

Abstract
The PI3K/PDK1/PKB signaling pathway plays essential roles in regulating neuronal survival, differentiation and plasticity in response to neurotrophic factors, neurotransmitters and ion channels. Both PDK1 and PKB can interact at the plasma membrane with a phosphoinositide synthesized by PI3K, the second messenger PtdIns(3,4,5)P3, enabling PDK1 to phosphorylate and activate PKB. In the PDK1 K465E knock-in mice expressing a mutant form of PDK1 incapable of phosphoinositide binding, activation of PKB was markedly affected, but not totally abolished. It has been recently proposed that in the absence of PtdIns(3,4,5)P3 binding, PDK1 can still moderately activate PKB due to a docking site-mediated interaction of these 2 kinases. A recent report has uncovered that in the PDK1 K465E mice neurons, a PKB signal threshold was sufficient to support neuronal survival responses, whereas neuritogenesis, neuronal polarization and axon outgrowth were severely impaired. We propose here that the low-efficiency mechanism of PKB activation observed in the PDK1 K465E mice might represent the ancestral mechanism responsible for the essential functions of this pathway, while the phosphoinositide-dependent activation should be considered an evolutionary innovation that enabled the acquisition of novel functions.
Keywords: knock-in mice, neuronal survival, neuronal morphogenesis, PDK1, PKB/Akt, PH-domain, phosphoinositides
PDK1 is a master kinase that activates 203 different kinases of the AGC family including PKB/Akt, RSK, S6K, SGK and PKC isoforms, which in turn control cell growth, survival, proliferation and metabolism responses to diverse extracellular signals.1 Since PDK1 is constitutively active in non-stimulated cells, regulating the accessibility of PDK1 to the different substrates becomes the critical control-point of this signaling pathway. PKB/Akt isoforms are activated downstream of PI3K activation and PtdIns(3,4,5)P3 production. This second messenger promotes the translocation and co-localization of both PDK1 and PKB at the plasma membrane due to the ability of the pleckstrin homology (PH) domains present in both kinases to specifically interact with phosphoinositides.2-4 Moreover, the binding of the PKB PH-domain to PtdIns(3,4,5)P3 also induces a conformational change that converts PKB onto a substrate that can be readily phosphorylated by PDK1 at Thr308 within the activation loop,5,6 while mTORC2 phosphorylates Ser473 at the hydrophobic motif,7 rendering PKB fully active. Since the rest of PDK1 substrates lack PH-domains to sense phosphoinositides levels, their mechanism of activation rely instead on the prior activation of the kinase phosphorylating the residue within the hydrophobic motif equivalent to that of PKB Ser473. Phosphorylation of the hydrophobic motif creates a substrate docking site that can then be specifically recognized by a small cavity located within the PDK1 catalytic domain termed the PIF binding pocket. Once bound to the substrate, PDK1 gains access to the phosphorylation of the activation loop of these set of enzymes.8,9 While PDK1 is the common upstream kinase phosphorylating the activation loop, the identity of the hydrophobic motif kinase is different among the different AGC family members, where mTORC1 phosphorylates the hydrophobic motif of S6K10,11 and novel PKCs,12 RSK becomes phosphorylated downstream of ERK,13 while mTORC2 is the hydrophobic motif kinase for PKB7 and SGK,14 allowing in this way differential responses to particular stimuli.15,16
