

Abstract
KDM4D is a lysine demethylase that removes tri- and di- methylated residues from H3K9 and is involved in transcriptional regulation and carcinogenesis. We recently showed that KDM4D is recruited to DNA damage sites in a PARP1-dependent manner and facilitates double-strand break repair in human cells. Moreover, we demonstrated that KDM4D is an RNA binding protein and mapped its RNA-binding motifs. Interestingly, KDM4D-RNA interaction is essential for its localization on chromatin and subsequently for efficient demethylation of its histone substrate H3K9me3. Here, we provide new data that shed mechanistic insights into KDM4D accumulation at DNA damage sites. We show for the first time that KDM4D binds poly(ADP-ribose) (PAR) in vitro via its C-terminal region. In addition, we demonstrate that KDM4D-RNA interaction is required for KDM4D accumulation at DNA breakage sites. Finally, we discuss the recruitment mode and the biological functions of additional lysine demethylases including KDM4B, KDM5B, JMJD1C, and LSD1 in DNA damage response.
Keywords: chromosomal instability, and cancer, DNA damage response (DDR), double strand break (DSB), KDM4D, lysine demethylases (KDM), PARP1, poly(ADP-ribose)ylation (PARylation)
Introduction
Our genome is highly susceptible to the action of endogenous and exogenous DNA damaging agents.1,2 Defective DNA damage response (DDR) could lead therefore to accumulation of mutations and genetic instability promoting tumorigenesis.3-5Double-strand breaks (DSBs) are considered the most cytotoxic form of DNA damage, as a single unrepaired DSB can trigger cell death.6-8 Vertebrate cells use at least 2 distinct pathways for DSB repair.9,10 The first is non-homologous end-joining (NHEJ), an error-prone process that functions throughout the cell cycle.11-13 The second is homology-directed repair (HDR); an error-free process that functions only in late S phase and G2, when an intact chromatid is available and serves as a template for repairing the broken DNA.14,15 In the course of DDR, DSBs are translated into a molecular signal, which is substantially amplified, allowing the recruitment, retention and activation of downstream DDR proteins at DNA lesions3,16-19 One common feature of the DDR proteins is their recruitment to DNA damage sites and the formation of microscopically visible foci.17 Beside DDR proteins, emerging evidence implicate non-coding RNAs (ncRNAs) in DDR.20 For example, a potential template role for RNA in DNA repair events has been recently described.21-23 ncRNAs can also regulate the expression of various DDR genes such as ATM, BRCA1, H2AX, RAD51 and p53.24-28 In addition, it has been shown that DSBs trigger the expression of ncRNAs (called diRNAs) from sequences surrounding the damage sites. These diRNAs regulate the recruitment of DDR proteins and promote DSB repair.20,29-32 However, the mode of action of most diRNAs remains to be discovered. Interestingly, some diRNAs are processed by Dicer and Drosha or by Dicer-like proteins into smaller RNAs.31,32 On the other hand, CU1276, a tRNA derived 22nt RNA, that modulates DDR 33 can be generated in a Drosha- and Dicer-independent manner, suggesting that additional RNA-processing enzymes are implicated in processing diRNAs.34-36
One main characteristic of DNA damage repair is the rapid sensing and initiation of the DDR, which is mediated by posttranslational modifications (PTMs) of histones and non-histone proteins. Several PTMs are involved in the DDR, including phosphorylation, ubiquitylation, SUMOylation, acetylation, ADP-ribosylation and methylation (reviewed in16,37-40). Accumulating evidence suggest that lysine methylation is a highly dynamic modification owing to the interplay between lysine methyltransferases (KMTs) and lysine demethylases (KDMs).41,42 KDMs consist of 2 protein families: the first is LSD1/KDM1A, which contains a flavin adenine dinucleotide (FAD)-dependent amine oxidase domain that demethylates H3K4me2 and H3K4me1.43 The second KDM family includes the Jumonji C (JmjC)-domain containing proteins. The JmjC catalytic domain forms an enzymatically active pocket that coordinates the 2 co-factors, ferrous oxide (Fe(II)) and α-ketoglutarate, that are needed for the radical-based oxidative demethylation reaction (reviewed in44-47).
