

Abstract
The apical damage kinase, ATR, is activated by replication stress (RS) both in response to DNA damage and during normal S-phase. Loss of function studies indicates that ATR acts to stabilize replication forks, block cell cycle progression and promote replication restart. Although checkpoint failure and replication fork collapse can result in cell death, no direct cytotoxic pathway downstream of ATR has previously been described. Here, we show that ATR directly reduces survival by inducing phosphorylation of the p50 (NF-κB1, p105) subunit of NF-кB and moreover, that this response is necessary for genome maintenance independent of checkpoint activity. Cell free and in vivo studies demonstrate that RS induces phosphorylation of p50 in an ATR-dependent but DNA damage-independent manner that acts to modulate NF-кB activity without affecting p50/p65 nuclear translocation. This response, evident in human and murine cells, occurs not only in response to exogenous RS but also during the unperturbed S-phase. Functionally, the p50 response results in inhibition of anti-apoptotic gene expression that acts to sensitize cells to DNA strand breaks independent of damage repair. Ultimately, loss of this pathway causes genomic instability due to the accumulation of chromosomal breaks. Together, the data indicate that during S-phase ATR acts via p50 to ensure that cells with elevated levels of replication-associated DNA damage are eliminated.
Keywords: ATR, DNA damage, NF-κB, p50, replication stress, S-phase
Abbreviations
- ATM
ataxia telangiectasia mutated
- ATR
ataxia telangiectasia mutated and Rad3-related
- Bax
BCL2-associated X protein
- Bclxl
Bcl-2-like protein
- ChIP
chromatin immunoprecipitation
- Chk1
checkpoint kinase 1
- DSBs
double-strand breaks
- H2AX
histone 2AX
- HR
homologous recombination
- Hu
hydroxyurea
- IκB
inhibitor kappaB
- IκK
inhibitor kappaB kinase
- IR
ionizing radiation
- NF-κB
nuclear factor-kappaB
- RS
replication stress
- RT-PCR
reverse transcriptase polymerase chain reaction
- siRNA
short interfering RNA
- TAM
tamoxifen
- TMZ
temozolomide
- TopBP1
topoisomerase-binding protein-1
Introduction
During the normal cell cycle, endogenous or acquired impediments to DNA replication result in the appearance of stretches of single-stranded DNA, or single-strand/double-strand DNA junctions, that are the basic substrate of replication stress (RS).1,2 To modulate the genotoxic effects of RS, cells induce a checkpoint response coordinated by the apical damage kinase, ATR.3 ATR is activated following the co-localization of a series of proteins including the critical co-activator, topoisomerase-binding protein-1 (TopBP1), a protein that contains an activation domain that is able to induce ATR on its own.4,5 Although the ATR response attenuates the amount of DNA double-strand breaks (DSBs) that accumulate as a result of RS,6 low levels of strand breaks are often tolerated, possibly because DSB repair is inefficient below a certain threshold.7 In order to prevent the propagation of such potentially harmful damage, multi-cellular species activate a variety of pathways that eliminate cells with raised levels of DNA damage.
The transcription factor NF-κB plays a pivotal role in modulating the response to DNA damage.8 While genotoxic stress activates NF-κB by a well-described mechanism involving DNA DSBs and ATM,9 the response of NF-κB to ATR and RS is poorly understood. Preliminary data suggest that ATR can inhibit NF-κB,10,11 yet the full mechanism and physiological role of this pathway is unclear. NF-κB proteins are composed of 5 subunits: p50 (NF-κB1, p105), p52 (NF-κB2, p100), p65 (relA), c-rel, and relB that bind to DNA as dimers and are retained in the cytoplasm through interaction with inhibitor-κB (IκB) proteins.12 Although the NF-κB system is generally regarded as a stimulus-induced transcription factor, significant levels of DNA bound NF-κB dimers are found in resting cells. The most abundant form of DNA bound NF-κB in unstimulated cells consists of the p50/p65 heterodimer. Interestingly, during the unperturbed cell cycle, a complex exchange of NF-κB dimers and subunit post-translational modifications occur,13 suggesting that NF-κB proteins likely play a fundamental role in the basal homeostatic process.
We recently reported that certain DNA damaging agents inhibit NF-κB by inducing Chk1-mediated phosphorylation of p50 Ser329.14 Given the association between ATR/Chk1 and RS, we set out to examine the role of p50 in response to RS. Our results demonstrate that ATR inhibits NF-κB and induces p50 phosphorylation in response to RS. Moreover, this pathway is induced during S-phase and acts to maintain cellular genome stability by facilitating elimination of damaged cells.
Results
Replication stress inhibits NF-κB DNA binding in an ATR- and p50-dependent manner
To examine NF-κB in the setting of RS, cells were administered the ribonucleotide reductase inhibitor, hydroxyurea (Hu), and NF-κB examined by gel shift assay. As previously reported,10 Hu activates NF-κB in ATM+/+ cells (Fig. 1A). However, in the absence of ATM, we note that Hu not only fails to induce but actually inhibits NF-κB within about 1 hour of treatment (Fig. 1A; Fig. S1A). Supershift analysis demonstrates that the NF-κB dimer in these cells is comprised of p50 and p65 (Fig. S1B). While anti-p50 and anti-p65 antibodies do not result in a discernable supershifted band, they break up the NF-κB-DNA complex, an observation not seen with a non-specific control antibody. In addition, in the absence of ATM, other genotoxic agents that induce RS, including aphidicolin and etoposide, also inhibit NF-κB (Fig. S1C). Inhibition of NF-κB by RS is blocked by caffeine, a general ATM/ATR inhibitor (data not shown). Therefore, to examine whether ATR specifically is involved in the inhibition of NF-κB, ATR was depleted using siRNA. Loss of ATR blocks the ability of Hu to inhibit NF-κB (Fig. 1B). Moreover, siRNA studies also demonstrate that p50 is required for inhibition of NF-κB by RS (Fig. 1B). Importantly, the inhibition of NF-κB is independent of changes in either IκBα protein level or nuclear p50 and p65 levels (Fig. 1C) suggesting that RS and ATR specifically affect NF-κB DNA binding and not nuclear translocation. Consistent with the ability of RS to activate Chk1, a previously identified p50 S329 kinase,14 Hu induces S329-phosphorylation with kinetics similar to the inhibition of NF-κB DNA binding (Fig. 1C). The inhibition of DNA binding by Hu is reflected by a decrease in NF-κB transcriptional activity, as measured using a reporter bearing 3 repeats of the immunoglobulin κ-light chain κB-site, a finding also noted to be ATR- and p50-dependent (Fig. 1D).
Figure 1.