The physiological functions of this signaling pathway have been dissected in vivo with the generation of 2 PDK1 knock-in mice expressing 2 mutant forms of PDK1 specifically affecting either the substrate binding pocket or the phosphoinositide binding pocket.17 In the PDK1 L155E knock-in mice, which express a mutant form of PDK1 in which Leucine 155 within the PIF-pocket was substituted by Glutamic acid, PDK1 cannot recognize the phosphorylated hydrophobic motif in the substrate and, as a consequence, activation of the different PDK1 targets with the exception of PKB was severely damaged.18,19 Conversely, in the PDK1 K465E mice, in which Lysine at position 465 within the PH-domain was mutated to Glutamic acid, PDK1 cannot interact with phosphoinositides. While the mechanism of activation of RSK, S6K, PKC and SGK was intact, PKB could surprisingly also be activated in these second knock-in mice model, albeit to a lesser extent when compared to control mice.20 Najafov and co-workers recently proposed that the reduced activation of PKB that was still observed in the PDK1 K465E cells was dependent upon the binding of PDK1 to the hydrophobic motif of PKB when phosphorylated at Ser473.21 While characterizing the GSK2334470 PDK1 inhibitor, the authors found this compound more efficient at inhibiting S6K or SGK than at inhibiting PKB. Only under conditions that moderately activated the PI3K signaling, or in cells expressing mutant forms of either PDK1 or PKB incapable of PtdIns(3,4,5)P3 binding, the inhibitor blocked PKB activation with the same efficiency than that observed for S6K or SGK.22 Moreover, pharmacological inhibition of PKB Ser473 phosphorylation in several cancer cell lines, or genetic inactivation of different mTORC2 components in MEF cells, sensitized PKB to the PDK1 inhibitor. Also, the reduced activation levels of PKB observed in the PDK1 K465E cells were totally blocked by mTOR inhibitors.21
Therefore, in the absence of PtdIns(3,4,5)P3 binding, PDK1 can still take advantage of the docking site to bind and activate PKB, albeit less efficiently. This new uncovered ability of PDK1 to interact also with the phosphorylated PKB Ser473 hydrophobic motif indicates that the substrate docking site-mediated activation is shared by all the PDK1-regulated kinases and therefore should be considered as an ancestral mechanism of PDK1 regulation, which was conserved during eukaryotic evolution.23 In contrast, the mutual co-localization of PDK1 and PKB at PtdIns(3,4,5)P3-rich surfaces might represent a functional innovation relying on the convergent acquisition of phosphoinositide binding domains by these 2 kinases, which in turn allowed faster and acute responses to particular second messengers. Protein sequence analysis indicates that the PKB and PDK1 PH-domains are different in origin.24 Also, the PDK1 PH-domain is located C-terminal to the kinase domain, whereas the PKB-PH domain is situated at the N-terminus of the protein. Moreover, the lipid binding specificity is also distinct between these 2 domains, with the PKB PH-domain exhibiting a marked selectivity for the PI3K product PtdIns(3,4,5)P3, while the PDK1 PH-domain binding a more broad range of phospholipids as well as inositol phosphates. Therefore, the hypothesis of a primitive AGC kinase possessing a PH-domain giving origin to PDK1 and PKB is not favored. Indeed, PH-domain-containing PDK1 orthologues have been identified in all the major eukaryotic phyla and were only secondarily lost in particular groups such as S. cerevisiae, but not S. pombe, and in most non-photosynthetic plants.25 By contrast, the presence of bona fide PKB homologues containing PH-domains is only reported in metazoans as well as in Dictyostelium, thereby indicating that a PH-domain-containing PKB ortholog was originated before the divergence of amoebozoans and metazoans.26 It is therefore intriguing to speculate that the adoption of this more efficient mechanism of activating PKB in response to PtdIns(3,4,5)P3 raises allowed functional and morphological innovations that accompanied the major transitions in the evolution toward the multicellularity within the animal kingdom and, among them, the development of a complex nervous system.