KDM proteins are involved in a plethora of cellular processes including gene expression regulation,48-53 DNA replication,41,54 DNA damage response,55-58 worm development and germ cell apoptosis,59 cell differentiation and renewal of embryonic stem cells.60 Interestingly, several KDM proteins show oncogenic activity and are overexpressed in various types of human cancer (reviewed in 41,61).
Recently, we showed, for the first time, that the KDM4D lysine demethylase is an RNA binding protein and mapped KDM4D residues that mediate its interaction with RNA. Additionally, we generated KDM4D mutant that lost its ability to bind RNA and demonstrated that KDM4D-RNA interactions are critical for KDM4D association with chromatin and subsequently for H3K9me3 demethylation in vivo.62 Additionally, we have previously described a novel function of KDM4D in DDR. We found that KDM4D lysine demethylase is transiently recruited to DNA damage sites in a PARP1-dependent manner. Further, we showed that the DNA damage-induced ADP-ribosylation (PARylation) of KDM4D C-terminal region mediates its recruitment to DNA damage sites. Finally, we showed that KDM4D demethylase activity promotes double-strand break repair by facilitating the ATM-dependent phosphorylation of the DNA damage markers through the regulation of ATM chromatin localization.58
Here, we further dissect the recruitment mode of KDM4D to DNA damage sites by addressing whether KDM4D binds poly(ADP-ribose) (PAR); and whether KDM4D-RNA interaction is required for KDM4D accumulation at DNA damage sites. Moreover, we discuss the recruitment mode and the emerging roles of other lysine demethylases in DDR.
Results
KDM4D binds poly(ADP-ribose) (PAR) in vitro via its C-terminal region
During DDR, PARP1 is recruited to sites of DNA damage and mediates the local PARylation of DDR proteins and histones. This promotes the rapid recruitment of PAR-binding proteins to DNA damage sites, which is important for efficient damage repair.63 We hypothesize therefore that KDM4D binds PAR moieties. To check directly the PAR-binding capacity of KDM4D, purified 6xHis-tag fused to a full-length KDM4D was blotted on a membrane and incubated with radiolabelled PAR. Results show that KDM4D protein binds PAR moieties. Histone H3 and 6xHis-Rpn8 proteins were used as positive and negative controls, respectively (Fig. 1A). To identify KDM4D region that binds PAR, we performed deletion-mapping analysis and the observed deletion mutants were tested for their ability to bind PAR. Results show that the PAR-binding domain is located in the C-terminal region (Fig. 1B, C) spanning amino acids 350–474 of KDM4D (Fig. 1D). Interestingly, KDM4D PAR-binding domain includes 4 residues (E357, R450, R451 and R455) that were substituted to alanine to generate KDM4D mutant (KDM4D-4M) that can neither undergo PARylation nor accumulate at laser-microirradiated sites.58 This observation prompted us to address whether KDM4D-4M mutant can still bind PAR moieties in vitro. Results show that KDM4D-4M binds PAR, suggesting that the region between 350–474 amino acids has 2 different motifs: the first binds PAR and the second contains PARylated residues (Fig. 1E).
Figure 1.