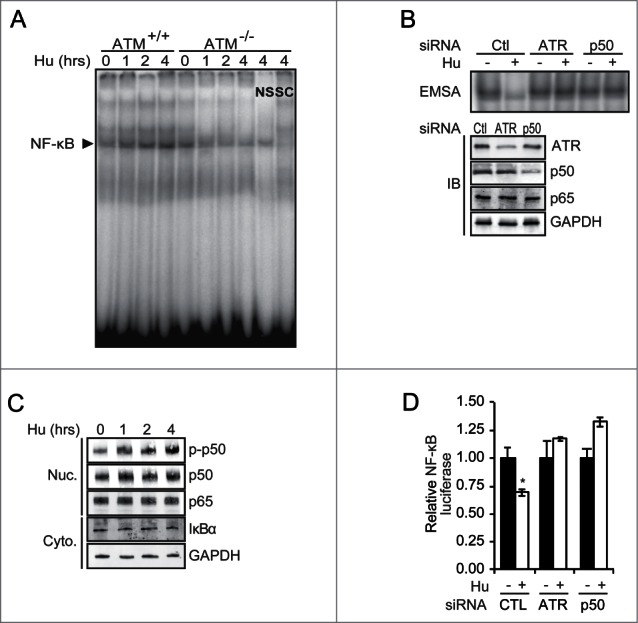
RS inhibits NF-κB in an ATR- and p50-dependent manner. (A) Human ATM−/− and ATM+/+ cells were treated with Hu (2 mM) and EMSA performed after isolation of nuclear extracts. Competition was performed with specific (SC) and non-specific (NS) cold DNA. (B) NF-κB EMSA (upper) and immunoblot (IB, lower) using nuclear extract from ATM−/− cells transfected with the indicated siRNA and treated with Hu (2 mM, 4 hrs). (C) IB in nuclear and cytoplasmic fractions from ATM−/− cells treated with Hu (2 mM) (p-p50: anti-phospho-S329-p50 antibody). (D) NF-κB-dependent luciferase assay in ATM−/− cells transfected with siRNA and treated with vehicle or 2 mM Hu (4 hrs). Data show mean value relative to renilla, normalized to control, ± SD of triplicate samples. *P < 0.05 relative to untreated.
As chemotherapeutics like Hu have pleotropic effects and also induce DNA DSBs, we next sought to examine NF-κB in response to a non-chemotherapeutic model of RS. Synthetic poly(dA)70-poly(dT)70 oligonucleotides (TA oligos) that form the minimal replicative intermediates previously used to study RS signaling in Xenopus and mammalian cell extracts15,16 were obtained and used in vitro. Nuclear extracts were isolated from U87 human glioma cells that have stably-suppressed endogenous p105,14 the parental protein of p50. Extracts were supplemented with purified p50WT or p50S329A, a mutant that cannot be phosphorylated by Chk1. As initially noted using these oligos,15,16 TA but not monomeric poly(dT)70 oligonucleotides (T oligos) activate Chk1 by phosphorylation (Fig. 2A). Moreover, consistent with the findings using Hu, we find that TA oligos both induce p50 S329 phosphorylation and inhibit NF-κB binding in a p50-S329-dependent fashion (Fig. 2A). These responses are abrogated by caffeine and the Chk1 inhibitor, Gö6976, but not by IκK inhibition (Fig. 2A). In these cells, the NF-κB dimer is comprised of p50/p65 as shown by supershift analysis (Fig. S2A). The requirement of ATR for inhibition of NF-κB by TA oligos is demonstrated using extracts from cells expressing wildtype, or kinase-dead, ATR,17 and the role of Chk1 is shown using Gö6976 (Fig. 2B). In addition, studies using living, immortal Nfkb1−/− MEFs stably expressing p50WT or p50S329A transfected with the oligos recapitulate the cell-free experiments (Fig. S2B) and also demonstrate that TA oligos inhibit NF-κB transcriptional activity in a manner dependent on p50 S329 (Fig. S2C). Importantly, the p50S329A mutant binds DNA (Fig. S2D), and hetero-dimerizes with p65 as efficiently as p50WT (Fig. S2E). Consistent with the latter observation, the primary NF-κB dimer in stable Nfkb1−/− MEFs expressing p50WT or p50S329A is made of p50/p65 (Fig. S2F). These results support the findings with pharmacological RS-inducing agents and indicate that RS inhibits NF-κB in an ATR-dependent manner.
Figure 2.
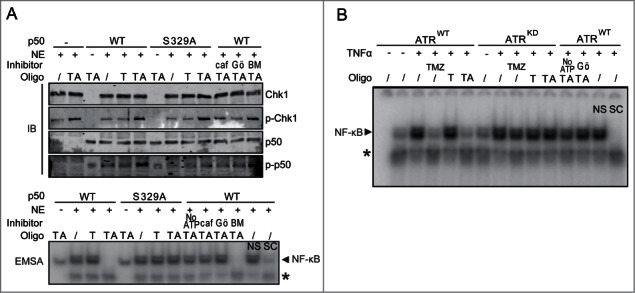
Oligonucleotide mimetics of RS inhibit NF-κB DNA binding in a p50 S329-dependent manner. (A) Nuclear extract (NE) isolated from U87-sh-p105 cells were supplemented with bacterially expressed p50WT or p50S329A (or no p50). IB (upper) or EMSA (lower) was performed following stimulation of extract with TA or T oligos under kinase conditions. Extracts were supplemented with caf-caffeine (0.5 mM); Gö- Gö6976 (1 µM) or BM-BMS-345541 (IKK inhibitor) (1 µM) where shown. p-Chk1: anti-phospho-Ser345 Chk1. Specific (SC) and non-specific (NS) competitor. * Non-specific band present in nuclear extract. (B) NE was isolated from vehicle or TNFα-stimulated (10 ng/ml, 30 min) U2OS cells stably expressing tet-on wild-type ATR (ATRWT) or kinase-dead ATR (ATRKD) 48 hrs after stimulation with 1 mg/ml doxycycline. EMSA was performed after incubation of extract with TA or T oligos (or vehicle). EMSA was also run following treatment of cells with TMZ (100 µM, 16 hrs, lanes 4 and 9). Controls were also performed in the presence of Gö- Gö6976, in the absence of ATP or with cold competitors as shown. *Non-specific band.
ATR inhibits NF-κB activity in the absence of DNA damage
While TA oligos simulate replication forks and activate ATR, these oligos can also induce ATM and have primarily been used for in vitro studies.18 Therefore, to more specifically induce ATR and examine NF-κB in vivo, a tamoxifen (TAM)-inducible system that incorporates the activation domain of TopBP1 fused to the estrogen receptor (TopBP1ER) was used.5 Importantly, this system allows ATR induction in the absence of DNA DSBs and without activation of ATM.5 Exposure of 293T cells stably expressing TopBP1ER to TAM leads to activation (phosphorylation) of ATR within 30 minutes (Fig. 3A). This response subsequently results in p50 S329 phosphorylation and inhibition of NF-κB binding, both of which occur in an ATR- and Chk1-dependent manner as demonstrated using ATR-specific siRNA and Chk1 inhibitor (Fig. 3A; Fig. S3A). Again, the basal, NF-κB dimer present in these cells is comprised of p50 and p65, as demonstrated by supershift analysis (Fig. S3B). Neither TAM in the absence of TopBP1ER nor TopBP1ER alone result in inhibition of NF-κB (Fig. S3C). Exposure to TAM also leads to inhibition of NF-κB transcriptional activity in a manner dependent upon ATR and p50 (Fig. 3B). Subsequently, to examine the role of S329 in this pathway, Nfkb1−/− MEFs stably expressing p50WT or p50S329A were infected with a retrovirus expressing TopBP1ER. Administration of TAM to these cells leads to inhibition of NF-κB DNA binding, and transcriptional activity, specifically in a p50 S329-dependent manner (Fig. 3C; Fig. S3D). To examine whether the effect on NF-κB is reflected by a change in endogenous gene expression, we examined the NF-κB-regulated anti-apoptotic gene, BCLXL, whose inhibition is required for cytotoxicity by S-phase specific alkylating agents.19 As we previously noted, this gene has p50/p65 heterodimers bound to its promoter at baseline.14,20 In both human and murine cells, ATR induced by TopBP1ER leads to a decrease in BCLXL expression, a finding that is specifically dependent on p50 S329 (Fig. 3D; Fig. S3E). Of note, TAM inhibits NF-κB trans-activity in the absence of p65, as seen using P65−/− MEFs (Fig. S3F). Also, BCLXL mRNA expression can still be inhibited by ATR in human cells depleted of p65 with si-RNA (Fig. S3G). Interestingly, despite inhibition of NF-κB activity and Bclxl mRNA expression, activation of ATR in the absence of additional DNA damage does not affect overall clonal survival (Fig. S3H).
