

Abbreviations
- CO
carbon monoxide
- HD
Huntington's disease
- Keap1
Kelch-like ECH-associated protein 1
- NO
nitric oxide
- Nrf2
nuclear factor erythroid 2-related factor 2
- TAF4
TBP associated factor 4
- TBP
TATA box binding protein
Huntington's disease (HD) is an autosomal dominant lethal neurologic disorder. Its pathophysiology involves massive, highly selective degeneration of the corpus striatum of the brain, which regulates motor activity, and whose loss in HD leads to the characteristic choreiform movements of patients. The unique degradation of the striatum appears to reflect the binding of mutant huntingtin to a striatal-selective small G protein called Rhes.1 Recently, we have identified specific molecular mechanisms whereby huntingtin elicits neuronal destruction.
Earlier studies of gasotransmitters such as nitric oxide (NO) and carbon monoxide (CO) had led to an interest in hydrogen sulfide (H2S). H2S can be physiologically formed by cystathionine β-synthase (CBS), cystathionine γ-lyase (CSE) (Fig. 1A) and 3-mercaptopyruvate sulfurtransferase.2 We had characterized CSE depleted mice focusing on their hypertension, implicating H2S as a physiologic vasorelaxant.3 CSE mutant mice also displayed neurologic abnormalities such as hind limb clasping which resembled mouse models of HD. These behavioral observations prompted us to explore the possibility of CSE abnormalities in HD.
Figure 1.
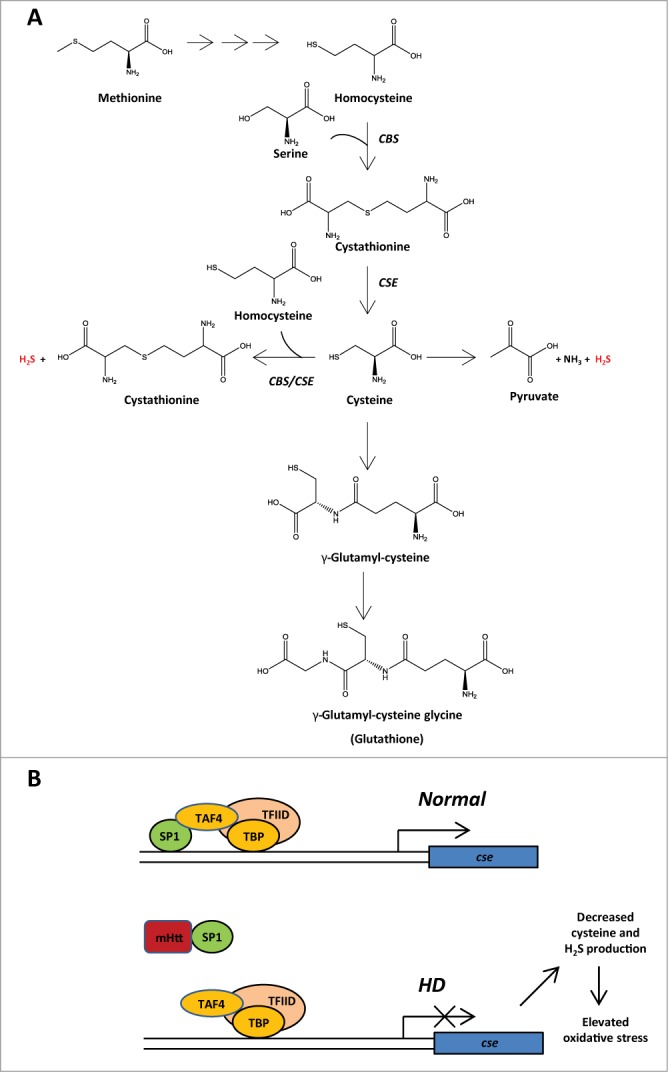
Cystathionine γ-lyase (CSE), the biosynthetic enzyme for cysteine is depleted in Huntington's disease (HD) at the transcriptional level. (A) CSE is a key enzyme in the reverse transsulfuration pathway which generates cysteine from its precursor, cystathionine. Cystathionine is synthesized by cystathionine β-synthase (CBS) by condensing homocysteine derived from dietary methionine with serine. Both CSE and CBS generate the major gasotransmitter hydrogen sulfide (H2S) from cysteine as well as homocysteine. In addition, cysteine serves as a building block for biosynthesis of the antioxidant glutathione. (B) Under normal conditions, CSE transcription is regulated by the transcription factor specificity protein 1 (SP1) in conjunction with associated coactivators such as TATA box binding protein (TBP), TBP-associated factor 4 (TAF4), subunits of the general transcription factor, transcription factor II D (TFIID). In HD, mutant huntingtin (mHtt)sequesters SP1 leading to a diminished expression of CSE, which results in lowered cysteine levels, elevated oxidative stress and neurodegeneration.