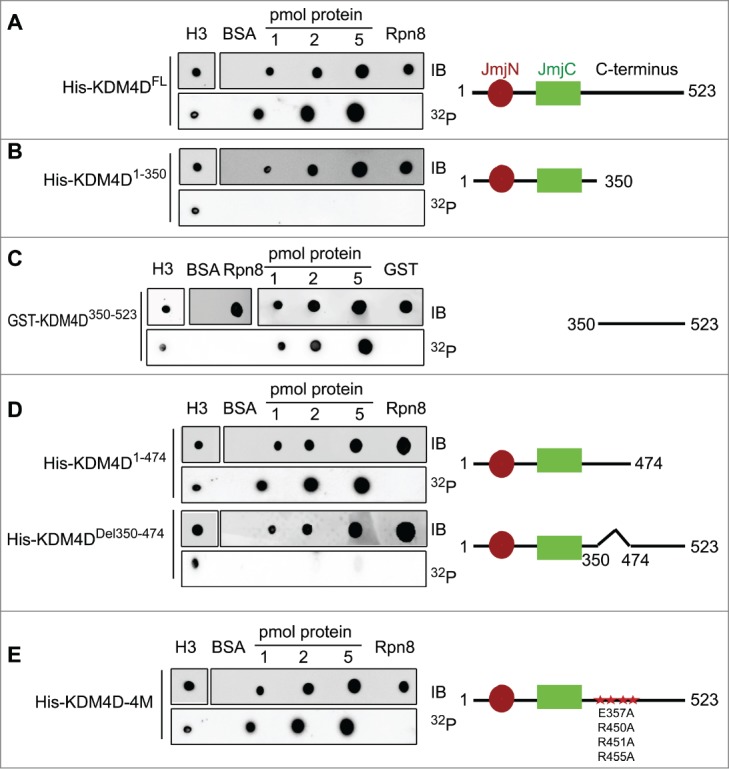
KDM4D region spanning 350–474 amino acids binds PAR in vitro. PAR-binding assay with 6xHis tagged KDM4D full-length (FL) protein (523aa) (A), deletion mutants: N-terminal (1–350aa) (B), GST-tagged C-terminal (350–523aa) (C), truncated C-terminal (1–474aa) and internal deletion 350–474aa (D), and 6xHis tagged KDM4D-4M mutant (contains 4 mutations: E357A, R450A, R451A and R455A) (E). 6xHis-Rpn8, GST-only and BSA are used as negative controls and H3 as a positive control. Right: schematic representation of KDM4D mutants. IB: Immunoblot. 32P: radiolabelled PAR.
KDM4D PAR-binding domain is essential for KDM4D accumulation at DNA damage sites
We sought to characterize the role of KDM4D PAR-binding domain in regulating KDM4D recruitment to DNA damage sites. Toward this end, U2OS cells expressing EGFP-KDM4D1-474aa fusion, which contains the PAR-binding domain, were subjected to laser-microirradiation. Results show a minor and transient increase in the fluorescence intensity of EGFP-KDM4D1-474aa fusion at laser-microirradiated sites, compared to wild type KDM4D (Fig. 2). This result suggests that KDM4D N-terminal region containing the PAR-binding domain is able to recruit KDM4D to DNA damage sites, however the C-terminal region spanning amino acids 475–523 is needed to facilitate KDM4D recruitment to DNA damage sites. To address whether the PAR-binding domain is essential for KDM4D recruitment, we tested the recruitment of EGFP-KDM4DΔ350-474aa fusion, which lacks its PAR-binding domain, at laser-microirradiated sites. As shown in Figure 2, EGFP-KDM4DΔ350–474aa completely lost its ability to accumulate at DNA damage sites. Altogether, we concluded that PAR-binding domain of KDM4D is essential but not sufficient for intact recruitment of KDM4D to DNA damage sites.
Figure 2.
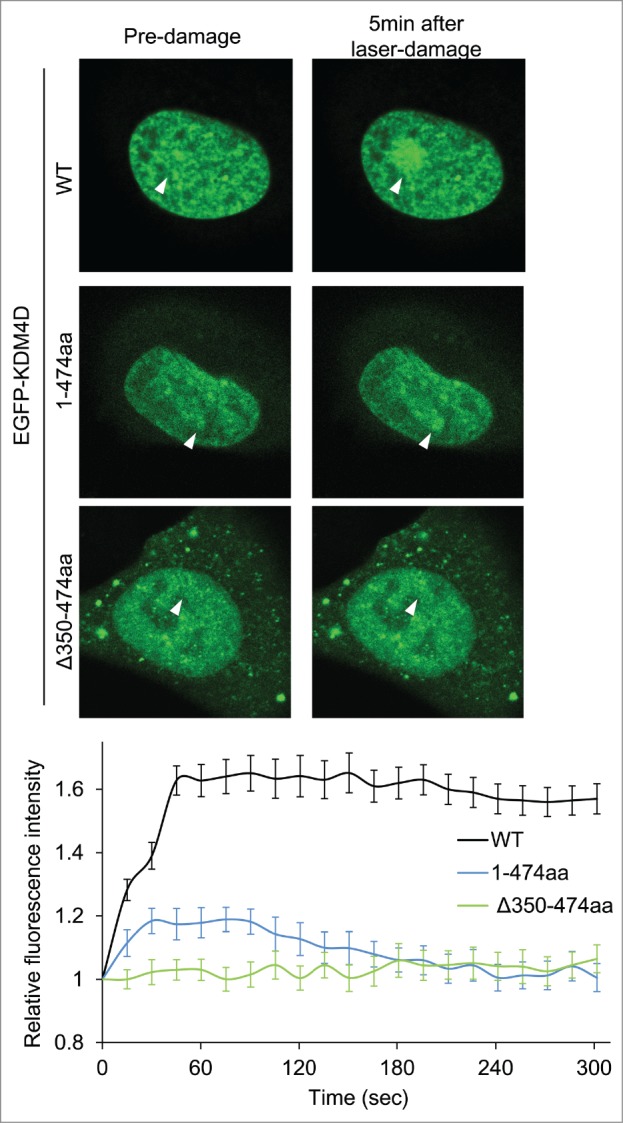
