

The aryl hydrocarbon receptor (AhR), a ligand-activated transcription factor, acts as a heterodimeric transcriptional regulator. Widely expressed in a variety of animal species as well as in humans, AhR has constitutive functions that are only now beginning to be appreciated. The main focus is now shifting from the role of AhR in the hepatic clearance of xenobiotics toward the nature of its physiological ligands as well as its mode of action in response to functionally distinct molecules, disparate in nature as to their endogenous source and chemical structure. Animal and human data indicate that AhR is involved in various signaling pathways critical to cell normal homeostasis, which covers multiple aspects of physiology, such as cell proliferation and differentiation, gene regulation, cell motility and migration, inflammation and others.1,2
Signal molecules function as ligands for AhR, and activated AhR forms heterodimers at promoter recognition sequences of the target genes. The AhR/AhR nuclear translocator (ARNT) complex may then require coactivators (including members of other families of transcription factors)3 in order to initiate transcription and to unwind histone-bound DNA for exposing additional promoter recognition sites via their histone acetyltransferase function. Within this scenario, 3 major factors appear to contribute to the outcome of gene transcriptional regulation by AhR, namely, nature of the ligand, local tissue microenvironment, and presence of coactivators in the cell. Prototypical examples are represented by AhR activation in gut innate lymphoid cells by microbiota-derived indole-3-aldehyde (IAld),4 in skin keratinocytes by endogenous 6-formylindolo[3,2-b]carbazole (FICZ),5 and in lymphoid tissue dendritic cells by a product of tryptophan catabolic enzymes, l-kynurenine.6
The functions of AhR in T cells depend on the specific ligand bound to the receptor. For instance, binding of dioxin to AhR suppresses experimental autoimmune encephalomyelitis by promoting the development of Foxp3+ T regulatory cells, whereas FICZ enhances encephalomyelitis by inducing the differentiation of IL-17–producing T cells.1 Therefore, in determining the qualitative effect of AhR engagement, it is not the potency (dictated in turn by affinity) and the efficacy of the ligand that matter so much as the ligand's ability to select a specific conformation of the receptor. When contextualized to the widely accepted conformation-based operational model of agonism—which considers multiple active receptor conformations, agonist efficacy and maximum effect of the system—it is likely that different AhR ligands preferentially bind distinct conformations of the AhR complex—each having a distinct set of fingerprint residues—thus initiating different pathways of downstream signaling and transcriptional events. Along this direction, we have recently demonstrated that the AhR fingerprint residues required for activation by dioxin are distinct from those necessary for activation by l-kynurenine, even when the response being measured is the same, namely, transcription of a gene (Cyp1a1) whose promoter contains xenobiotic response elements. A mutated form of the receptors that does not bind l-kynurenine will, instead, bind dioxin with increased potency and likely affinity.6 This suggests that structurally and/or functionally distinct AhR ligands have distinct affinities for distinct conformations, each active conformation of the receptor recruiting a unique set of downstream signaling molecules. Thus both ligand-intrinsic and cell-intrinsic factors contribute to diversifying the effects of AhR activation in a tissue. The cell-intrinsic factors include competence for substrates—in the downstream signaling cascade—that can be phosphorylated via a nongenomic function of AhR, namely, AhR complex-associated Src kinase activity. It is interesting in this regard that l-kynurenine, on engaging AhR, also activates Src kinase activity.6
Xenobiotics including dietary phytochemicals, products of microbiota, and ubiquitous environmental pollutants may have shaped this system in intestinal epithelia during millions of years of evolution. In the gut, the ligand for AhR can be derived or generated from diet, microbiome, and/or host cells. Mice exposed to a tryptophan-enriched diet expand a population of lactobacilli in the gut that produce IAld, which promotes AhR-dependent transcription of the IL-22–encoding gene by host innate lymphoid cells and thus prevents microbial infections and local inflammation.4 In the skin, AhR ligation controls oxidation/antioxidation, epidermal barrier function, photo-induced response, melanogenesis, and innate immunity. Dioxin-mediated skin and intestinal inflammation is associated with deregulated T-cell differentiation. In contrast, AhR activation by endogenous FICZ in keratinocytes dampens the severity of inflammatory skin conditions.5 In overinflammatory systemic responses induced by infection and other noxae, 2 distinct tryptophan catabolic enzymes, hepatic tryptophan 2,3-dioxygenase and ubiquitous indoleamine 2,3-dioxygenase 1, produce an amino acid, l-kynurenine, that suppresses inflammatory cytokine gene transcription, and induces, instead, transcription of the genes encoding anti-inflammatory IL-10 and TGF-β. This mechanism requires contribution by noncanonical NF-κB family members and other molecules, which are recruited via AhR complex-associated Src kinase activity6 (Fig. 1).
