

Abstract
DNA damage activates the cell cycle checkpoint to regulate cell cycle progression. The checkpoint clamp (Rad9-Hus1-Rad1 complex) is recruited to damage sites, and is required for checkpoint activation. While functions of the checkpoint clamp in checkpoint activation have been well studied, its functions in DNA repair regulation remain elusive. Here we show that Rad9 is required for efficient homologous recombination (HR), and facilitates DNA-end resection. The role of Rad9 in homologous recombination is independent of its function in checkpoint activation, and this function is important for preventing alternative non-homologous end joining (altNHEJ). These findings reveal novel function of the checkpoint clamp in HR.
Keywords: cell cycle checkpoint, DNA repair, homologous recombination
Abbreviations
- DSB
Double-strand break
- HR
homologous recombination
- altNHEJ
alternative non-homologous recombination
- ATM
ataxia-telangiectasia-mutated
- ATR
ataxia telangiectasia and Rad3-related
Introduction
Double-strand breaks (DSBs) are generated by exogenous agents such as ionizing radiation and mutagenic chemicals. In addition, they arise endogenously from oxidative damage and replication fork collapse. Accurate repair of DSBs in chromosomal DNA is integral to the maintenance of genomic integrity in all cells and is essential for early development in vertebrates. The checkpoint regulation by the checkpoint clamp has been well studied. The roles of the checkpoint clamp in DNA repair regulation remain elusive, however.1 It is thought that the checkpoint clamp functions in the ATR-dependent replication checkpoint pathway to activate CHK1. However, the checkpoint clamp mutants are sensitive to ionizing irradiation (IR) although the mutants can activate CHK2, indicating its functions in DNA repair. Our recent studies have provided new insights into ATM (ataxia-telangiectasia-mutated) regulation of repair pathways through phosphorylation of the checkpoint clamp (Rad9-Rad1-Hus1 complex). This discovery was unexpected because it has been believed that the checkpoint clamp is regulated by ATR (ataxia telangiectasia and Rad3-related), not ATM. Moreover, this regulation is independent of its function in checkpoint activation.2 The checkpoint clamp complex is recruited to near DSB sites. Nevertheless, functions of the checkpoint clamp in DSB repair are largely unknown. Biochemical analyses have shown that the checkpoint clamp preferentially binds to 5′ recessed DNA,3 and that single-strand DNA areas on double-strand DNA seem to be required for checkpoint activation.4 The 5′ recessed structures could be generated in many biological processes in response to many types of genotoxic stresses. The checkpoint clamp is recruited to chromatin in response to these stresses, including DNA replication inhibition, ultraviolet light, alkylation, and IR.3 Rad9−/− and Rad9 knockdown cells are sensitive to these genotoxic treatments.2,5 Therefore, Rad9 plays a role in response to DSBs as well as to replication perturbation. Interestingly, however, Rad9−/− cells are not defective in CHK2 phosphorylation that is activated in response to DSBs. Furthermore, phosphorylations at the C-terminal tail are not required for resistance to IR, implying that the “tailless” clamp might play a direct role in DSB repair.
Results and Discussion
To investigate roles of Rad9 in DSB repair, we performed GFP-based repair assays. First, we investigated whether the checkpoint clamp is involved in controlling the HR process. Indeed, knockdown of Rad9 reduced the HR frequency detected by the GFP-based HR assay system6,7 (Fig. 1a). This HR defect was rescued by full-length Rad9 expression (Fig. 1a). Total NHEJ frequency was slightly reduced by Rad9 knockdown (Fig. S1). In contrast, frequency of altNHEJ was increased by Rad9 knockdown, implying that the cells were not able to commit to HR but redirected to a mutagenic altNHEJ pathway (Fig. 1b). Presumably it is due to failure of longer resection process that occurs after short resection by the BRCA1-CtIP complex. It has been shown that CtIP is required for altNHEJ.8 Indeed, CtIP knockdown reduced altNHEJ frequency (Fig. S2). These results imply that the checkpoint clamp functions after the short-resection process by CtIP. These phenotypes are not caused by changes in the cell cycle states in the Rad9-knockdown cells, since they showed similar cell cycle profiles to the wild-type controls (data not shown). Note that the checkpoint clamp is not required for DSB-induced checkpoint particularly in G1 phase (the reporter cells are mainly in G1).9 Therefore, it is unlikely that deregulation of DSB repair is caused by the checkpoint defect. In order to confirm this notion, we performed experiments to see whether tailless Rad9 can rescue the phenotypes. Tailless Rad9 allows distinguishing between the checkpoint defect (the C-terminal tail of Rad9 is required for checkpoint function) and DNA repair defects. Indeed, tailless Rad9 expression rescued HR defect and suppressed elevated altNHEJs in the Rad9 knockdown cells (Fig. 1).
