1093/molbev/msm057) [DOI] [PubMed] [Google Scholar]
- 37.Sezgin E, Duvernell DD, Matzkin LM, Duan Y, Zhu CT, Verrelli BC, Eanes WF. 2004. Single-locus latitudinal clines and their relationship to temperate adaptation in metabolic genes and derived alleles in Drosophila melanogaster. Genetics 168, 923–931. ( 10.1534/genetics.104.027649) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lavington E, Cogni R, Kuczynski K, Koury S, Behrman EL, O'Brien KR, Schmidt PS, Eanes WF. 2014. A small system—high resolution study of metabolic adaptation in the central metabolic pathway to temperate climates in Drosophila melanogaster. Mol. Biol. Evol. 31, 2032–2041. ( 10.1093/molbev/msu146) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cogni R, Kuczynski K, Lavington E, Koury S, Schmidt PS, Eanes WF. 2015. Variation in Drosophila melanogaster central metabolic genes appears driven by natural selection both within and between populations. Proc. R. Soc. B 282, 20142688 ( 10.1098/rspb.2014.2688) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alvarez-Ponce D, Aguade M, Rozas J. 2009. Network-level molecular evolutionary analysis of the insulin/TOR signal transduction pathway across 12 Drosophila genomes. Genome Res. 19, 234–242. ( 10.1101/gr.084038.108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alvarez-Ponce D, Guirao-Rico S, Orengo DJ, Segarra C, Rozas J, Aguade M. 2012. Molecular population genetics of the insulin/TOR signal transduction pathway: a network-level analysis in Drosophila melanogaster. Mol. Biol. Evol. 29, 123–132. ( 10.1093/molbev/msr160) [DOI] [PubMed] [Google Scholar]
- 42.Eanes WF. 2011. Molecular population genetics and selection in the glycolytic pathway. J. Exp. Biol. 214, 165–171. ( 10.1242/jeb.046458) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Min KJ, Flatt T, Kulaots I, Tatar M. 2007. Counting calories in Drosophila diet restriction. Exp. Gerontol. 42, 247–251. ( 10.1016/j.exger.2006.10.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Merritt TJ, Sezgin E, Zhu CT, Eanes WF. 2006. Triglyceride pools, flight and activity variation at the Gpdh locus in Drosophila melanogaster. Genetics 172, 293–304. ( 10.1534/genetics.105.047035) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Burnett C, et al. 2011. Absence of effects of Sir2 overexpression on lifespan in C. elegans and Drosophila. Nature 477, 482–U136. ( 10.1038/nature10296) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eanes WF, Merritt TJ, Flowers JM, Kumagai S, Sezgin E, Zhu CT. 2006. Flux control and excess capacity in the enzymes of glycolysis and their relationship to flight metabolism in Drosophila melanogaster. Proc. Natl Acad. Sci. USA 103, 19 413–19 418. ( 10.1073/pnas.0607095104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Merritt TJ, Kuczynski C, Sezgin E, Zhu CT, Kumagai S, Eanes WF. 2009. Quantifying interactions within the NADP(H) enzyme network in Drosophila melanogaster. Genetics 182, 565–574. ( 10.1534/genetics.109.100677) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Merritt TJ, Duvernell D, Eanes WF. 2005. Natural and synthetic alleles provide complementary insights into the nature of selection acting on the Men polymorphism of Drosophila melanogaster. Genetics 171, 1707–1718. ( 10.1534/genetics.105.048249) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eanes WF, Merritt TJS, Flowers JM, Kumagai S, Zhu CT. 2009. Direct evidence that genetic variation in glycerol-3-phosphate and malate dehydrogenase genes (Gpdh and Mdh1) affects adult ethanol tolerance in Drosophila melanogaster. Genetics 181, 607–614. ( 10.1534/genetics.108.089383) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moser D, Johnson L, Lee C-Y. 1980. Multiple forms of Drosophila hexokinases: purification, biochemical and immunological characterization. J. Biol. Chem. 255, 4673–4679. [PubMed] [Google Scholar]
- 51.Hay RE, Armstrong FB. 1976. Biochemical characterization of allelic forms of soluble malate dehydrogenase of Drosophila melanogaster. Insect Biochem. 6, pp. 367–376. [Google Scholar]
- 52.Geer BW, Krochko D, Williamson JH. 1979. Ontogeny, cell distribution, and the physiological role of NADP- malic enzyme in Drosophila melanogaster. Biochem. Genet. 17, 867–879. ( 10.1007/BF00504309) [DOI] [PubMed] [Google Scholar]
- 53.Houtkooper RH, Canto C, Wanders RJ, Auwerx J. 2010. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 31, 194–223. ( 10.1210/er.2009-0026) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oakhill JS, Steel R, Chen ZP, Scott JW, Ling N, Tam S, Kemp BE. 2011. AMPK is a direct adenylate charge-regulated protein kinase. Science 332, 1433–1435. ( 10.1126/science.1200094) [DOI] [PubMed] [Google Scholar]