KDM4D PAR-binding region is essential for its recruitment to laser-microirradiated sites. Representative cells showing the localization of EGFP-KDM4D-WT EGFP-KDM4D1-474aa and EGFP-KDM4DΔ350-474aa fusions before and 5 minutes after the induction of laser-microirradiation to a single region, marked with a white arrow. Each cell is representative of at least 20 different cells. The graph shows the increase in the relative fluorescence intensity of KDM4D fusions at laser-microirradiated sites. Error bars represent SD of 10 different cells.
KDM4D-RNA interactions are essential for KDM4D recruitment to DNA damage sites
Given that KDM4D is recruited to DNA damage sites,57,58 and ncRNAs promote the recruitment of DDR proteins to DNA damage sites,30,31 we sought to address whether KDM4D-RNA interactions affect KDM4D recruitment to laser-microirradiated sites. Toward this end, we took advantage of KDM4D-1H4R-HRK mutant that lost its ability to bind RNA molecules and shows defective chromatin localization.62 Laser microirradiation assay, performed on U2OS cells expressing EGFP-KDM4D-1H4R-HRK mutant, shows no detectable accumulation of the mutant at DNA breakage sites (Fig. 3). This observation further confirms the defective association of KDM4D-1H4R-HRK mutant with chromatin and implicates KDM4D-RNA interactions in regulating KDM4D accumulation at DNA damage sites. It should be noted that KDM4D-1H4R-HRK mutant has an intact C-terminal region, which is essential and sufficient for KDM4D recruitment to DNA damage sites.58 On the other hand, KDM4D-1H4R-HRK shows no accumulation at DNA damage sites (Fig. 3). One possible explanation for these apparently contradictory results is that the defective accumulation of KDM4D-1H4R-HRK at DNA damage sites results from the fact that KDM4D-1H4R-HRK is found in the nuclear soluble fraction but not in the chromatin-bound fraction.62 In other words, 1H4R-HRK mutations exhibit dominant negative effect and suppress the ability of the C-terminal region to recruit KDM4D to damage sites.
Figure 3.