Figure 1.
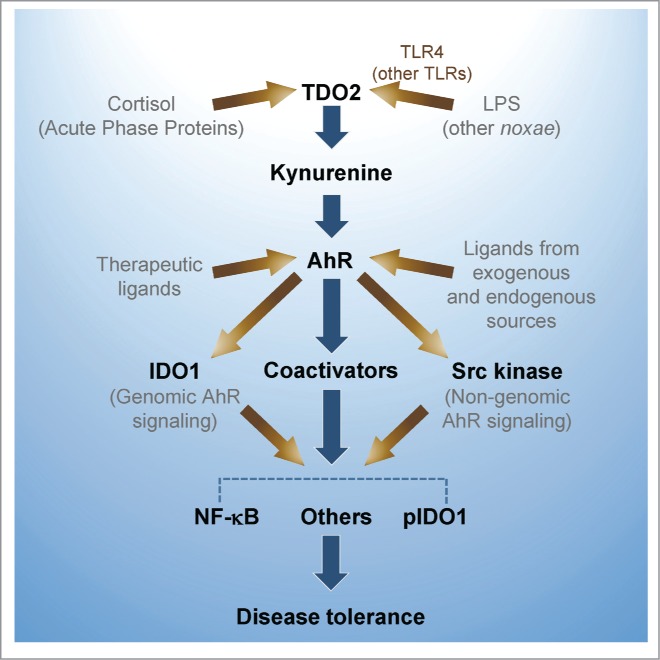
AhR controls hyperinflammatory responses to lipopolysaccharide (LPS) and other noxae, and contributes to "disease tolerance." An LPS sublethal dose activates TDO2 leading to kynurenine production from tryptophan. Kynurenine, by acting as an AhR ligand increases IL-10, and decreases IL-1β, TNF-α and IL-6. High-dose LPS rechallenge in primed mice triggers IDO1 phosphorylation by AhR complex-associated Src kinase activity and TGF-β production. IDO1 further increases kynurenine production, phosphorylated IDO1 acts a signaling molecule, and AhR, in association with several transcriptional partners, contributes to reprogramming gene expression and chromatin remodeling. LPS-tolerant mice challenged with either gram-negative or gram-positive bacteria are less prone to inflammatory pathology.
In conclusion, there is now strong evidence to support the concept that the outcome of AhR activation is largely dictated by the nature of the ligand—which initiates a specific sequence of downstream signaling events—as well as by the specific tissue (e.g., gut, skin, and lymphoid tissue) in which AhR engagement occurs. In addition to controlling the production and degradation of AhR ligands, the local tissue may indeed provide a specific set of coactivators and functions bridging the basic transcriptional machinery to the target genes.
References
- 1. Quintana FJ, et al. Pharmacol Rev 2013; 65:1148-61; PMID:23908379; http://dx.doi.org/ 10.1124/pr.113.007823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stockinger B, et al. Annu Rev Immunol 2014; 32:403-32; PMID:24655296; http://dx.doi.org/ 10.1146/annurev-immunol-032713-120245 [DOI] [PubMed] [Google Scholar]
- 3. Vogel CF, et al. J Biol Chem 2014; 289:1866-75; PMID:24302727; http://dx.doi.org/ 10.1074/jbc.M113.505578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zelante T, et al. Immunity 2013; 39:372-85; PMID: 23973224; http://dx.doi.org/ 10.1016/j.immuni.2013.08.003 [DOI] [PubMed] [Google Scholar]
- 5. Di Meglio P, et al. Immunity 2014; 40:989-1001; PMID:24909886; http://dx.doi.org/ 10.1016/j.immuni.2014.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bessede A, et al. Nature 2014; 511:184-90; PMID: 24930766; http://dx.doi.org/ 10.1038/nature13323 [DOI] [PMC free article] [PubMed] [Google Scholar]