Figure 1.
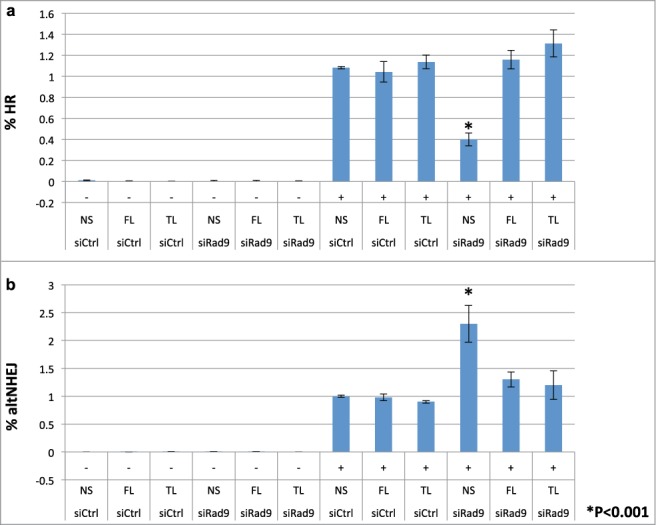
Rad9 is required for efficient HR and suppression of altNHEJ. (A) the HR frequency was assayed using a GFP-based HR assay system. Knockdown of Rad9 inhibited HR, and tailless Rad9 rescued the HR defect caused by Rad9 knockdown. (B) the altNHEJ frequency was assayed using a GFP-based altNHEJ assay system. Knockdown of Rad9 increased altNHEJs, and tailless Rad9 suppressed the increased level of altNHEJ caused by Rad9 knockdown.
RPA32-S4/S8 has been utilized to detect DNA-end resection. As expected, CtIP knockdown inhibited IR-induced RPA32-S4/S8 phosphorylation that is an indication of DSB-end resection defect.10,11 Rad9 knockdown also significantly inhibited RPA32-S4/S8 phosphorylation, implying that the checkpoint clamp is required for the DSB-end resection process (Fig. 2). RPA32-S4/S8 are phosphorylated by DNA-PK.12-14 Therefore it is unlikely that this phenotype is caused by defect in ATR-dependent signaling, but possible. To clarify this, we tested whether tailless Rad9 can rescue this phenotype or not. Rad9-C-terminal tail is required for ATR signaling. Expression of tailless-Rad9 rescued the phenotype, indicating that function of Rad9 in the DNA-end resection process is independent of the ATR-dependent pathway (Fig. 2).
Figure 2.
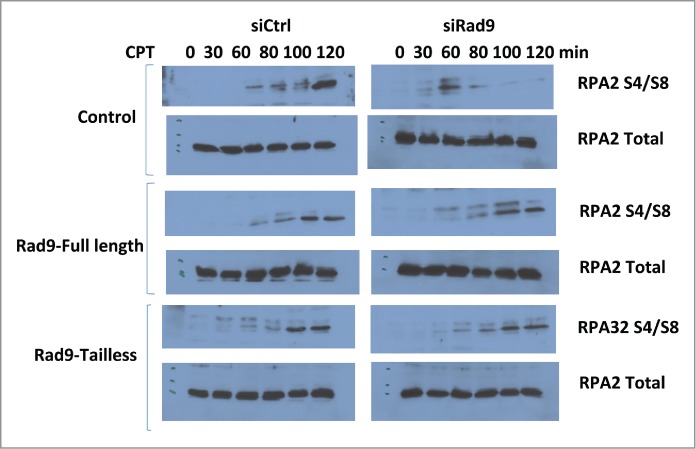
Rad9 knockdown inhibits RPA32 S4/S8 phosphorylation. RPA S4/S8 phosphorylation was monitored after CPT treatment (500 μM) for the indicated times.