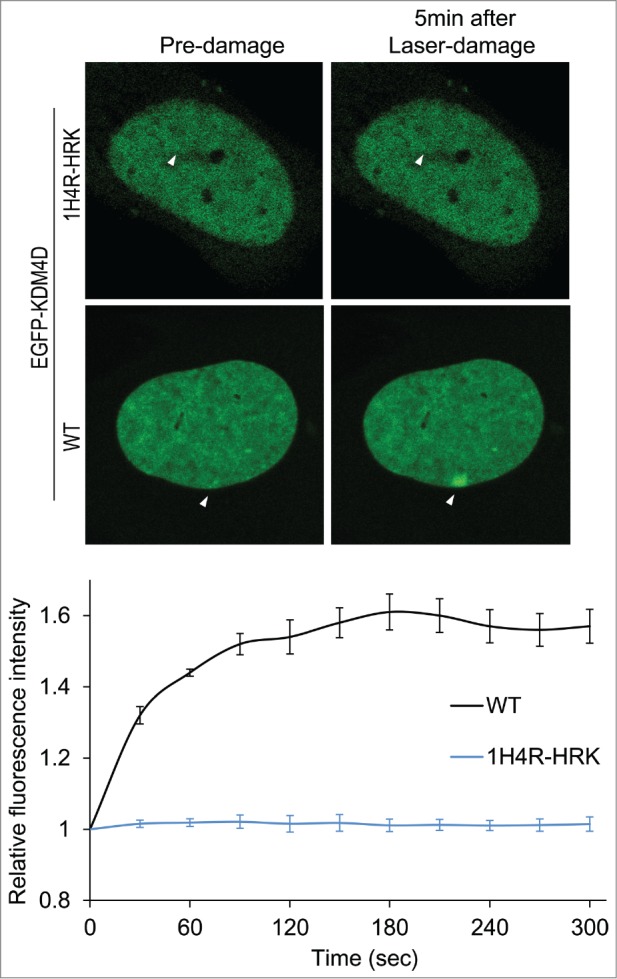
KDM4D-RNA interactions are essential for KDM4D accumulation at DNA damage sites. Representative cells showing the localization of EGFP-KDM4D-WT and EGFP-KDM4D-1H4R-HRK fusions before and 5 minutes after the induction of laser-microirradiation to a single region, marked by a white arrow. Each cell is representative of at least 20 cells. The graph shows the increase in the relative fluorescence intensity of EGFP-KDM4D-WT and EGFP-KDM4D-1H4K-HRK at laser-microirradiated sites. Error bars represent SD of 10 different cells.
Discussion
Here, we further characterized the recruitment mode of KDM4D to DNA damage sites. We showed that KDM4D binds PAR moieties and mapped the KDM4D PAR-binding domain. Also, we demonstrated that KDM4D-RNA interactions are essential for KDM4D recruitment to laser-microirradiated sites.
The fact that KDM4D-4M mutant (cannot undergo PARylation) can still bind PAR (Fig. 1) suggests that KDM4D has 2 different motifs; the first binds PAR and the second includes residues that undergo PARylation. Similar to KDM4D, various DDR proteins, such as DNA-PK and XRCC1, were shown to undergo PARylation and also bind PAR moieties.63-68 The significance of having a distinct PAR binding domain and PARylation domain in KDM4D could be to facilitate the recruitment of DDR proteins to DNA damage sites. Indeed, PAR moieties provide binding sites for recruiting DDR proteins containing PAR-binding domain. In addition, part of these DDR proteins undergoes PARylation to promote the recruitment of additional DNA damage-responsive proteins in a PAR-binding dependent manner. In support of this, the accumulation of several DDR proteins at DNA breakage sites depends either on their ability to undergo damage-induced PARylation and/or binding PAR moieties.65,69-75 For example, mutating the PAR-binding motif of ALC1 and APLF disrupts their ability to accumulate at DNA damage sites.69,71 These findings are in line with our data showing that internal deletion of amino acids 350–474 (KDM4DΔ350-474aa) abolishes KDM4D binding to PAR and abrogates KDM4D accumulation at DNA damage sites (Fig. 2).
The role of KDMs in DDR is extensively studied as evident by the increasing number of reports describing new functions of KDM in DDR.