HD stems from mutations of the gene encoding huntingtin with polyglutamine repeats that mediate neurotoxicity. One useful HD model employs a striatal cell line harboring mutant huntingtin with 111 glutamine repeats.4 CSE levels in these cells were reduced by more than 90%.5 We also explored R6/2 mice and Q175 mice, which harbor expanded polyglutamine repeats. In both strains CSE levels were substantially diminished. Most importantly, in the striatum of human HD patients CSE levels were reduced 85–90% with greater reductions in patients displaying more severe clinical manifestations of the disease. Also, CSE depletion was selective for the striatum with diminished loss in the cortex and normal levels of CSE in the cerebellum, paralleling the relative susceptibility of these brain regions to HD damage. The CSE depletion does not simply reflect influences of generalized neurodegeneration, as we found no alteration in CSE levels in other neurodegenerative diseases such as amyotrophic lateral sclerosis, multiple sclerosis or spinocerebellar ataxia. Interestingly, CSE levels were diminished in the liver and pancreas of R6/2 HD mice, consistent with the body-wide expression of huntingtin as well as the presence of gastrointestinal symptoms in HD patients.
What molecular mechanisms might underlie the CSE depletion? We observed substantial reductions of mRNA for CSE in Q111 HD cell lines indicating that the deficit is at a transcriptional level. Striatal Q111 cells are highly susceptible to hydrogen peroxide stress, a defect that is reversed by overexpressing CSE.5 Mutant huntingtin is known to bind and inhibit specificity protein 1 (Sp1), a well characterized transcription factor for CSE.6 Overexpressing Sp1 and its co-activator TATA box binding protein (TBP)-associated factor 4 (TAF4) reverses the diminished mRNA and protein levels of CSE in Q111 cells. Accordingly, we conclude that the CSE depletion in HD stems from mutant huntingtin inhibiting the CSE transcription factor Sp1 (Fig. 1B).
If CSE depletion is responsible for the phenotype of HD, then CSE depleted mice should display neurologic symptoms. Indeed, the CSE mutants manifest neurobehavioral alterations closely resembling those of HD mice. CSE knockout mice are highly susceptible to neurotoxic actions of 3-nitropropionic acid, further emphasizing a role for CSE in neuroprotection.5
Our findings indicate that CSE is physiologically cytoprotective against the neurodegenerative features of HD. How might CSE exert cytoprotection? CSE is a major source of cysteine, which is generated from cystathionine. Cysteine is an important component of proteins and a precursor of the antioxidant glutathione. Moreover, cysteine is a precursor of the gasotransmitter H2S, which activates cytoprotective enzymes by sulfhydrating them, i.e. attaching a thiol group to the target proteins. Sulfhydration of parkin stimulates its catalytic activity and appears to provide neuroprotection of the striatum in Parkinson Disease.7 H2S sulfhydrates Kelch-like ECH-associated protein 1 (Keap1), a repressor of the transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2), which regulates enzymes in the antioxidant defense pathway.8
If CSE depletion and the loss of its products, such as cysteine, are responsible for clinical deficiencies in HD, then restoration of cysteine should be therapeutic. Cystamine, a decarboxylated derivative of cystine (a disulfide of cysteine) is neuroprotective in rodent models of HD.9,10 In our own experiments treating R6/2 HD mice with N-acetylcysteine in their drinking water along with a cysteine-enriched diet delays the onset of motor abnormalities and partially reverses the decreases in striatal weight of the mutant mice.