Actually, it is widely believed that the PI3K/PKB signaling axis plays essential roles in regulating neuronal survival by inhibiting the apoptotic cell death machinery in response to neurotrophic factors and synaptic activity. The PI3K/PKB pathway plays also fundamental roles in regulating neuronal differentiation by defining the axon-dendrite axis. We recently characterized the neuronal phenotype of the PDK1 K465E mice,27 which express a single-aminoacid mutant form of PDK1 incapable of phosphoinositide binding but with intact catalytic activity and preserved substrate docking-site binding abilities. In embryonic primary neurons derived from these mice, the activation of PKB induced by BDNF (Brain-Derived Neurotrophic Factor) was markedly decreased due to reduced phosphorylation of PKB at the PDK1 site. Unexpectedly, we found that the low levels of PKB activation achieved in the PDK1 K465E neurons were sufficient to support cell survival and, in agreement with that, no differences in the total number of neurons were detected in different brain areas of the mutant mice including the cortex and the hippocampus. By contrast, the differentiation capacity of primary cortical and hippocampal neuronal cultures was markedly affected by the PDK1 K465E mutation, resulting in deficient neuritogenesis, abnormal cell polarization and reduced axonal outgrowth. These phenotypes correlated with the fact that the low levels of PKB activity observed in the mutant neurons were rate-limiting for phosphorylation and inhibition of PRAS40 and TSC2, two mTORC1 inhibitory proteins, resulting in incomplete mTORC1 activity release and reduced synthesis of particular proteins that are important for neuronal morphogenesis, such as the BRSK brain-specific kinases. By contrast, phosphorylation of GSK3 and FOXO, which are involved in the inhibition of multiple pro-apoptotic proteins, proceeded normally. Analogous observations were also made in the PDK1 K465E mice immune system, where low levels of PKB activity were sufficient for T-cell survival and proliferation but insufficient to initiate the migratory program of effector T cells.28
Since the activation of PKB in the PDK1 K465E tissues most likely arises from the ability of the PDK1 PIF-pocket to recognize and bind to the PKB hydrophobic motif when phosphorylated at Ser473, we propose that the cellular responses elicited by the low levels of PKB activity uncovered by the PDK1 K465E mutation might reflect the ancient functions of this signaling branch (Fig. 1). In basal conditions, or under stimuli that does not induce strong activation of the PI3K signaling pathway, PKB can still become phosphorylated to some extent at Ser473 within the hydrophobic motif, creating a substrate binding site for PDK1 that will gain access in this manner for phosphorylation of the Thr308 residue within the PKB activation loop. This entails the sequential rather than simultaneous phosphorylation of PKB at the 2 activation sites, and is therefore in essence less rapid and inefficient. This might represent the ancestral mechanism by which the first PKB-like orthologues lacking PH-domains were activated. In this scenario, regulation of the phosphorylation levels of PKB at Ser473 should be the critical control point in regulating PKB activity. It is worthy to mention that both the Sin1 subunit of mTORC2 complex as well as PHLPP, the kinase and the phosphatase regulating PKB Ser473 phosphorylation respectively, possess PH-domains able to interact with phospholipids. In the PDK1 K465E mice, the PIF-pocket dependent activation of PKB was proved to be sufficient to guarantee essential functions such as cell survival and proliferation through the phosphorylation of certain cellular substrates such as GSK3 or FOXO1. Mutual binding of both PDK1 and PKB to PtdIns(3,4,5)P3 dramatically increases the rate of PKB activation, exceeding in this way a threshold of signal that enables efficient phosphorylation of other cellular substrates, namely PRAS40 or TSC2, controlling particular physiological responses such as neuronal morphogenesis27 or T-cell migration.28
Figure 1.

A model for the evolution of the PKB activating mechanisms. In ancestral eukaryotes lacking PH domain-containing PKB-like orthologues, interaction of PDK1 with PKB relied on the PIF-pocket mechanism. This promoted limited PKB activation that was responsible for essential functions such as supporting cell survival, a situation that is reproduced in the PDK1 K465E knock-in mice neurons. At the amoebozoans-metazoans origin, the acquisition of a PH domain allowed PKB to co-localize with PDK1 in response to PtdIns(3,4,5)P3 raises. This enabled activating PKB with high efficiency, which allowed the recruitment of additional cellular substrates regulating novel functions, such as complex neuronal morphogenesis.