Figure 3.
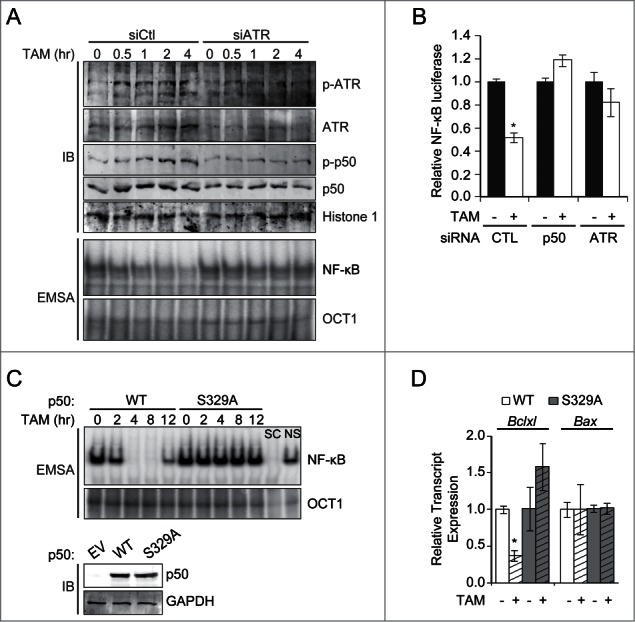
ATR inhibits NF-κB activity independent of DNA damage. (A) 293T cells stably expressing TopBP1ER were transfected with the indicated siRNA and stimulated with TAM (500 nM). IB and EMSA (NF-κB and Oct1) were performed on nuclear extracts isolated at the indicated time. p-ATR (anti-phospho-Ser428 ATR). (B) NF-κB-dependent luciferase assay in 293T-TopBP1ER cells expressing the indicated siRNA following stimulation with TAM (8 hrs). *P < 0.01. (C) Nfkb1−/− MEFs stably expressing p50WT or p50S329A were infected with TopBP1ER and stimulated with TAM (500 nM). EMSA was performed on nuclear extracts using the indicated probe. IB shows equal p50 expression in cells. (D) qPCR of Bclxl and Bax mRNA expression in cells from (C) following stimulation with TAM (8 hrs). Data show mean normalized value, ± SEM of triplicate samples from 3 experiments. *P < 0.05 relative to untreated.
In sum, the data generated using both in vivo and cell free systems, demonstrate that the ATR response to RS leads to inhibition of NF-κB p50/p65 DNA binding and NF-κB transcriptional activity that is dependent on p50-S329.
p50 S329 phosphorylation occurs during S-phase and enables inhibition of NF-κB
ATR is activated during the unperturbed S-phase,21,22 therefore, we examined whether NF-κB and p50 are also modified at this time. U87 glioma cells were synchronized at G2/M using nocodazole and nuclear extracts isolated at the indicated times following release (Fig. 4A). Maximal p50 S329-phosphorylation occurs when the greatest percentage of cells are in S-phase, identified by FACS analysis and cyclin E expression to be approximately 12 hours after release from synchronization (Fig. 4A). Moreover, EMSA reveals that it is during this phase that NF-κB DNA binding is at its lowest level (Fig. 4A, lower blot). As we previously noted, in these glioma cells NF-κB dimers are comprised of p50 and p65.14 Similarly, following synchronization of cells at G1 by double thymidine block, NF-κB DNA binding is also noted to nadir during S-phase, indicating that the NF-κB changes are not merely a consequence of the synchronization method (Fig. 4B). Moreover, the reduced DNA binding during S-phase is Chk1- and caffeine-dependent (Fig. 4B). Consistent with the finding that ATR inhibits BCLXL expression, S-phase-dependent p50 phosphorylation and reduced DNA binding is mirrored by a decrease in p50 recruitment to the BCLXL promoter at this time (Fig. 4C). Furthermore, BCLXL mRNA expression is also attenuated at this time (Fig. 4D), ultimately leading to a modest but reproducible decrease in Bclxl protein expression in late S-phase (Figs. 4A and E). To investigate the role of p50 S329 in the cell cycle-dependent regulation of NF-κB, Nfkb1−/− MEFs stably expressing p50WT or p50S329A were synchronized by thymidine block and examined following release. As with human cells, p50 phosphorylation and the nadir of NF-κB DNA binding occur specifically during S-phase (Fig. 4F), findings that are blocked by mutation of S329. Similarly, the S-phase-dependent decrease in Bclxl mRNA expression is blocked by S329 mutation (Fig. S4A). Together, these results demonstrate that p50 is phosphorylated during S-phase resulting in an S329-dependent decrease in NF-κB DNA binding and Bclxl expression.
Figure 4.

p50 is phosphorylated and NF-κB binding inhibited during S-phase. (A) U87 glioma cells were released from nocodazole synchronization, nuclear lysates isolated and IB performed on lysate or following IP. NF-κB EMSA (lower blot) was also performed. Cell cycle phase at the corresponding time following release is shown above. Asynchronous (A). (B) U87 cells were released from thymidine block and EMSA performed following isolation of nuclear lysates. Cells were pre-incubated with caffeine (0.5 mM) and Gö6976 (1 µM) where shown. (C) qChIP in U87 cells at the indicated phase following thymidine release. Data show enrichment of p50 relative to IgG, histone H1 and input, ± SEM of 3 separate experiments. *P < 0.03 relative to A. (D) qPCR of BCLXL mRNA in U87 cells at the indicated phase. Mean ± SEM of triplicate samples from 3 separate experiments. *P < 0.05 relative to G1. (E) IB with anti-Bclxl antibody in U87 cells following release from thymidine. (F) Nfkb1−/−MEFs stably expressing p50WT or p50S329A were released from thymidine block, nuclear lysates isolated and IB performed on lysate or following IP. NF-κB EMSA (lower blot) was performed using the same samples. Se- early S-phase. Images are representative of 2 or 3 separate experiments.
p50 phosphorylation during S-phase sensitizes cells to DNA strand breaks independent of damage repair
It was previously noted that p50 signaling sensitizes cells to cytotoxicity in response to DNA damaging agents.14 We therefore examined whether this pathway also acts downstream of ATR to modulate survival during the cell cycle. First, Nfkb1−/− MEFs expressing TopBP1ER and either p50WT or p50S329A were exposed to TAM, to induce ATR signaling, and also to 0.5 Gy ionizing radiation (IR), to induce low levels of DNA strand breaks. As TopBP1ER activation does not induce appreciable levels of DNA damage,5 IR delivery can be used to induce strand breaks at a specific time. IR was administered either 8 hours before or after exposure to TAM. Importantly, activation of ATR for 8 hours is sufficient to induce p50-signaling but does not alter cell survival (Fig. 3; Fig. S3F). A decrease in survival is noted when the p50 is activated prior to IR, compared to the reverse, and specifically in cells expressing p50WT but not p50S329A (Fig. 5A) suggesting that p50 phosphorylation sensitizes cells to DNA strand breaks. To examine whether p50 phosphorylation in S-phase mediates a similar effect, Nfkb1−/− MEFs stably expressing p50WT or p50S329A were synchronized by thymidine block and then treated at the indicated time following release with low dose IR. Despite the minor levels of damage induced by 0.5 Gy IR, a significant decrease in surviving fraction is noted in cells expressing p50WT and specifically at the time when p50 is phosphorylated (Fig. 5B). Together, these results indicate that p50 phosphorylation in response to ATR sensitizes cells to DNA damage during S-phase.