PARP-dependent recruitment of KDMs to DNA damage sites
A recent study showed that in addition to KDM4D, KDM4B (another KDM4 family member), but not KDM4A and KDM4C, is recruited to laser-microirradiated sites in a PARP1-dependent manner. Consistent with our findings, the recruitment of KDM4B is independent of ATM, ATR, DNA-PK and γH2AX.57 Unlike KDM4D, KDM4B recruitment is dependent on its catalytic activity. This difference may be attributed to the difference in the structure and the substrates specificity of KDM4B and KDM4D. While both proteins contain JmjC and JmjN domains, only KDM4B contains 2 PHD and 2 Tudor domains. Additionally, both KDM4D and KDM4B demethylate H3K9me2/me3; but KDM4B can also demethylate H3K36me2/me3, a modification that has been recently implicated in DSB repair.76 A third JmjC-domain containing protein, KDM5B, that removes di and tri-methylations of lysine 4 of histone H3 (H3K4me2/3)77-79 was shown to accumulate at I-SceI-induced DSBs in a PARP1- and macroH2A1.1-dependent manner. Further, it was also shown that KDM5B-PARP1 interaction is enhanced upon DNA damage.80
RNF8 and RNF168-dependent recruitment of KDMs to DNA damage sites
In addition to the PARP-dependent recruitment of KDMs, Bartek and colleagues showed that JMJD1C lysine demethylase is also recruited to DNA damage sites and this recruitment depends on its physical interaction with RNF8 and RNF168 ubiquitin ligases. Depletion of both RNF8 and RNF168 impaired JMJD1C recruitment to DNA damage sites. Similarly, JMJD1C mutant that lost its ability to interact with RNF8 and RNF168 failed to accumulate at DNA damage sites. Unlike KDM4D, the catalytic activity of JMJD1C is also required for its recruitment to DNA breakage sites.81 Future studies will be required to address whether PARP1 activity is involved in regulating JMJD1C recruitment and whether RNF8 and RNF168 regulate KDM4B, KDM4D and KDM5B recruitment to DNA damage sites.
In addition to JMJD1C, Yang Shi and colleagues reported that KDM1A/LSD1 demethylase, which removes H3K4me2/me1 marks, is recruited to both UV-microirradiated sites and to DSBs generated by IPpoI endonuclease. Similar to JMJD1C, LSD1 recruitment is mediated by physical interaction with RNF168 via LSD1 N-terminal domain. Moreover, LSD1 recruitment is independent of 53BP1, ATM, ATR and PARP proteins.82
Role of KDMs in double-strand break repair
We have demonstrated that KDM4D demethylase promotes double-strand break repair by facilitating the ATM-dependent phosphorylation of DNA damage markers through regulating ATM chromatin localization.58 Human KDM4B was also shown to promote DSB repair, as cells overexpressing KDM4B are associated with decreased numbers of γH2AX foci following γ-irradiation, as well as increased cell survival.57 Previous work has also implicated the drosophila KDM4B in both UV- and γ irradiation-induced DNA damage.56 They showed that upon exposing drosophila salivary gland cells to UV irradiation, KDM4B protein is upregulated in a p53-dependent manner and this was accompanied by a decrease in H3K9me3 levels, which occurs preferentially in heterochromatin. Importantly, drosophila flies heterozygous for KDM4B mutant are more sensitive to UV irradiation and are deficient in the removal of Cyclobutane-Pyrimidine-Dimers (CPDs) from damage sites. Additionally, depletion of the C. elegans KDM4 homolog, JMJD-2, leads to a significant increase in CEP-1/p53-dependent germ cell apoptosis and altered progression of meiotic DSB repair, as evident by RAD51 foci persistence in mitotic cells.59
Similar to KDM4D, KDM5B is also required for proper repair of the I-SceI-induced DSBs by both HDR and NHEJ. Accordingly, KDM5B depletion impairs the accumulation of the NHEJ and HDR mediator proteins, ku70 and BRCA1, respectively, at DNA damage sites.80 On the other hand, JMJD1C is primarily required for DSB repair by HDR as its depletion reduced the levels of RNF8 and polyubiquitination at DSBs and impaired the recruitment of RAP80–BRCA1, but not 53BP1.81 Finally, LSD1 is also implicated in DSB repair as its depletion sensitizes cells to γ-irradiation. Accordingly, LSD1 demethylase activity facilitates 53BP1 foci formation at DNA damage sites mainly in late S/G2 of the cell cycle. Furthermore, LSD1 activity promotes the damage-induced H2A and H2AX ubiquitylation and consequently enhances the recruitment of BRCA1 and RAP80 to DNA damage sites.