- 55.Hardie DG, Carling D. 1997. The AMP-activated protein kinase—fuel gauge of the mammalian cell? Eur. J. Biochem. 246, 259–273. ( 10.1111/j.1432-1033.1997.00259.x) [DOI] [PubMed] [Google Scholar]
- 56.Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC, Elliott PJ, Puigserver P, Auwerx J. 2009. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458, 1056–U140. ( 10.1038/nature07813) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Braco JT, Gillespie EL, Alberto GE, Brenman JE, Johnson EC. 2012. Energy-dependent modulation of glucagon-like signaling in Drosophila via the AMP-activated protein kinase. Genetics 192, 457–466. ( 10.1534/genetics.112.143610) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Johnson EC, Kazgan N, Bretz CA, Forsberg LJ, Hector CE, Worthen RJ, Onyenwoke R, Brenman JE. 2010. Altered metabolism and persistent starvation behaviors caused by reduced AMPK function in Drosophila. PLoS ONE 5, e12799 ( 10.1371/journal.pone.0012799) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stenesen D, Suh JM, Seo J, Yu K, Lee KS, Kim JS, Min KJ, Graff JM. 2013. Adenosine nucleotide biosynthesis and AMPK regulate adult life span and mediate the longevity benefit of caloric restriction in flies. Cell Metab. 17, 101–112. ( 10.1016/j.cmet.2012.12.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miwa S, St Pierre J, Partridge L, Brand MD. 2003. Superoxide and hydrogen peroxide production by Drosophila mitochondria. Free Radic. Biol. Med. 35, 938–948. ( 10.1016/S0891-5849(03)00464-7) [DOI] [PubMed] [Google Scholar]
- 61.Lin SJ, Guarente L. 2003. Nicotinamide adenine dinucleotide, a metabolic regulator of transcription, longevity and disease. Curr. Opin. Cell Biol. 15, 241–246. ( 10.1016/S0955-0674(03)00006-1) [DOI] [PubMed] [Google Scholar]
- 62.Houtkooper RH, Pirinen E, Auwerx J. 2012. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 13, 225–238. ( 10.1038/nrn3209) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhu CT, Rand DM. 2012. A hydrazine coupled cycling assay validates the decrease in redox ratio under starvation in Drosophila. PLoS ONE 7, e47584 ( 10.1371/journal.pone.0047584) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maechler P, Wollheim CB. 1999. Mitochondrial glutamate acts as a messenger in glucose-induced insulin exocytosis. Nature 402, 685–689. ( 10.1038/45280) [DOI] [PubMed] [Google Scholar]
- 65.Brosnan JT. 2000. Glutamate, at the interface between amino acid and carbohydrate metabolism. J. Nutr. 130, 988S–990S. [DOI] [PubMed] [Google Scholar]
- 66.Wagner A, Fell DA. 2001. The small world inside large metabolic networks. Proc. R. Soc. Lond. B 268, 1803–1810. ( 10.1098/rspb.2001.1711) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dick KB, Ross CR, Yampolsky LY. 2011. Genetic variation of dietary restriction and the effects of nutrient-free water and amino acid supplements on lifespan and fecundity of Drosophila. Genet. Res. 93, 265–273. ( 10.1017/S001667231100019X) [DOI] [PubMed] [Google Scholar]
- 68.Liao CY, Johnson TE, Nelson JF. 2013. Genetic variation in responses to dietary restriction—an unbiased tool for hypothesis testing. Exp. Gerontol. 48, 1025–1029. ( 10.1016/j.exger.2013.03.010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schleit J, Wasko BM, Kaeberlein M. 2012. Yeast as a model to understand the interaction between genotype and the response to calorie restriction. FEBS Lett. 586, 2868–2873. ( 10.1016/j.febslet.2012.07.038) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schleit J, et al. 2013. Molecular mechanisms underlying genotype-dependent responses to dietary restriction. Aging Cell 12, 1050–1061. ( 10.1111/acel.12130) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mair W, Piper MD, Partridge L. 2005. Calories do not explain extension of life span by dietary restriction in Drosophila. PLoS Biol. 3, e223 ( 10.1371/journal.pbio.0030223) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Grandison RC, Piper MDW, Partridge L. 2009. Amino-acid imbalance explains extension of lifespan by dietary restriction in Drosophila. Nature 462, 1061–1064. ( 10.1038/nature08619) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tatar M. 2011. The plate half-full: status of research on the mechanisms of dietary restriction in Drosophila melanogaster. Exp. Gerontol. 46, 363–368. ( 10.1016/j.exger.2010.12.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Teleman AA. 2010. Molecular mechanisms of metabolic regulation by insulin in Drosophila. Biochem. J. 425, 13–26. ( 10.1042/BJ20091181) [DOI] [PubMed] [Google Scholar]
- 75.Kim SK, Rulifson EJ. 2004. Conserved mechanisms of glucose sensing and regulation by Drosophila corpora cardiaca cells. Nature 431, 316–320. ( 10.1038/nature02897) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw data for all the lifespan experiments is deposited in Dryad under http://dx.doi.org/10.5061/dryad.r620q.