Figure 5.
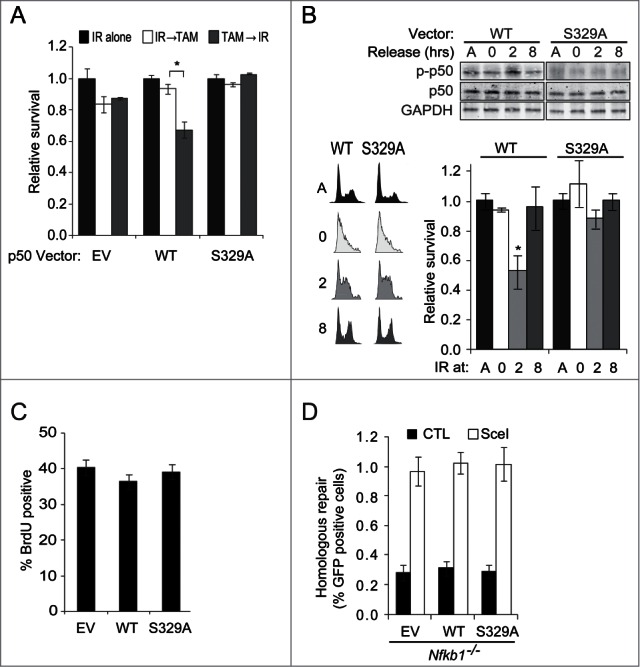
p50 phosphorylation in response to ATR sensitizes cells to DNA strand breaks. (A and B) Clonogenic assays. (A) Nfkb1−/−MEFs expressing TopBP1ER and the p50 vector shown were exposed to 0.5 Gy IR and TAM, in the order indicated, separated by 8 hrs. Inset: IB showing p50 expression. Data is normalized to IR alone, ± SEM of duplicate samples from 3 experiments (TAM alone does not alter survival). *P < 0.02. (B) Thymidine synchronized Nfkb1−/−MEFs stably expressing p50WT or p50S329A were exposed to 0.5 Gy IR at indicated time following release. A- asynchronous cells. IB of nuclear lysates (upper) and FACS analysis (left) from the same experiment is shown (anti-p50 IB in asynchronous shows equal p50 expression). *P < 0.02, relative to p50S329A. (C) FACS analysis of BrdU labeling in asynchronous Nfkb1−/−MEFs expressing empty vector, p50WT or p50S329A. Mean ± SD of triplicate samples from 3 experiments shown. n ≥ 105 cells per group. (D) HR assay in Nfkb1−/−MEFs stably expressing empty vector (EV), p50WT or p50S329A following co-transfection with DR-GFP and either control vector (CAG) or pC⇓ASce (SceI). Data show value ± SD of triplicate samples (n = 3000− 5000 cells per group).
Although the data suggest that the mechanism of sensitization to strand breaks occurs via ATR/p50-induced inhibition of anti-apoptotic gene expression, it is possible that the ATR-mediated checkpoint or damage repair response also contribute to modulating survival. However, the observation that Nfkb1−/− MEFs stably expressing p50 constructs cycle at similar rates (Fig. 5B, inset) and have similar percentages of cells in S-phase (Fig. 5C) indicates that p50 phosphorylation does not affect the replication checkpoint. With regard to strand break repair, homologous recombination (HR), the major DSB repair pathway associated with replication,23 is unaffected by either loss or mutation of p50 (Fig. 5D; Fig. S4B). In addition, the finding that IR induces similar levels of breaks in p50WT and p50S329A-expressing cells (Fig. S4C), suggests that p50 phosphorylation does not affect other pathways that act to repair DSBs. Taken together, these data indicate that in response to ATR, p50 sensitizes cells to DNA strand breaks by a mechanism independent of checkpoint or damage repair and that this pathway is functional during S-phase.
Phosphorylation of p50 is necessary for maintaining genome stability
If cell cycle-dependent p50 phosphorylation plays a physiological role in sensitizing to damage, then loss of this response in proliferating cells would be expected to result in damage accumulation. To examine this hypothesis, freshly isolated primary Nfkb1−/− MEFs were infected with p50 constructs and DNA DSB levels assessed following serial passage by analysis of γ-H2AX focus formation. The percentage of γ-H2AX-positive cells rises with increasing passage and expression of p50WT significantly reduces the increase relative to empty vector (Fig. 6A; Fig. S5). The observation that cells expressing p50S329A have higher endogenous γ-H2AX foci than p50WT cells indicates that p50 phosphorylation is necessary to attenuate spontaneous replication associated damage build up. Given the well-described association between DNA damage accumulation and senescence,24 we examined senescence in serially passaged primary MEFs. Nfkb1−/− MEFs expressing p50S329A have a significantly higher level of β-galactosidase staining at late passage than isogenic cells expressing p50WT (Fig. 6B). These data suggest that p50 phosphorylation is required to maintain genomic stability. To directly examine genome integrity, metaphase spreads were studied in MEFs infected with the p50 constructs. Despite initially having equivalent levels of DNA breaks and gaps, primary Nfkb1−/− MEFs expressing p50WT accumulate significantly less breaks and gaps than p50S329A-expressing cells following serial passage (Fig. 6C). These findings demonstrate that p50 phosphorylation is necessary to attenuate spontaneous DNA damage accumulation and to maintain genomic integrity.
Figure 6.
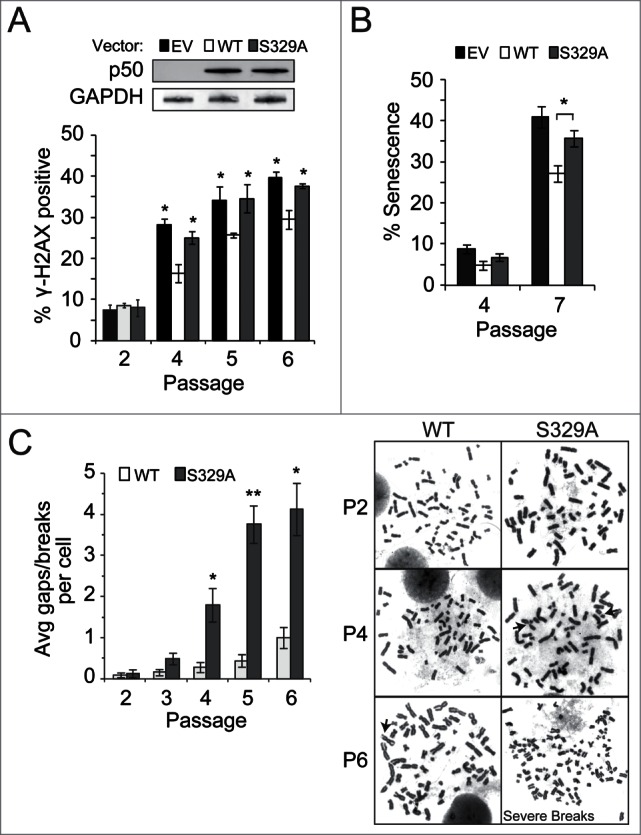
p50 S329 is necessary to maintain genome stability. (A–C) Primary Nfkb1−/−MEFs were infected at passage 1 with the indicated p50 construct and analyses made after serial passage. Experiments were performed using at least 3 separate MEF isolations/ infections expressing equivalent levels of p50. (A) γH2AX foci. *P < 0.001 relative to p50WT-expressing cells. Inset: IB with anti-p50. Data from (A and B) show mean ± SD (n ≥ 150 cells). (B) β-gal staining. *P < 0.005. (C) Spontaneous breaks and gaps. n = 25 metaphase spreads per sample. Data show mean ± SEM. *P < 0.005 relative to p50WT-expressing cells. Representative metaphase spreads shown (right). Arrows indicate chromosomal gaps/breaks. P6 cells expressing p50S329A have multiple/severe breaks.