DNA damage-induced substrates of KDMs
We have shown that KDM4D, which demethylates H3K9me2/me3, is rapidly recruited to DNA damage sites. However, we were unable to visualize reproducible changes in the levels of H3K9me3 at laser microirradiated sites using immunofluorescence-based techniques. Moreover, we rigorously measured the levels of H3K9me2/me3 methylation at 5 minutes intervals after DNA damage using western blot and no detectable changes in H3K9me2/me3 were observed.58,83 These observations may suggest the following scenarios: (i) H3K9 demethylation is restricted to few nucleosomes surrounding the damaged sites. Alternatively, the methylation/demethylation of H3K9 is highly dynamic at sites of DNA damage. In both cases, new sensitive approaches should be established in order to track these delicate changes in methylation of H3K9 at sites of damage. (ii) The lack of changes in the levels of H3K9me2/3 marks despite the recruitment of KDM4D may result from the binding of Tip6084 to H3K9me3 and thus protecting it from demethylation via KDM4D. (iii) KDM4D might be required for demethylating lysine residues other than H3K9. In agreement with this, it was recently shown that KDM4D demethylates H1.4K26me2/3 85 and H3K56me3, which is enriched at heterochromatic regions.86 Future work will be required to determine H1.4K26me2/3 and H3K56me3 levels at sites of DNA damage. (iv) KDM4D might be essential for demethylating DNA-damage-responsive proteins that accumulate at sites of DNA damage. In support of this, it was found that JMJD1C demethylates MDC1-K45 in response to DNA damage. This demethylation enhances RNF8-MDC1 interaction and subsequently facilitates RNF8, BRCA1-RAP80 recruitment to DNA damage sites.81 (v) The demethylation of H3K9me2/me3 after DNA damage might favorably occur at heterochromatic regions. In support of this, a decrease in H3K9me3 levels, which occurs preferentially at heterochromatic chromocenter regions, was documented in drosophila.56 Moreover, decrease in the levels of H3K9me2/3 at γH2AX-positive regions was reported at 20 minutes after IR in mammalian cells.87 Similarly, Young and colleagues observed local reduction in H3K9me3 levels at sites of damage in cells expressing low to moderate levels of EGFP-KDM4B fusion.57 In contrast, a recent study reported local increase in H3K9me2/3 at DSB sites,88 suggesting that H3K9 methylation levels at DNA damage sites are highly dynamics and might be also influenced by the chromatin context at the DNA breakage sites.83,89-93 In light of these observations, future in depth analysis will be required to track the fluctuations in the levels of H3K9 methylation at several time points after DSB induction within different chromatin structures.
Two recent works reported decrease in H3K4me2/me3 at DNA damage sites. The first showed that KDM5B recruitment to DNA damage sites is accompanied by a local decrease in H3K4me3 at I-SceI-Induced DSBs.80 The second showed that the recruitment of LSD1 to UV damage sites and to DSB induced by IPpoI endonuclease causes demethylation of H3K4me2 mainly at late S/G2 of the cell cycle.82 Moreover, it was previously shown that LSD1 demethylates K370me2 of the tumor suppressor gene product, P53.94 P53 is methylated on 3 different lysine residues, K370, K372 and K382 that regulate its activity, stability and sub-cellular localization in response to different stimuli such as apoptosis and DNA damage.94-96 These observations suggest that, similar to JMJD1C, LSD1 can exert its function in DDR by demethylating also non-histone DDR proteins.
Role of KDMs in maintaining genomic stability
Interestingly, various types of human cancer show misregulated expression of KDM4A-D members suggesting that dysregulated expression of KDMs is associated with genomic instability and carcinogenesis (for recent reviews see97,98). The mechanism by which KDM4 dysregulation promotes genomic instability could be related to their emerging functions DNA lesions repair. In addition, we and other groups have revealed DNA damage-independent pathways by which KDM misregulation can lead to genomic instabilities. For example, we demonstrated that either depletion or overexpression of KDM4C leads to a significant increase in the frequency of abnormal mitotic cells showing either misaligned chromosomes at metaphase, anaphase-telophase lagging chromosomes or anaphase-telophase bridges. These results highlight a causative role of KDM4C lysine demethylase in regulating the fidelity of mitotic chromosome segregation.99 Furthermore, it was shown that KDM2A depletion promotes genomic instability as evident by the destabilization of centromeric chromatin during mitosis100 and the increase in the percentage of cells showing micronuclei or chromosome bridges.101 Finally, an important study showed that overexpression of a catalytically active KDM4A protein induces copy number gains of specific genomic regions, which are known to contain oncogenes. This KDM4A-dependent copy gain can be induced in less than 24 hr and requires cells progression through S phase. In addition, tumors with amplified KDM4A show increased copy gains for the same regions. These data altogether suggest that KDM4A catalytic activity provides a potential enzymatic link for generating copy number alterations through replication abnormalities of regions amplified in human tumors.97
Given the tight correlation between lysine methylation and regulation of gene expression, forthcoming research should address whether KDM proteins exert their functions in preserving genomic integrity by determining the transcription state of the damaged chromatin before and after DNA repair. In addition, future studies are required to identify additional KDM enzymes involved in DDR, map their damage-induced substrates and investigate their role in repairing DNA lesions other than DSBs.