In order to test this model, we explored whether pharmacological inhibition of PKB Ser 473 phosphorylation affected the biochemical and cellular responses of the PDK1 K465E knock-in mice neurons. We employed the AZD8055 compound, a novel ATP-competitive mTORC1 and mTORC2 inhibitor, with at least 1,000-fold differential in potency against class I and class III PI3Ks as well as PIKK family members ATM and DNA-PK.29 Treatment of cortical primary cultures with the AZD8055 compound reduced the BDNF-induced phosphorylation of PKB at Ser473 to basal levels in both PDK1 wild type and PDK1 K465E cortical neurons in a dose-dependent manner, while totally inhibited the phosphorylation of PKB at Thr308 in the PDK1 K465E mutant neurons at doses that did not affect PKB Thr308 phosphorylation in the control cells. The differential inhibition of PKB resulted in impaired phosphorylation of the PKB substrates PRAS40, TSC2, as well as GSK3 and FOXO, in the mutant cells (Fig. 2B). As a consequence, the BDNF-elicited survival responses of the PDK1 K465E cortical neurons were further decreased when compared to the control neurons (Fig. 2A). We also observed that the AZD8055 inhibitor modestly compromised the viability of the PDK1 wild type neurons at doses that did not affected PKB activation. Since the mTORC1 specific inhibitor rapamycin has not effect on neuronal viability,27 and the contribution of other PI3K classes poorly targeted by the inhibitor to neuronal viability is highly unlikely, while the mTORC2 complex acts also as the hydrophobic motif kinase for the SGK isoforms, these genotype-independent effects of the AZD8055 compound on cell viability point out to a role of SGK in promoting neuronal survival. Indeed, we and other members of the group have observed that in the PDK1 L155E mice expressing a mutant form of PDK1 incapable of activating SGK, a synergistic role of both PKB and SGK in controlling neuronal survival can be envisaged (Lluis Cordon-Barris and Jose R Bayascas, unpublished data). Altogether, these results further support the notion of low levels of PKB activity attained by means of the PIF-pocket mechanism being sufficient to guarantee essential functions such as neuronal viability, whereas more specialized functions necessitate the acute and rapid activation of PKB that is achieved upon phosphoinositide dependent co-localization with PDK1 in response to variations in the levels of the PtdIns(3,4,5)P3 second messenger.
Figure 2.
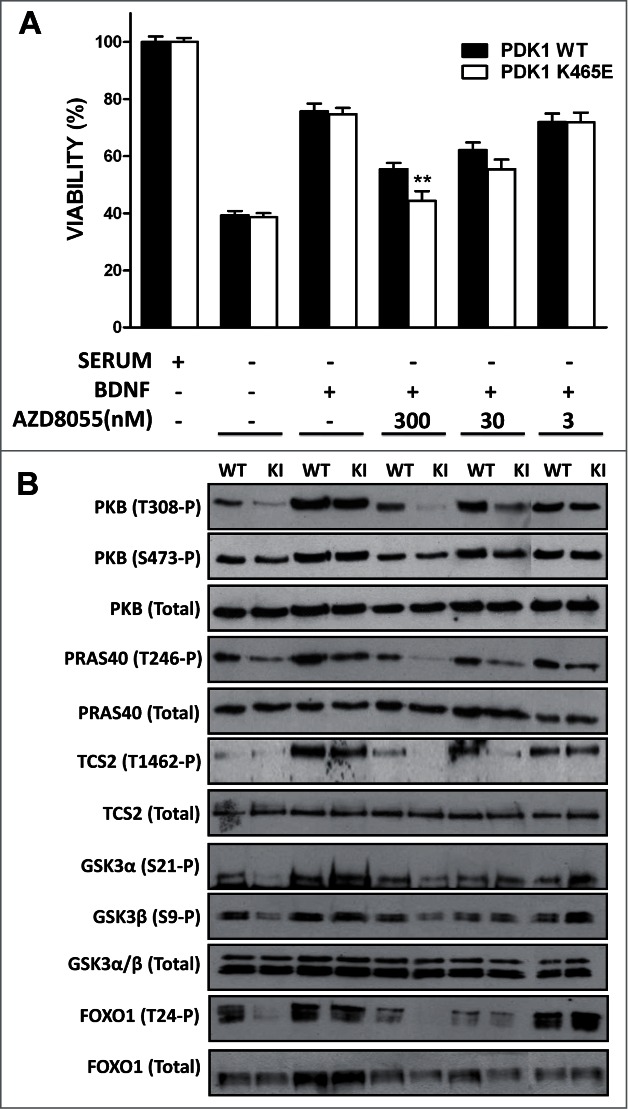