An inducible-DSB system was established recently.15 This system utilizes a LacI repressor-operator system integrated at a single chromosomal site in a human osteosarcoma U2OS cell line. LacI is fused with FokI nuclease to induce DSBs on the lac operator sequence and with fluorescent mCherry for detection of DSBs. As reported we have observed perfect co-localization of γH2AX and mCherry signals (data not shown).15 Induction of DSBs is regulated by ER (estrogen receptor) and DD (destabilization domain). Upon addition of 4OHT (4-Hydoxytaminofen) and Shield1, mCherry-LacI-FokI expression is induced and generates DSBs within less than 6 hours, and DSBs can be detected for over 30 hours.15 We detected clear co-localization of Rad9 and mCherry in this system (Fig. 3). Approximately 60% of both Rad9 and the mutant Rad9 foci co-localized with mCherry (data not shown).
Figure 3.
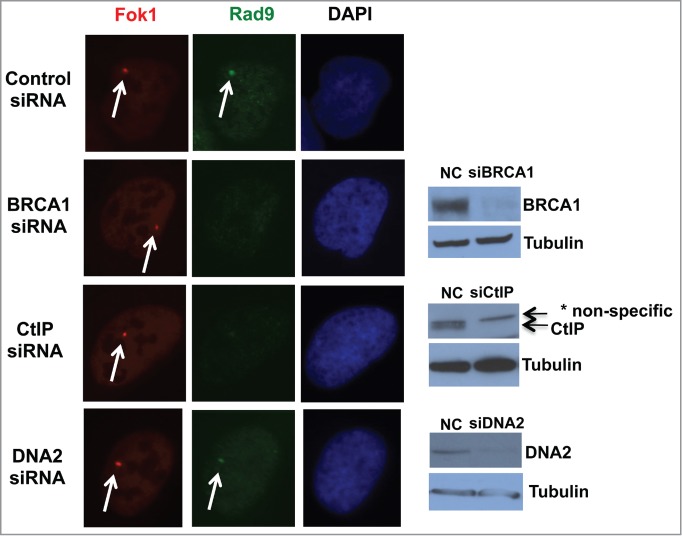
BRCA1 and CtIP are required for recruitment of Rad9 to the DSB site. DSBs were induced upon addition of 4OHT and Shield1 (6 hours), and DSBs and Rad9 were detected by mCherry and anti-Rad9 antibody respectively. * Nonspecific band.
The checkpoint clamp recruitment requires single-stranded DNA area. Therefore, we speculated that recruitment of Rad9 to DSB sites require short resection by BRCA1-CtIP. Indeed, knockdown of BRCA1 and CtIP inhibited Rad9 foci formation at DSB sites, whereas DNA2 knockdown did not (Fig. 3). These data indicate that BRCA1 and CtIP are required for Rad9 localization to DSB site, but DNA2 is not.
Given that Rad9 knockdown cells show HR defect and reduced RPA32-S4/S8 phosphorylation upon DNA damage (Figs. 1 and 2), it is likely that the checkpoint clamp is required for DSB-end resection for proper HR. DNA2 mutant cells exhibit reduced and slow resection.16,17 Indeed, it has been shown in yeast that the checkpoint clamp and clamp loader mutations increase break-induced replication: a phenomenon seen in mutants that are involved in the DSB-end resection process.18 To further confirm this notion, we performed anti-RPA ChIP experiments using the primers on the transgene.19 As expected, CtIP knockdown inhibited RPA loading to the DSB site (Fig. S5). Rad9 knockdown also inhibited RPA loading to the DSB site, indicating that Rad9 is required for DSB-end resection (Fig. 4).