Discussion
In this report, we demonstrate that RS, induced either pharmacologically or by oligonucleotide fork mimetics, causes phosphorylation of p50 and inhibition NF-κB activity. This response is shown to require the damage kinase ATR and, in contrast to the activation of NF-κB by DNA DSBs, is independent of ATM. A previous report noted a propensity of ATR to block NF-κB and suggested that ATR acts in opposition to ATM to block NF-κB nuclear translocation by inhibiting inhibitor-κB kinase (IκK).10 While our work also shows that ATR inhibits NF-κB, we demonstrate that ATR acts on nuclear p50-containing NF-κB dimers to modulate NF-κB activity independent of IκBα. Interestingly, in response to p14ARF, ATR was also reported to inhibit NF-κB activity in an IκBα-independent fashion by inducing phosphorylation of p65 T505.11 Although we note that inhibition of NF-κB activity by ATR can occur even in the absence of p65, given that c-rel cross-compensates when p65 is lost,25 it is likely that in the physiological setting p65-dependent signaling is also required for the ATR response. Taken together, these previous studies and ours indicate that the ATR pathway incorporates several mechanisms to modulate NF-κB signaling and moreover, that this occurs in a promoter-specific manner. Most importantly, these studies arrive at the same conclusion, namely, that the functional role of the NF-κB response to ATR acts to decrease survival and promote cell death (Fig. 7).
Figure 7.
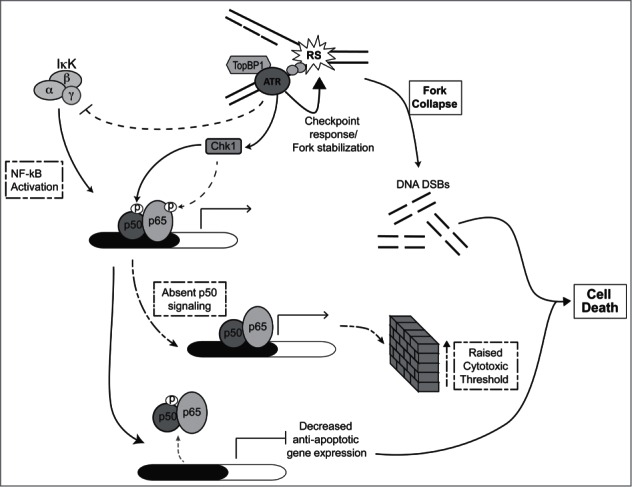
Model demonstrating role of p50 in the ATR-mediated response to RS. In this model, RS-induced ATR signaling leads to phosphorylation of nuclear p50 resulting in promoter-specific modulation of NF-κB (p50/p65) binding. This pathway, activated in parallel to the ATR-mediated checkpoint response, leads to attenuation of anti-apoptotic Bclxl expression. ATR signaling also modulates NF-κB activity by inhibiting the IκK complex and by inducing phosphorylation of p65. Together, the NF-κB-dependent response to ATR promotes cell death. Loss of p50 signaling leads to an elevation in the threshold for cell death resulting in survival of cells harboring higher levels of DNA strand breaks.
Our data indicate that the p50-mediated response to ATR, while not sufficient on its own to induce cytotoxicity, facilitates loss of survival by enabling inhibition of anti-apoptotic gene expression. These findings, coupled with the observation that ATR is activated and p50 phosphorylated during replication, suggest that p50 acts during S-phase to facilitate death of damaged cells. Consistent with this hypothesis, we note that loss of the p50 signaling due to S329 mutation, results in an increase in the accumulation of cells with DNA breaks. These data suggest a model in which ATR activation during S-phase induces a pathway separate from the checkpoint that acts on p50 to modulate NF-κB activity and facilitate cell elimination (Fig. 7). In the presence of normal p50 signaling, cells that acquire elevated levels of DNA strand breaks (i.e. levels that are harmful to the genome) are removed from the population. In the absence of p50, the cytotoxic threshold is raised resulting in DNA damage tolerance and genomic instability. Importantly, if replication-associated damage is adequately addressed, then p50-mediated signaling has no deleterious effects and the cell continues to cycle. This last point is emphasized by the observation that the p50 response, involving a decrease in Bclxl expression, while significant, is small and not great enough in magnitude to affect survival in the absence of additional damage.
It is recognized that during S-phase ATR functions to stabilize replication forks and coordinate the checkpoint response to ensure faithful DNA replication prior to mitosis.3,26 However, if the overall role of ATR is to maintain genomic stability,27 it is likely that this master-regulator of the RS response would directly modulate survival in the event that replication-induced damage is not adequately repaired. Such a direct ATR-induced cytotoxic pathway, independent of the checkpoint, has not been previously reported firstly because ATR is an essential gene that plays a dominant role in modulating the checkpoint,28-31 as do downstream mediators of cytotoxicity, such as p53;32 and secondly because yeast, the organism in which much of the information on ATR/Mec1 has been obtained,33-35 is unicellular and lacks NF-κB.
Given the imperfect nature of DSB repair,36 even a slight rise in DNA DSB levels can lead to increased mutation formation and tissue dysfunction. The p50/NF-κB response to RS can be considered a pathway that addresses the minor levels of damage encountered during replication. p50 is ideally situated to modulate the response to RS not only because it is found in virtually all tissues, but also because it constitutes a primary DNA-bound NF-κB subunit that is present at baseline. Importantly, as DNA-bound p50 can be modified quickly, in comparison to latent NF-κB pools that must first translocate to the nucleus, it can relatively rapidly mediate signaling during replication. Although NF-κB is best known as a stimulus-induced transcription factor, the presence of basal activity suggests that NF-κB also contributes to more routine cellular processes. Consistent with a role in regulating low endogenous damage levels, loss of p50/Nfkb1 does not lead to overwhelming damage accumulation or embryonic lethality;37 however, loss of this subunit does result in chronic disease as demonstrated by the propensity of Nfkb1−/− mice to develop premature age-related findings (see refs. 38− 40 and unpublished data, BY).
In summary, we propose that p50-signaling acts as a fail-safe mechanism by which ATR ensures that cells with raised levels of DNA damage do not survive. Ultimately, this response functions in multicellular species to maintain overall organismal health at the expense of individual cellular integrity.
Materials and Methods
Antibodies, reagents and plasmids
Cellular lysates were subjected to SDS-PAGE and western blotting performed as described.20 The antibodies used include: anti-p50 (Santa Cruz, sc7178 and sc8414); anti-phospho-S329-p50 (YenZym Antibodies, San Francisco, CA);14 anti-ATR and anti-phospho-S428-ATR (Cell Signaling, 2790 and 2853, respectively); anti-p65 (Santa Cruz, sc-8008); anti-IkBα (Cell Signaling, 9242); anti-Flag (Cell Signaling, 2368); anti-Chk1 and anti-phospho-S345-Chk1 (Cell Signaling, 2345 and 2341s, respectively); anti-histone 1 (Santa Cruz; sc-8030); anti-Gapdh (Santa Cruz, sc-137179); anti-Bclxl (Cell Signaling, 2762); anti-cyclin E (Santa Cruz, sc-198); anti-phospho-S140-H2AX (Cell Signaling, 9718). Hydroxyurea, tamoxifen, caffeine, Gö6976, BMS-345541, TNFα, etoposide, thymidine, nocodazole, aphidicolin and cyclohexamide were obtained from Sigma. TMZ was obtained from the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, National Cancer Institute, NIH. TMZ was dissolved in DMSO (final concentration <0.1% v/v). The TopBP1ER construct was a generous gift from Dr. Oscar Fernandez-Capetillo.