Primary cultures of cortical neurons derived from PDK1 wild type (WT) and PDK1 K465E knock-in (KI) E15,5 embryo littermates were generated as described.27 After 6 d in culture, cells were incubated in complete medium or deprived of serum for 24 h in the absence or presence of 50 ng/ml of Brain-Derived Neurotrophic Factor (BDNF) and the indicated concentrations of the AZD8055 inhibitor. Cell viability was determined by the MTT (1-(4,5-Dimethylthiazol-2-yl)-3,5-diphenylformazan) reduction assay and is represented as the mean ± standard errors of the mean from 5 independent embryos per genotype, with each sample assayed in quadriplicate. As a control for the different treatments, cell lysates from matched PDK1 wild type and PDK1 mutant littermate mice were immunoblotted with the indicated antibodies, where the first 8 lanes and the last 2 lanes were generated from separate gels. **, P < 0.01 between genotypes as determined by the Student's t test.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
A Ramon y Cajal contract from the Spanish Ministerio de Educación y Ciencia supported JRB and the China Scholarship Council founded XZ. This work was financed by the Ministerio de Sanidad y Consumo (Grant FIS-PI070101) and Ministerio de Ciencia y Innovación (Grant AES-PI10/00333).
References
- 1. Mora A, Komander D, Van Aalten DM, Alessi DR. PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol 2004; 2:161-70; PMID:15209375; http://dx.doi.org/ 10.1016/j.semcdb.2003.12.022 [DOI] [PubMed] [Google Scholar]
- 2. Alessi DR, Deak M, Casamayor A, Caudwell FB, Morrice N, Norman DG, Gaffney P, Reese CB, MacDougall CN, Harbison D, et al. 3-Phosphoinositide-dependent protein kinase-1 (PDK1): structural and functional homology with the Drosophila DSTPK61 kinase. Curr Biol 1997; 10:776-89; PMID:9368760; http://dx.doi.org/ 10.1016/S0960-9822(06)00336-8 [DOI] [PubMed] [Google Scholar]
- 3. Stephens L, Anderson K, Stokoe D, Erdjument-Bromage H, Painter GF, Holmes AB, Gaffney PR, Reese CB, McCormick F, Tempst P, et al. Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science 1998; 5351:710-14; http://dx.doi.org/ 10.1126/science.279.5351.710 [DOI] [PubMed] [Google Scholar]
- 4. Currie RA, Walker KS, Gray A, Deak M, Casamayor A, Downes CP, Cohen P, Alessi DR, Lucocq J. Role of phosphatidylinositol 3,4,5-trisphosphate in regulating the activity and localization of 3-phosphoinositide-dependent protein kinase-1. Biochem J 1999; 575-83; PMID:9895304; http://dx.doi.org/ 10.1042/0264-6021:3370575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Thomas CC, Deak M, Alessi DR, Van Aalten DM. High-resolution structure of the pleckstrin homology domain of protein kinase b/akt bound to phosphatidylinositol (3,4,5)-trisphosphate. Curr Biol 2002; 14:1256-62; PMID:12176338; http://dx.doi.org/ 10.1016/S0960-9822(02)00972-7 [DOI] [PubMed] [Google Scholar]
- 6. Milburn CC, Deak M, Kelly SM, Price NC, Alessi DR, Van Aalten DM. Binding of phosphatidylinositol 3,4,5-trisphosphate to the pleckstrin homology domain of protein kinase B induces a conformational change. Biochem J 2003; Pt 3:531-38; http://dx.doi.org/ 10.1042/BJ20031229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005; 5712:1098-101; http://dx.doi.org/ 10.1126/science.1106148 [DOI] [PubMed] [Google Scholar]