Figure 4.
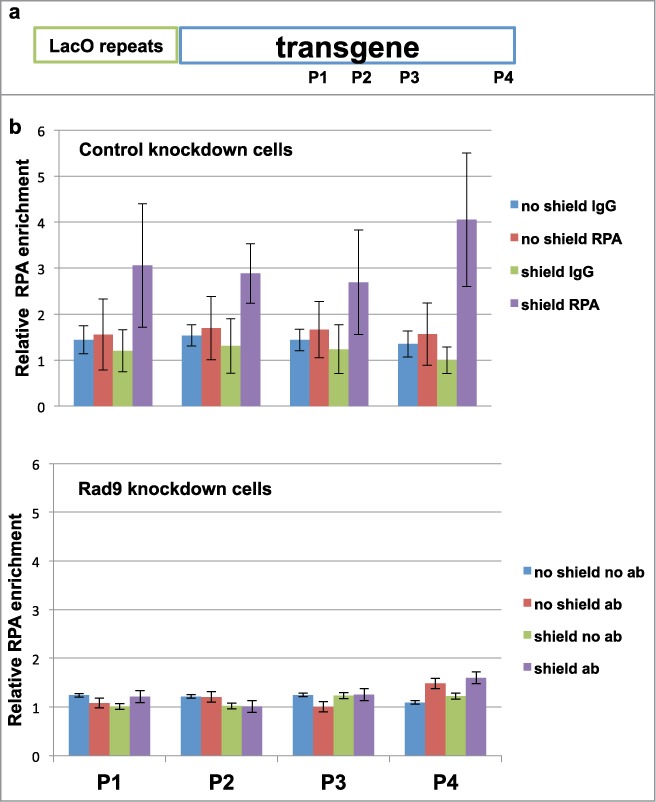
Rad9 is required for DSB-end resection. (A) Schematic of inducible DSB system and locations of primers of qPCR. (B and C) ChIP-qPCR was performed with anti-RPA32 antibody to detect DSB-end resection.
BRCA1 and DNA2 were identified as a damage-enhanced Rad9 interacting protein by mass-spectrometry analysis. We confirmed BRCA1-Rad9 interaction by IP analysis. We did not detect damage-enhanced interaction consistently (Fig. S4A). The damage-induced interaction might be transient or too weak to be detected with the condition of IP. As we found interaction between Rad9 and DNA2 that was enhanced by DNA damage (Fig. S4B).
One possible cause of HR defect of Rad9 knockdown cells is that Rad9 phosphorylation is required for proper DNA2 activity; that is, DNA2 is not fully functional in the Rad9 knockdown cells. The checkpoint clamp stimulates flap-end nuclease activity of FEN1 in vitro.20 Therefore, it is highly possible that the checkpoint clamp affects flap-endonuclease activities of other FEN1-family nucleases DNA2 and EXO1. Based on our results, we propose a model that the checkpoint clamp is recruited to single-stranded area at DSB sites that are created by BRCA1 and CtIP complex. BRCA1 secures the checkpoint clamp at the DSB sites by preventing the clamp to slide away in order for the clamp to support further resection by DNA2. This process allows the cells to commit to HR, preventing altNHEJ that can be initiated after short resection by CtIP (Fig. S6). While this manuscript was under review, similar results were published with yeast checkpoint clamp.21,22
Materials and Methods
Materials, antibodies and cell lines
The Rad9-flag- expressing plasmids were created by inserting Rad9 cDNA into p3xFLAG-CMV-14 (Sigma-Aldrich) and pBMN vectors. Rad9 shRNA (V3LHS_401433, TRCN0000018881) constructs were obtained from OpenBio Systems. The Rad9 cell lines (stably express exogenous wild-type, and endogenous Rad9 expression is stably knocked down) were created first by selecting Rad9-flag-expressing single clones and then infecting a shRad9 construct in the DR-293 cell line. Finally, stable Rad9-knockdown cells were selected. The tailless Rad9 construct was created by replacing the full length Rad9-flag cDNA with the truncated Rad9-flag cDNA (aa1-272) with directional cloning. The siCtIP-6 (Cat. no. GS5932), siBRCA1–9 (Cat. no. GS672), siDNA2-6 (Cat. no. GS1763), siRad9-5 (Cat. No. GS5883) and negative control siRNAs (Cat. no. SI03650318) were obtained from QIAGEN. Twenty-four nM of siRNAs were used for the experiments. Antibodies against Rad9 (BD Biosciences, 1:1000), DNA2 (Proteintech, 1:800), BRCA1 (Cell Signaling, 1:1000), CtIP (Abcam, 1:1000), RPA32 (Santa Cruz, 1:1000), RPA32 S4/S8 (Bethyl, 1:4000) and Flag (Sigma-Aldrich, 1:1000) were used for western blotting.