To create p50 retroviral constructs, p50 was liberated from pcDNA3.1-p50 previously described,20 by digesting with PmeI and XhoI and ligated into the BglII site of pMSCV-MigR1 containing an IRES-GFP insert. The QuikChange Lightning Site Directed Mutagenesis Kit (Stratagene) was used to create individual site mutants in the MigR1-p50 backbone.
Cells, RNA interference and retroviral infection
Experiments were performed using the following cell lines: HEK293T, U87, human ATM−/−and the corresponding isogenic ATM+/+ controls, tet-on ATRKD-expressing U2OS, immortal Nfkb1+/+, Nfkb1−/− and P65−/− MEFs, and primary Nfkb1+/+ and Nfkb1−/− MEFs, cultured as previously described.20 Primary Nfkb1+/+ and Nfkb1−/− MEFs were harvested at embryonic day 13.5. Stable re-expression of p50 isoforms was performed in Nfkb1−/− MEFs as previously described,20 or using retroviral infection. Stable 293T-TopBP1ER cells were constructed by selecting in puromycin following transfection with TopBP1ER. ATM+/+ and ATM−/− cells were purchased from Coriell Cell Repositories. The following siRNA constructs were obtained from Dharmacon: p50 (sense: GUCACUCUAACGUAUGCAAUU), ATR (M003202-05) and scrambled control (sense: CCUACGCCACCAAUUUCGUUU). Cells were transfected with siRNA using TransIT-TKO (Mirus) 24 hours prior to treatment or harvest for IB. U87 cells stably expressing sh-RNA targeting the C-terminal of p105, or control sequence, were previously described.14
For retroviral production, the pantropic platinum-GP retroviral packaging cell line (PlatGP) was transfected with retroviral vector containing various MigR1-p50 isoforms or TopBP1ER using XtremeGENE transfection reagent (Roche). Target cells were then spinoculated with the virus/polybrene-containing supernatant. After infection, colonies were either sorted by FACS or selected in medium containing 0.5 μg/mL puromycin. Studies using Nfkb1−/− MEFs expressing p50 mutants represent data from at least 2 separate clones.
Poly(dA) 70-poly(dT) 70 oligos
TA oligos were obtained from IDT as poly(dA)70 and poly(dT)70 and pre-annealed prior to use. Studies in extracts were performed based on previous descriptions.15,16 Briefly, nuclear extracts from immortal Nfkb1−/− MEFs were supplemented with the indicated bacterially expressed p50 protein, purified as previously described,14 and incubated with either single-stranded poly(dA)70, or double-stranded poly(dA)70-poly(dT)70 oligonucleotides (25 pM) in kinase reaction buffer (50 mM Tris-HCl pH 7.5, 1 mM DTT, 10 mM MgCl2, 10 mM MnCl2,0.2 µM ATP) at 30°C for 1 hour. Reactions were then used for EMSA and immunoblotting. For in vivo studies, cells were transfected, using oligofectamine (Invitrogen), with the indicated concentration of oligo and EMSA or luciferase assay performed.
Synchronization and cell cycle analysis
Cell cycle analysis was made by evaluation of DNA content following incubation with propidium iodide (PI) as previously described.41 For G2/M synchronization, cells were treated with 40 ng/ml nocodazole for 16 hours and then released. For thymidine synchronization, cells were grown in the presence of 2 mM thymidine (Sigma) for 16 hours, released for 8 hours into medium without thymidine and then re-incubated for 16 hours in thymidine. Harvested cells were then analyzed for cell cycle progression as above. For clonogenic assays in synchronized cells, cells were treated at the indicated time following release from thymidine block with 0.5 Gy IR. BrdU labeling was performed by pre-labeling cells with 10 μM BrdU for 60 minutes. Cells were then fixed and permeabilized according to the manufacturers’ instructions (Cell Signaling). Flow cytometric analysis was performed after staining with anti-BrdU antibody (Santa Cruz, sc-32323).
Immunoprecipitation
Cells were treated as indicated and nuclear extracts prepared. IP was performed in buffer: [50 mM Tris (pH 8.), 125 mM NaCl, 1% Nonidet P-40 (NP-40), 10% glycerol and 2 mM EDTA] supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF). Extracts were pre-cleared with protein A/G conjugated Sepharose beads (GE Healthcare) and IP performed with the indicated antibodies and beads. Antigen-antibody complexes were precipitated with protein G beads and immunoblot performed following SDS-PAGE.
Luciferase reporter assay
Transfection was normalized using renilla reniformis and luciferase assay performed with the indicated NF-κB firefly reporter construct as previously described.20 Data is plotted as firefly/renilla (F/R) relative luciferase. For luciferase assays with oligo substrates, cells were initially transfected with Ig-кB luciferase/ renilla and 24 hours later treated with the indicated amount of oligo substrate in the presence of Oligofectamine (Invitrogen). Luciferase activity was quantified 6 hours after oligo administration. Assays were performed in triplicate at least thrice unless otherwise indicated.
Real time/quantitative reverse transcriptase (RT)-PCR
Total RNA was isolated from cells following treatment. The Protoscript M-MuLV Taq RT-PCR Kit (New England Biolabs) with poly-T primers was used for reverse transcriptase reaction. Transcripts were quantified using SYBR Green PCR (Bio-Rad), and normalized to Gapdh (mouse and human: sense 5′-CTTCACCACCATGGAGAAGGC-3′, antisense 5′-GGCATGGACTGTGGTCATGAG-3′). Primers included Bclxl (human: sense, 5′-GGTGAGTCGGATCGCAGC-3′; antisense, 5′-CAGCGGTTGAAGCGTTCC-3′; mouse: 5′-TTTGAATCCGCCACCATGTCTCAGAGCAACCGGGAGCTG-3′; antisense, 5′-TTTCTCGAGCTTTCCGACTGAAGAGTGAGCCCA-3′), Bax (sense, 5′-CCAGGATGCGTCCACCAAGAAG-3′, antisense, 5′-GGAGTCCGTGTCCACGTCAGC-3′). Data are averages of ≥ 3 independent experiments, each in triplicate.
Quantitative chromatin immunoprecipitation (qChIP)
Cells were left untreated or treated as indicated and IP performed with anti-p50, anti-Histone H1 or anti-mouse IgG to control for nonspecific binding. qPCR was performed with the BCLXL promoter specific primers: sense, 5′-GCACCACCTACATTCAAATCC-3′ and antisense, 5′-CGATGGAGGAGGAAGCAAGC-3′. Amplification was performed as described above and quantification of the change in DNA enrichment for each IP condition was determined relative to input DNA as previously described.14
Electrophoretic mobility shift assays (EMSA)
EMSA was performed following nuclear fraction isolation, or with purified protein, including treatment and supershift analysis as previously described.14 A double-stranded oligonucleotide (5′-TCGAGTTAGATGGGGACTTTCCAGGCAC-3′) (IDT) containing the decameric кB-consensus sequence (underlined) was end labeled with [γ-32P] ATP and used as a probe. Reaction mixtures were incubated at room temperature for 20 min prior to resolving on a 5% polyacrylamide gel (0.5x TBE) at ∼10 V/cm for 2–4 hours at 4°C and assayed by autoradiography and PhosphorImager analysis (Molecular Dynamics). EMSA are representative of at least 3 independent experiments.
Clonogenic assays
Cells were plated and allowed to attach overnight. Following treatment, colony formation assay was performed as previously described.14 The surviving fraction was calculated based on the plating efficiency of untreated cells.