In summary, a substantial portion of HD neurotoxicity appears to be attributable to CSE deficiency. This deficiency arises presumably by the binding of mutant huntingtin to the transcriptional activator Sp1 diminishing its activity and thereby lowering CSE levels. The beneficial effects of cysteine supplementation in murine HD models suggests that treatment such as N-acetylcysteine may be therapeutically relevant in HD patients.
References
- 1. Subramaniam S, Sixt KM, Barrow R, Snyder SH. Rhes, a striatal specific protein, mediates mutant-huntingtin cytotoxicity. Science 2009; 285(27): 20428-32; PMID: 19498170; http://dx.doi.org/ 10.1126/science.1172871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Paul BD, Snyder SH. H2S signalling through protein sulfhydration and beyond. Nat Rev Mol Cell Biol 2012; 13:499-507; PMID: 22781905; http://dx.doi.org/ 10.1038/nrm3391 [DOI] [PubMed] [Google Scholar]
- 3. Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, et al. . H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science 2008; 322:587-90; PMID: 18948540; http://dx.doi.org/ 10.1126/science.1162667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, MacDonald ME. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet 2000; 9:2799-809; PMID: 11092756; http://dx.doi.org/ 10.1093/hmg/9.19.2799 [DOI] [PubMed] [Google Scholar]
- 5. Paul BD, Sbodio JI, Xu R, Vandiver MS, Cha JY, Snowman AM, Snyder SH. Cystathionine γ-lyase deficiency mediates neurodegeneration in Huntington's disease. Nature 2014; 509:96-100; PMID: 24670645; http://dx.doi.org/ 10.1038/nature13136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ishii I, Akahoshi N, Yu XN, Kobayashi Y, Namekata K, Komaki G, Kimura H. Murine cystathionine gamma-lyase: complete cDNA and genomic sequences, promoter activity, tissue distribution and developmental expression. Biochem J 2004; 381:113-123; PMID: 15038791; http://dx.doi.org/ 10.1042/BJ20040243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vandiver MS, Paul BD, Xu R, Karuppagounder S, Rao F, Snowman AM, Ko HS, Lee YI, Dawson VL, Dawson TM, et al. . Sulfhydration mediates neuroprotective actions of parkin. Nat Commun 2013; 4:1626; PMID: 23535647; http://dx.doi.org/ 10.1038/ncomms2623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang G, Zhao K, Ju Y, Mani S, Cao Q, Puukila S, Khaper N, Wu L, Wang R. Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf. Antioxid Redox Signal 2013; 18:1906-19; PMID: 23176571; http://dx.doi.org/ 10.1089/ars.2012.4645 [DOI] [PubMed] [Google Scholar]
- 9. Karpuj MV, Becher MW, Springer JE, Chabas D, Youssef S, Pedotti R, Mitchell D, Steinman L. Prolonged survival and decreased abnormal movements in transgenic model of Huntington disease, with administration of the transglutaminase inhibitor cystamine. Nat Med 2002; 8:143-9; PMID: 11821898; http://dx.doi.org/ 10.1038/nm0202-143 [DOI] [PubMed] [Google Scholar]
- 10. Fox JH, Barber DS, Singh B, Zucker B, Swindell MK, Norflus F, Buzescu R, Chopra R, Ferrante RJ, Kazantsev A, et al. . Cystamine increases L-cysteine levels in Huntington's disease transgenic mouse brain and in a PC12 model of polyglutamine aggregation. J Neurochem 2004; 91:413-22; PMID: 15447674; http://dx.doi.org/ 10.1111/j.1471-4159.2004.02726.x [DOI] [PubMed] [Google Scholar]