GFP-based HR assay
Analysis of the HR efficiency was carried out as previously described.7 I-SceI expression plasmids (100 μg) were electroporated (250 V, 950 μF) into DR293 cells (5 × 106 cells). After electroporation, cells were plated in non-selective media for 72 h and were recovered and concentrated by centrifugation and resuspended in Opti-MEM media (Invitrogen) prior to flow cytometry. Cells were analyzed in FACSCalibur (BD Biosciences).
GFP-based NHEJ assay
Analysis of the NHEJ repair efficiency was carried out as previously described.7 Briefly, NHEJ reporter cells were transfected with indicated shRNAs. Transfected cells were selected with puromycin (1 μg/ml) for one week to achieve efficient knockdown. Following the selection, cells were electroporated with I-SceI expression plasmids, recovered and analyzed as above.
GFP-based altNHEJ assay
Analysis of the altNHEJ repair efficiency was carried out as previously described.7 AltNHEJ reporter cells were transfected with indicated siRNAs. On the following day, electroporation of I-SceI expression plasmids was carried out, and cells were recovered and analyzed as above.
Inducible DSB system and ChIP-PCR
DSBs were induced in cells integrated with the inducible DSB reporter construct as previously described.15,19 Briefly, cells were treated with 1 μM Shield-1 (Clontech) and 1 μM 4-hydroxytaminofen (Sigma) for 6 hr. Induction of DSB was verified by checking the mCherry-LacI-Fok1 foci formation with fluorescence microscopy. ChIP assay was carried out as previously described.15,19 Briefly, cells were harvested and washed once in 1XPBS, then cross-linked for 15 minutes at room temperature in 1% formaldehyde with occasional swirling. The crosslink was quenched with glycine, and cells were lysed in cell lysis buffer (10 mM Tris pH8.0, 10 mM NaCl, 0.2% NP-40), followed by centrifugation of the nuclei. The nuclei were lysed in nucleus lysis buffer (50 mM Tris pH8.0, 10 mM EDTA, 1% SDS) and sonicated to ∼500 bp chromatin fragments. The fragmented chromatin were immunoprecipitated with 2 μg of anti-RPA32 antibodies (Santa Cruz) or control rabbit IgG (Abcam) for 2 hours followed by protein G beads (GE Life Science) incubation for 1 hr at 4°C. To reverse crosslink, immunoprecipitates were incubated in 250 μl of elution buffer (100 mM NaHCO3/1%SDS) containing 240 mM NaCl at 65°C overnight, followed by proteinase K treatment at a concentration of 240 μg/ml for 2 hr. DNA was extracted with phenol-chloroform and amplified with qPCR using the ABI 7900HT instrument and the SYBR green chemistry. Initial denaturing was carried out at 95°C for 15 min and the following condition was run for 40 cycles: 95°C for 15 seconds and 60°C for 60 seconds.