γH2AX focus formation
Cells cultured on glass coverslips were fixed in 4% paraformaldehyde. Following permeabilization, cells were immunostained with phospho-S140-H2AX (Cell Signaling, 9718) and nuclei counterstained with 1 mg/ml 4’,6-diamidino-2-phenylindole (DAPI) solution. Slides were imaged on a Zeiss microscope and the percentage of cells positive for γH2AX foci quantified in at least 200 cells per group.
Senescence associated (SA)-β-galactosidase staining
Primary MEFs at the indicated passage were plated onto 35 mm glass bottom plates (MatTek Corporation, Ashland, MA, USA) and at 60% confluency were fixed with formaldehyde for 20 minutes at 4°C. SA-β-galactosidase staining was performed in the standard fashion. Cells were incubated overnight with freshly made staining solution and the percentage of cells positive for SA-β-gal was calculated from a total of 200 cells per plate. Experiments were repeated in triplicate.
Metaphase spreads
Total breaks, gaps or constrictions on chromosomes were quantified on metaphase spreads stained by Giemsa (Sigma) essentially as previously described42 but without aphidicolin pre-treatment to obtain a homogeneous staining of chromosomes. Analysis of samples was made in a blinded fashion by 2 independent observers.
Single-cell gel electrophoresis (comet) assay
Alkaline comet assay was performed as described previously.43 Nuclei were visualized on a Zeiss microscope and images analyzed with ImageJ (NIH). DNA damage was quantified for 50 cells by determining the percentage of the cellular DNA within the tail using Comet Score (TriTek).
Homologous recombination analysis
The assay was performed essentially as described.44 HEK293 cells stably expressing the recombination substrate, DR-GFP, were a generous gift from Dr. from Phillip Connell and were grown to 70% confluence prior to transfection with p50 or scrambled siRNA. Cells were grown in complete growth medium for 24 hours and then re-transfected with the I-SceI expression vector, pCβASce, or the control vector, pCAGGS. Cells were incubated for an additional 24 hours in normal medium and then analyzed with a Becton-Dickinson FACScan. Alternatively, Nfkb1−/− MEFs stably expressing p50WT, p50S329A or empty vector were co-transfected with the pHR-DR-GFP reporter vector and either pCβASce or pCAGGS. Cells were grown in complete growth medium for 24 hours in normal medium and then analyzed with a Becton-Dickinson FACScan. For HR analysis, live cells were collected based on size/complexity and propidium iodide (PtdIns) exclusion and the fraction of live cells exhibiting GFP positivity displayed ± SEM of triplicate samples repeated.
Statistical analysis
Statistical analyses were performed using a 2-tailed Student's t-test.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to Oscar Fernandez-Capetillo, Warner Greene and Phillip Connell for plasmids, P. Nghiem for tet-on U2OS cells, Elizabeth Davis for assistance with metaphase spreads and Adam Schmitt for critical reading of the manuscript.
Funding
This work was supported by National Institutes of Health (NIH) grant R01CA136937 to B.Y. and the Ludwig Center for Metastasis Research.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Lopez-Contreras AJ, Fernandez-Capetillo O. The ATR barrier to replication-born DNA damage. DNA Repair (Amst) 2010; 9: 1249-55; PMID:21036674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Garner E, Costanzo V. Studying the DNA damage response using in vitro model systems. DNA Repair (Amst) 2009; 8: 1025-37; PMID:19482562 [DOI] [PubMed] [Google Scholar]
- 3.Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol 2008; 9: 616–27; PMID:18594563; http://dx.doi.org/10.1038/nrm2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kumagai A, Lee J, Yoo HY, Dunphy WG. TopBP1 activates the ATR-ATRIP complex. Cell 2006; 124: 943-55; PMID:16530042; http://dx.doi.org/10.1016/j.cell.2005.12.041 [DOI] [PubMed] [Google Scholar]
- 5.Toledo LI, Murga M, Gutierrez-Martinez P, Soria R, Fernandez-Capetillo O. ATR signaling can drive cells into senescence in the absence of DNA breaks. Genes Dev 2008; 22: 297-302; PMID:18245444; http://dx.doi.org/10.1101/gad.452308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown EJ, Baltimore D. Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev 2003; 17: 615-28; PMID:12629044; http://dx.doi.org/10.1101/gad.1067403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rothkamm K, Lobrich M. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc Natl Acad Sci U S A 2003; 100: 5057-62; PMID:12679524; http://dx.doi.org/10.1073/pnas.0830918100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Janssens S, Tschopp J. Signals from within: the DNA-damage-induced NF-kappaB response. Cell Death Differ 2006; 13: 773-84; PMID:16410802; http://dx.doi.org/10.1038/sj.cdd.4401843 [DOI] [PubMed] [Google Scholar]
- 9.Wu ZH, Shi Y, Tibbetts RS, Miyamoto S. Molecular linkage between the kinase ATM and NF-kappaB signaling in response to genotoxic stimuli. Science 2006; 311: 1141-6; PMID:16497931; http://dx.doi.org/10.1126/science.1121513 [DOI] [PubMed] [Google Scholar]
- 10.Wu ZH, Miyamoto S. Induction of a pro-apoptotic ATM-NF-kappaB pathway and its repression by ATR in response to replication stress. Embo J 2008; 27: 1963-73; PMID:18583959; http://dx.doi.org/10.1038/emboj.2008.127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rocha S, Campbell KJ, Perkins ND. p53- and Mdm2-independent repression of NF-kappa B transactivation by the ARF tumor suppressor. Mol Cell 2003; 12: 15-25; PMID:12887889; http://dx.doi.org/10.1016/S1097-2765(03)00223-5 [DOI] [PubMed] [Google Scholar]
- 12.Hayden MS, Ghosh S. NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev 2012; 26: 203-34; PMID:22302935; http://dx.doi.org/10.1101/gad.183434.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barre B, Perkins ND. A cell cycle regulatory network controlling NF-kappaB subunit activity and function. Embo J 2007; 26: 4841-55; PMID:17962807; http://dx.doi.org/10.1038/sj.emboj.7601899 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Schmitt AM, Crawley CD, Kang S, Raleigh DR, Yu X, Wahlstrom JS, Voce DJ, Darga TE, Weichselbaum RR, Yamini B. p50 (NF-kappaB1) is an effector protein in the cytotoxic response to DNA methylation damage. Mol Cell 2011; 44: 785-96; PMID:22152481; http://dx.doi.org/10.1016/j.molcel.2011.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumagai A, Dunphy WG. Claspin, a novel protein required for the activation of Chk1 during a DNA replication checkpoint response in Xenopus egg extracts. Mol Cell 2000; 6: 839-49; PMID:11090622; http://dx.doi.org/10.1016/S1097-2765(05)00092-4 [DOI] [PubMed] [Google Scholar]
- 16.Clarke CA, Clarke PR. DNA-dependent phosphorylation of Chk1 and Claspin in a human cell-free system. Biochem J 2005; 388: 705-12; PMID:15707391; http://dx.doi.org/10.1042/BJ20041966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nghiem P, Park PK, Ys Kim YS, Desai BN, Schreiber SL. ATR is not required for p53 activation but synergizes with p53 in the replication checkpoint. J Biol Chem 2002; 277: 4428-34; PMID:11711532; http://dx.doi.org/10.1074/jbc.M106113200 [DOI] [PubMed] [Google Scholar]