- 8. Arencibia JM, Pastor-Flores D, Bauer AF, Schulze JO, Biondi RM. AGC protein kinases: from structural mechanism of regulation to allosteric drug development for the treatment of human diseases. Biochim Biophys Acta 2013; 7:1302-21; PMID:23524293; http://dx.doi.org/ 10.1016/j.bbapap.2013.03.010 [DOI] [PubMed] [Google Scholar]
- 9. Biondi RM, Komander D, Thomas CC, Lizcano JM, Deak M, Alessi DR, Van Aalten DM. High resolution crystal structure of the human PDK1 catalytic domain defines the regulatory phosphopeptide docking site. EMBO J 2002; 16:4219-28; PMID:12169624; http://dx.doi.org/ 10.1093/emboj/cdf437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, Yonezawa K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 2002; 2:177-89; PMID:12150926; http://dx.doi.org/ 10.1016/S0092-8674(02)00833-4 [DOI] [PubMed] [Google Scholar]
- 11. Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002; 2:163-75; PMID:12150925; http://dx.doi.org/ 10.1016/S0092-8674(02)00808-5 [DOI] [PubMed] [Google Scholar]
- 12. Parekh D, Ziegler W, Yonezawa K, Hara K, Parker PJ. Mammalian TOR controls one of two kinase pathways acting upon nPKCdelta and nPKCepsilon. J Biol Chem 1999; 49:34758-64; PMID:10574945; http://dx.doi.org/ 10.1074/jbc.274.49.34758 [DOI] [PubMed] [Google Scholar]
- 13. Anjum R, Blenis J. The RSK family of kinases: emerging roles in cellular signalling. Nat Rev Mol Cell Biol 2008; 10:747-58; PMID:18813292; http://dx.doi.org/ 10.1038/nrm2509 [DOI] [PubMed] [Google Scholar]
- 14. Garcia-Martinez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem J 2008; 3:375-85; PMID:18925875; http://dx.doi.org/ 10.1042/BJ20081668 [DOI] [PubMed] [Google Scholar]
- 15. Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol 2010; 1:9-22; PMID:20027184; http://dx.doi.org/ 10.1038/nrm2822 [DOI] [PubMed] [Google Scholar]
- 16. Bayascas JR. PDK1: the major transducer of PI 3-kinase actions. Curr Top Microbiol Immunol 2010; 9-29; PMID:20563709; http://dx.doi.org/ 10.1007/82_2010_43 [DOI] [PubMed] [Google Scholar]
- 17. Bayascas JR. Dissecting the role of the 3-phosphoinositide-dependent protein kinase-1 (PDK1) signalling pathways. Cell Cycle 2008; 19:2978-82; PMID:18802401; http://dx.doi.org/ 10.4161/cc.7.19.6810 [DOI] [PubMed] [Google Scholar]
- 18. Collins BJ, Deak M, Arthur JS, Armit LJ, Alessi DR. In vivo role of the PIF-binding docking site of PDK1 defined by knock-in mutation. EMBO J 2003; 16:4202-11; PMID:12912918; http://dx.doi.org/ 10.1093/emboj/cdg407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bayascas JR, Sakamoto K, Armit L, Arthur JS, Alessi DR. Evaluation of approaches to generation of tissue-specific knock-in mice. J Biol Chem 2006; 39:28772-81; PMID:16887794; http://dx.doi.org/ 10.1074/jbc.M606789200 [DOI] [PubMed] [Google Scholar]