The following primers were used:
1F 5′ GGAAGATGTCCCTTGTATCACCAT
1R 5′ TGGTTGTCAACAGAGTAGAAAGTGAA
2F 5′ GCTGGTGTGGCCAATGC
2R 5′ TGGCAGAGGGAAAAAGATCTCA
3F 5′ GGCATTTCAGTCAGTTGCTCAA
3R 5′ TTGGCCGATTCATTAATGCA
4F 5′ CCACCTGACGTCTAAGAAACCAT
4R 5′ GATCCCTCGAGGACGAAAGG
RPA phosphorylation assay
RPA phosphorylation was examined as previously described with the following modifications.10 Cells were transfected with plasmids expressing wild-type Rad9-flag or Rad9 tailless-flag respectively. Then, each transfected cell line was transfected with siCtrl or siRad9 respectively. 72 hours after siRNA transfection, the cells were treated with 500 μM CPT (Sigma) for the indicated times and harvested. Cells were lysed in lysis buffer (50 mM Tris pH8.0, 150 mM NaCl, 10 mM EDTA) containing fresh protease inhibitor cocktail (Sigma) and phosphatase inhibitor cocktails 2 and 3 (Sigma). The samples were resuspended in SDS loading buffer and separated by SDS-PAGE electrophoresis followed by Western blotting.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to Drs. Roger Greenberg and Susan Janicki for the inducible DSB system, and Dr. Jeremy Stark for GFP-based repair assay systems.
Funding
This work was partly supported by an NIH Core Grant (P30CA006973).
Supplemental Materials
Supplemental materials for this article can be found on the publisher's website.
References
- 1. Kai M. Role of the checkpoint in DNA damage response. Biomolecules 2013; 3:75-84; PMID:24970157; http://dx.doi.org/ 10.3390/biom3010075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shin MH, Yuan M, Zhang H, Margolick J B, Kai M. ATM-dependent phosphorylation of the checkpoint clamp regulates repair pathways and maintains genomic stability. Cell Cycle 2012; 11:1796-803; PMID:22453082; http://dx.doi.org/ 10.4161/cc.20161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ellison V, Stillman B. Biochemical characterization of DNA damage checkpoint complexes: clamp loader and clamp complexes with specificity for 5′ recessed DNA. PLoS Biol 2003; 1:E33; PMID:14624239; http://dx.doi.org/ 10.1371/journal.pbio.0000033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lee JH, Paull TT. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 2007; 26:7741-8; PMID:18066086; http://dx.doi.org/ 10.1038/sj.onc.1210872 [DOI] [PubMed] [Google Scholar]
- 5. Roos-Mattjus P, Hopkins KM, Oestreich AJ, Vroman BT, Johnson KL, Naylor S, Lieberman HB, Karnitz LM. Phosphorylation of human Rad9 is required for genotoxin-activated checkpoint signaling. J Biol Chem 2003; 278:24428-37; PMID:12709442; http://dx.doi.org/ 10.1074/jbc.M301544200 [DOI] [PubMed] [Google Scholar]
- 6. Kass EM, Helgadottir HR, Chen CC, Barbera M, Wang R, Westermark UK, Ludwig T, Moynahan ME, Jasin M. Double-strand break repair by homologous recombination in primary mouse somatic cells requires BRCA1 but not the ATM kinase. Proc Nat Acad Sci U S A 2013; 110:5564-9; PMID:23509290; http://dx.doi.org/ 10.1073/pnas.1216824110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gunn A, Stark JM. I-SceI-based assays to examine distinct repair outcomes of mammalian chromosomal double strand breaks. Method Mol Biol 2012; 920:379-91; PMID:22941618; http://dx.doi.org/ 10.1007/978-1-61779-998-3_27 [DOI] [PubMed] [Google Scholar]
- 8. Lee-Theilen M, Matthews AJ, Kelly D, Zheng S, Chaudhuri J. CtIP promotes microhomology-mediated alternative end joining during class-switch recombination. Nat Struct Mol Biol 2011; 18:75-9; PMID:21131982; http://dx.doi.org/ 10.1038/nsmb.1942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lin JJ, Dutta A. ATR pathway is the primary pathway for activating G2M checkpoint induction after re-replication. J Biol Chem 2007; 282:30357-62; PMID:17716975; http://dx.doi.org/ 10.1074/jbc.M705178200 [DOI] [PubMed] [Google Scholar]