- 18.Zou L. Single- and double-stranded DNA: building a trigger of ATR-mediated DNA damage response. Genes Dev 2007; 21: 879-85; PMID:17437994; http://dx.doi.org/10.1101/gad.1550307 [DOI] [PubMed] [Google Scholar]
- 19.Crawley CD, Raleigh DR, Kang S, Voce DJ, Schmitt AM, Weichselbaum RR, Yamini B. DNA damage-induced cytotoxicity is mediated by the cooperative interaction of phospho-NF-kappaB p50 and a single nucleotide in the kappaB-site. Nucleic Acids Res 2013; 41: 764-74; PMID:23180782; http://dx.doi.org/10.1093/nar/gks1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamini B, Yu X, Dolan ME, Wu MH, Darga TE, Kufe DW, Weichselbaum RR. Inhibition of nuclear factor-kappaB activity by temozolomide involves O6-methylguanine induced inhibition of p65 DNA binding. Cancer Res 2007; 67: 6889-98; PMID:17638900; http://dx.doi.org/10.1158/0008-5472.CAN-06-4496 [DOI] [PubMed] [Google Scholar]
- 21.Hekmat-Nejad M, You Z, Yee MC, Newport JW, Cimprich KA. Xenopus ATR is a replication-dependent chromatin-binding protein required for the DNA replication checkpoint. Curr Biol 2000; 10: 1565-73; PMID:11137007; http://dx.doi.org/10.1016/S0960-9822(00)00855-1 [DOI] [PubMed] [Google Scholar]
- 22.Dart DA, Adams KE, Akerman I, Lakin ND. Recruitment of the cell cycle checkpoint kinase ATR to chromatin during S-phase. J Biol Chem 2004; 279: 16433-40; PMID:14871897; http://dx.doi.org/10.1074/jbc.M314212200 [DOI] [PubMed] [Google Scholar]
- 23.Arnaudeau C, Lundin C, Helleday T. DNA double-strand breaks associated with replication forks are predominantly repaired by homologous recombination involving an exchange mechanism in mammalian cells. J Mol Biol 2001; 307: 1235-45; PMID:11292338; http://dx.doi.org/10.1006/jmbi.2001.4564 [DOI] [PubMed] [Google Scholar]
- 24.Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev 2010; 24: 2463-79; PMID:21078816; http://dx.doi.org/10.1101/gad.1971610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hoffmann A, Leung TH, Baltimore D. Genetic analysis of NF-kappaB/Rel transcription factors defines functional specificities. Embo J 2003; 22: 5530-9; PMID:14532125; http://dx.doi.org/10.1093/emboj/cdg534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shechter D, Costanzo V, Gautier J. Regulation of DNA replication by ATR: signaling in response to DNA intermediates. DNA Repair (Amst) 2004; 3: 901-8; PMID:15279775 [DOI] [PubMed] [Google Scholar]
- 27.Flynn RL, Zou L. ATR: a master conductor of cellular responses to DNA replication stress. Trends Biochem Sci 2011; 36: 133-40; PMID:20947357; http://dx.doi.org/10.1016/j.tibs.2010.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brown EJ, Baltimore D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev 2000; 14: 397-402; PMID:10691732 [PMC free article] [PubMed] [Google Scholar]
- 29.de Klein A, Muijtjens M, van Os R, Verhoeven Y, Smit B, Carr AM, Lehmann AR, Hoeijmakers JH. Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr Biol 2000; 10: 479-82; PMID:10801416; http://dx.doi.org/10.1016/S0960-9822(00)00447-4 [DOI] [PubMed] [Google Scholar]
- 30.Cortez D, Guntuku S, Qin J, Elledge SJ. ATR and ATRIP: partners in checkpoint signaling. Science 2001; 294: 1713-6; PMID:11721054; http://dx.doi.org/10.1126/science.1065521 [DOI] [PubMed] [Google Scholar]
- 31.Nghiem P, Park PK, Kim Y, Vaziri C, Schreiber SL. ATR inhibition selectively sensitizes G1 checkpoint-deficient cells to lethal premature chromatin condensation. Proc Natl Acad Sci U S A 2001; 98: 9092-7; PMID:11481475; http://dx.doi.org/10.1073/pnas.161281798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev 2001; 15: 2177-96; PMID:11544175 [DOI] [PubMed] [Google Scholar]
- 33.Tercero JA, Longhese MP, Diffley JF. A central role for DNA replication forks in checkpoint activation and response. Mol Cell 2003; 11: 1323-36; PMID:12769855; http://dx.doi.org/10.1016/S1097-2765(03)00169-2 [DOI] [PubMed] [Google Scholar]
- 34.Desany BA, Alcasabas AA, Bachant JB, Elledge SJ. Recovery from DNA replicational stress is the essential function of the S-phase checkpoint pathway. Genes Dev 1998; 12: 2956-70; PMID:9744871; http://dx.doi.org/10.1101/gad.12.18.2956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Branzei D, Foiani M. The checkpoint response to replication stress. DNA Repair (Amst) 2009; 8: 1038-46; PMID:19482564 [DOI] [PubMed] [Google Scholar]
- 36.Malkova A, Haber JE. Mutations arising during repair of chromosome breaks. Annu Rev Genet 2012; 46: 455-73; PMID:23146099; http://dx.doi.org/10.1146/annurev-genet-110711-155547 [DOI] [PubMed] [Google Scholar]
- 37.Sha WC, Liou HC, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell 1995; 80: 321-30; PMID:7834752; http://dx.doi.org/10.1016/0092-8674(95)90415-8 [DOI] [PubMed] [Google Scholar]
- 38.Lu ZY, Yu SP, Wei JF, Wei L. Age-related neural degeneration in nuclear-factor kappaB p50 knockout mice. Neuroscience 2006; 139: 965-78; PMID:16533569; http://dx.doi.org/10.1016/j.neuroscience.2005.12.062 [DOI] [PubMed] [Google Scholar]
- 39.Fullard N, Moles A, O'Reilly S, van Laar JM, Faini D, Diboll J, Reynolds NJ, Mann DA, Reichelt J, Oakley F. The c-Rel subunit of NF-kappaB regulates epidermal homeostasis and promotes skin fibrosis in mice. Am J Pathol 2013; 182: 2109-20; PMID:23562440; http://dx.doi.org/10.1016/j.ajpath.2013.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Denis-Donini S, Dellarole A, Crociara P, Francese MT, Bortolotto V, Quadrato G, Canonico PL, Orsetti M, Ghi P, Memo M, et al. . Impaired adult neurogenesis associated with short-term memory defects in NF-kappaB p50-deficient mice. The Journal of neuroscience : the official journal of the Society for Neuroscience 2008; 28: 3911-9; PMID:18400889; http://dx.doi.org/10.1523/JNEUROSCI.0148-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yamini B, Yu X, Gillespie GY, Kufe DW, Weichselbaum RR. Transcriptional targeting of adenovirally delivered tumor necrosis factor alpha by temozolomide in experimental glioblastoma. Cancer Res 2004; 64: 6381-4; PMID:15374943; http://dx.doi.org/10.1158/0008-5472.CAN-04-2117 [DOI] [PubMed] [Google Scholar]
- 42.Glover TW, Berger C, Coyle J, Echo B. DNA polymerase alpha inhibition by aphidicolin induces gaps and breaks at common fragile sites in human chromosomes. Hum Genet 1984; 67: 136-42; PMID:6430783; http://dx.doi.org/10.1007/BF00272988 [DOI] [PubMed] [Google Scholar]
- 43.Singh NP, McCoy MT, Tice RR, Schneider EL. A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res 1988; 175: 184-91; PMID:3345800; http://dx.doi.org/10.1016/0014-4827(88)90265-0 [DOI] [PubMed] [Google Scholar]
- 44.Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev 1999; 13: 2633-8; PMID:10541549; http://dx.doi.org/10.1101/gad.13.20.2633 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.