- 20. Bayascas JR, Wullschleger S, Sakamoto K, Garcia-Martinez JM, Clacher C, Komander D, Van Aalten DM, Boini KM, Lang F, Lipina C, et al. Mutation of the PDK1 PH domain inhibits protein kinase B/Akt, leading to small size and insulin resistance. Mol Cell Biol 2008; 10:3258-72; PMID:18347057; http://dx.doi.org/ 10.1128/MCB.02032-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Najafov A, Shpiro N, Alessi DR. Akt is efficiently activated by PIF-pocket- and PtdIns(3,4,5)P3-dependent mechanisms leading to resistance to PDK1 inhibitors. Biochem J 2012; 2:285-95; PMID:23030823; http://dx.doi.org/ 10.1042/BJ20121287 [DOI] [PubMed] [Google Scholar]
- 22. Najafov A, Sommer EM, Axten JM, Deyoung MP, Alessi DR. Characterization of GSK2334470, a novel and highly specific inhibitor of PDK1. Biochem J 2011; 2:357-69; PMID:21087210; http://dx.doi.org/ 10.1042/BJ20101732 [DOI] [PubMed] [Google Scholar]
- 23. Silber J, Antal TL, Gammeltoft S, Rasmussen TE. Phosphoinositide-dependent kinase-1 orthologues from five eukaryotes are activated by the hydrophobic motif in AGC kinases. Biochem Biophys Res Commun 2004; 4:823-27; PMID:15358101; http://dx.doi.org/ 10.1016/j.bbrc.2004.07.031 [DOI] [PubMed] [Google Scholar]
- 24. Park WS, Heo WD, Whalen JH, O'Rourke NA, Bryan HM, Meyer T, Teruel MN. Comprehensive identification of PIP3-regulated PH domains from C. elegans to H. sapiens by model prediction and live imaging. Mol Cell 2008; 3:381-92; PMID:18471983; http://dx.doi.org/ 10.1016/j.molcel.2008.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dittrich AC, Devarenne TP. Perspectives in PDK1 evolution: insights from photosynthetic and non-photosynthetic organisms. Plant Signal Behav 2012; 6:642-49; PMID:22580698; http://dx.doi.org/ 10.4161/psb.20038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Herold M, Cikala M, MacWilliams H, David CN, Bottger A. Cloning and characterisation of PKB and PRK homologs from Hydra and the evolution of the protein kinase family. Dev Genes Evol 2002; 11:513-19; PMID:12459919; http://dx.doi.org/ 10.1007/s00427-002-0267-7 [DOI] [PubMed] [Google Scholar]
- 27. Zurashvili T, Cordon-Barris L, Ruiz-Babot G, Zhou X, Lizcano JM, Gomez N, Gimenez-Llort L, Bayascas JR. Interaction of PDK1 with phosphoinositides is essential for neuronal differentiation but dispensable for neuronal survival. Mol Cell Biol 2013; 5:1027-40; PMID:23275438; http://dx.doi.org/ 10.1128/MCB.01052-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Waugh C, Sinclair L, Finlay D, Bayascas JR, Cantrell D. Phosphoinositide (3,4,5)-triphosphate binding to phosphoinositide-dependent kinase 1 regulates a protein kinase B/Akt signaling threshold that dictates T-cell migration, not proliferation. Mol Cell Biol 2009; 21:5952-62; PMID:19703999; http://dx.doi.org/ 10.1128/MCB.00585-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chresta CM, Davies BR, Hickson I, Harding T, Cosulich S, Critchlow SE, Vincent JP, Ellston R, Jones D, Sini P, et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res 2010; 1:288-98; PMID:20028854; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-1751 [DOI] [PubMed] [Google Scholar]