- 10. Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP. Human CtIP promotes DNA end resection. Nature 2007; 450:509-14; PMID:17965729; http://dx.doi.org/ 10.1038/nature06337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas-Loba A, Sartori AA, Adams IR, Batista FD, Boulton SJ. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell 2013; 49:858-71; PMID:23333305; http://dx.doi.org/ 10.1016/j.molcel.2013.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shiotani B, Nguyen HD, Håkansson P, Maréchal A, Tse A, Tahara H, Zou L. Two distinct modes of ATR activation orchestrated by Rad17 and Nbs1. Cell Rep 2013; 3:1651-62; PMID:23684611; http://dx.doi.org/ 10.1016/j.celrep.2013.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Anantha RW, Vassin VM, Borowiec JA. Sequential and synergistic modification of human RPA stimulates chromosomal DNA repair. J Biol Chem 2007; 282:35910-23; PMID:17928296; http://dx.doi.org/ 10.1074/jbc.M704645200 [DOI] [PubMed] [Google Scholar]
- 14. Liu S, Opiyo SO, Manthey K, Glanzer JG, Ashley AK, Amerin C, Troksa K, Shrivastav M, Nickoloff JA, Oakley GG. Distinct roles for DNA-PK, ATM and ATR in RPA phosphorylation and checkpoint activation in response to replication stress. Nucleic Acids Res 2012; 40:10780-94; PMID:22977173; http://dx.doi.org/ 10.1093/nar/gks849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tang J, Cho NW, Cui G, Manion EM, Shanbhag NM, Botuyan MV, Mer G, Greenberg RA. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat Struct Mol Biol 2013; 20:317-25; PMID:23377543; http://dx.doi.org/ 10.1038/nsmb.2499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nimonkar AV, Genschel J, Kinoshita E, Polaczek P, Campbell JL, Wyman C, Modrich P, Kowalczykowski SC. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev 2011; 25:350-62; PMID:21325134; http://dx.doi.org/ 10.1101/gad.2003811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhu Z, Chung WH, Shim EY, Lee SE, Ira G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 2008; 134:981-94; PMID:18805091; http://dx.doi.org/ 10.1016/j.cell.2008.08.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Costelloe T, Louge R, Tomimatsu N, Mukherjee B, Martini E, Khadaroo B, Dubois K, Wiegant WW, Thierry A, Burma S, et al. The yeast Fun30 and human SMARCAD1 chromatin remodellers promote DNA end resection. Nature 2012; 489:581-4; PMID:22960744; http://dx.doi.org/ 10.1038/nature11353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shanbhag NM, Rafalska-Metcalf IU, Balane-Bolivar C, Janicki SM, Greenberg RA. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 2010; 141:970-81; PMID:20550933; http://dx.doi.org/ 10.1016/j.cell.2010.04.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang W, Brandt P, Rossi ML, Lindsey-Boltz L, Podust V, Fanning E, Sancar A, Bambara RA. The human Rad9-Rad1-Hus1 checkpoint complex stimulates flap endonuclease 1. Proc Nat Acad Sci U S A 2004; 101:16762-7; PMID:15556996; http://dx.doi.org/ 10.1073/pnas.0407686101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Blaikley EJ, Tinline-Purvis H, Kasparek TR, Marguerat S, Sarkar S, Hulme L, Hussey S, Wee BY, Deegan RS, Walker CA, et al. The DNA damage checkpoint pathway promotes extensive resection and nucleotide synthesis to facilitate homologous recombination repair and genome stability in fission yeast. Nucleic Acids Res 2014; 42:5644-56; http://dx.doi.org/ 10.1093/nar/gku190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ngo GH, Balakrishnan L, Dubarry M, Campbell JL, Lydall D. The 9-1-1 checkpoint clamp stimulates DNA resection by Dna2-Sgs1 and Exo1. Nucleic Acids Res 2014; 42(16):10516-28; PMID:25122752; http://dx.doi.org/10.1093nargku 746 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.