

Abstract
Kinases and phosphatases, two sides of the same coin; are they opposing forces that switch signals on and off or enzymes that work together to give the right type of response at the right time? It depends on how close you stand when you view the big picture. Up close and detailed, and you’ll see individual phosphorylation sites as binary switches - lights being toggled on/off by antagonistic forces. Take a step back and multiple copies of the same light are being toggled, perhaps leading to a range of intensities, or a flickering pattern, lights flashing in unison or at random. It depends what the signal requires. Stand even further back, let the story unfold, and you’ll see a dazzling multicolour array of different lights. A coordinated sequence of color that appears to burst into life at different times in different places, with a pace that is both frantic and serene. This is a vision of mitosis and what a true spectacle it is.
Kinases and phosphatases are the orchestrators of this show and from the vantage point at the back, it's impossible to separate them. They are inextricably linked and to understand the bigger picture we must view them together and learn how they work in unison. Not straight away of course, if you enter this show from the back you’re likely to be dazzled by the beauty before you ever get to the substance. The best way is to start up close and spend time understanding the individual components and how they work, safe in the knowledge that when you have this information you can take a step back and begin to appreciate the bigger picture. In this article we review our recent attempts to understand the details of spindle assembly checkpoint (SAC) signaling, which highlights how 2 mitotic kinases work in tandem with 2 phosphatases to ensure the SAC signal has the right type of rapidly switchable response.1
The SAC is an ancient mechanism, conserved from yeast to man, that protects against genomic instability by safeguarding against errors during mitosis. It works by delaying division until each and every chromosome has made effective attachments to microtubules.2 These attachments are bridged by the kinetochore, which also emits the inhibitory signal needed to delay division. Upon microtubule attachment this SAC signal is promptly extinguished and therefore a key feature of kinetochores is their ability to rapidly respond to changes in microtubule occupancy by switching localized SAC signaling either on or off.2,3
To understand how this switching is controlled in human cells, we performed an siRNA screen to identify phosphatases that regulate SAC silencing. This lead to the discovery that 2 kinetochore phosphatases were important; PP1 and PP2A-B56.1 PP1 is known to control SAC silencing in other species4 and so we were initially intrigued by the role of PP2A-B56, which is physically coupled to the SAC signal by virtue of its interaction with BUBR15,6 (BUBR1 is recruited to kinetochores by the SAC kinase MPS1). At first glance this appeared counterintuitive: why would the SAC activating signal also recruit a phosphatase that tries to extinguish it? The answer to this question appeared relatively obvious in hindsight for a signal that needs to switch off rapidly - it ensures that the phosphatase is primed and ready to switch the SAC off as soon as required (i.e. immediately following microtubule attachment). This was essentially the same conclusion as that reached by the Gruneberg group using similar approaches.7
On its own though, it was still difficult to conceptualise how the SAC signal would ever manage to establish efficiently if it recruited its own antagonising phosphatase from the outset. We believe the explanation is that PP2A-B56 doesn't antagonise the SAC directly, it antagonises Aurora B to allow PP1 to come in and shut down the SAC1 (Fig. 1). It is likely that this 2-step silencing mechanism has evolved so that Aurora B can work in concert with Mps1; MPS1 initiates the SAC signal and Aurora B restricts silencing of that signal. It also explains how the SAC can tolerate the presence of PP2A-B56 at unattached kinetochores, because Aurora B activity is high enough to brake the PP2A-B56/PP1 silencing axis, and why the SAC can silence so quickly upon microtubule attachment/tension, when Aurora B activity is lowered to release the brakes at a time when the activating signal from MPS1 is also lost. Finally, this intricate kinase-phosphatase coupling essentially allows Aurora B and MPS1 to rapidly initiate their respective signals unopposed; Aurora B can remove PP1 in the absence of PP2A-B56, freeing MPS1 to initiate the SAC without PP1. This is perhaps even easier at mitotic entry because BUBR1/PP2A-B56 kinetochore recruitment is restricted until after nuclear envelope breakdown, when both kinases have already maximally phosphorylated their targets on KNL1.1
Figure 1.
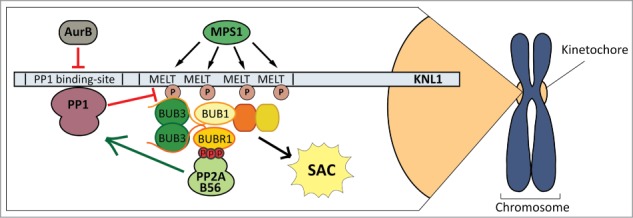
How kinases and phosphatases cooperate to shape a responsive SAC signal. PP2A-B56 is primed to silence the SAC by virture of its interaction with BUBR1. This allows PP2A-B56 to antagonise Aurora B and induce PP1 recruitment, which subsequently promotes MELT dephosphorylation and SAC silencing. Aurora B thus provides the brakes on SAC silencing until the appropriate time.
In conclusion, the SAC has evolved an intricate control network that allows it to start up and switch off incredibly rapidly, both key facets of localized SAC signaling. We hypothesize that this type of kinase-phosphatase coupling is likely to be used in other signaling systems that also rely on rapid signal switching. The key will be to determine the particular kinase and phosphatase inputs first and then take a step back to learn how the coupling of these inputs allows the right type of response.
References
- 1. Nijenhuis, et al. . Nat Cell Biol 2014; 16:1257-1264; PMID:25402682; http://dx.doi.org/ 10.1038/ncb3065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sacristan, et al. . Trends Cell Biol 2014; PMID:25220181; http://dx.doi.org/ 10.1016/j.tcb.2014.08.006 [DOI] [PubMed] [Google Scholar]
- 3. Funabiki, et al. . Chromosoma 2013; 122:135-158; PMID:23512483; http://dx.doi.org/ 10.1007/s00412-013-0401-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lesage, et al. . Curr Biol 2011; 21:R898-903; PMID:22075433; http://dx.doi.org/ 10.1016/j.cub.2011.08.063 [DOI] [PubMed] [Google Scholar]
- 5. Kruse, et al. . J Cell Sci 2013; 126:1086-1092; PMID:23345399; http://dx.doi.org/ 10.1242/jcs.122481 [DOI] [PubMed] [Google Scholar]
- 6. Suijkerbuijk, et al. . Dev Cell 2012; 23:745-755; PMID:23079597; http://dx.doi.org/ 10.1016/j.devcel.2012.09.005 [DOI] [PubMed] [Google Scholar]
- 7. Espert, et al. . J Cell Biol 2014; 206:833-842; PMID:25246613; http://dx.doi.org/ 10.1083/jcb.201406109 [DOI] [PMC free article] [PubMed] [Google Scholar]