Materials and Methods
PAR-binding assay
PAR-binding assay was performed as previously described.65 Briefly, 1–5pmol purified KDM4D protein fragments were blotted onto a nitrocellulose membrane and blocked with TBST buffer supplemented with 5% milk. Radioactively labeled PAR moieties were made from automodified PARP1 prepared by in vitro PARylation reaction. This reaction was carried out at room temperature for 20 min in a reaction buffer (50 mM Tris-HCl, pH 8, 25 mM MgCl2, 50 mM NaCl) supplemented with radiolabelled NAD+ (Perkin Elmer), activated DNA, and PARP1 enzyme (Trevigen). PAR moieties were detached from PARP1 using proteinase K and the blotted membrane was incubated for 2 hrs with the radiolabelled PAR diluted in 10 ml TBST. Membranes were then washed with TBST, and subjected to both autoradiography and protein gel blot using α-His, αGST and αH3 antibodies.
Laser-microirradiation
Laser-microirradiation was performed as previously described.58 Briefly, U2OS cells were grown on fluorodish and stained with 10 μM Hoechst 33342 for 10 min at 37°C. Then, laser microirradiation was performed using LSM-700 confocal microscope. Selected spot within the nucleus was microirradiated with 10 iterations of a 405 nm laser with 100% power to generate localized DNA damage. Then time-lamps images were acquired using 488 nm laser. Signal intensity at damaged sites was measured using Zen 2009 software.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This study is supported by grants from the Israel Cancer Research Fund (ICRF), Israel Science Foundation, the Israel Cancer association, and by the Binational Science Foundation (BSF).
References
- 1. Lindahl T. Instability and decay of the primary structure of DNA. Nature 1993; 362:709-15; PMID:8469282; http://dx.doi.org/ 10.1038/362709a0 [DOI] [PubMed] [Google Scholar]
- 2. Lindahl T, Barnes DE. Repair of endogenous DNA damage. Cold Spring Harbor Symp Quant Biol 2000; 65:127-33; PMID:12760027; http://dx.doi.org/ 10.1101/sqb.2000.65.127 [DOI] [PubMed] [Google Scholar]
- 3. Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009; 461:1071-8; PMID:19847258; http://dx.doi.org/ 10.1038/nature08467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature 2012; 481:287-94; PMID:22258607; http://dx.doi.org/ 10.1038/nature10760 [DOI] [PubMed] [Google Scholar]
- 5. Cassidy LD, Liau SS, Venkitaraman AR. Chromosome instability and carcinogenesis: insights from murine models of human pancreatic cancer associated with BRCA2 inactivation. Mol Oncol 2014; 8:161-8; PMID:24268522; http://dx.doi.org/ 10.1016/j.molonc.2013.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huang LC, Clarkin KC, Wahl GM. Sensitivity and selectivity of the DNA damage sensor responsible for activating p53-dependent G1 arrest. Proc Natl Acad Sci U S A 1996; 93:4827-32; PMID:8643488; http://dx.doi.org/ 10.1073/pnas.93.10.4827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bennett CB, Lewis AL, Baldwin KK, Resnick MA. Lethality induced by a single site-specific double-strand break in a dispensable yeast plasmid. Proc Natl Acad Sci U S A 1993; 90:5613-7; PMID:8516308; http://dx.doi.org/ 10.1073/pnas.90.12.5613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rich T, Allen RL, Wyllie AH. Defying death after DNA damage. Nature 2000; 407:777-83; PMID:11048728; http://dx.doi.org/ 10.1038/35037717 [DOI] [PubMed] [Google Scholar]
- 9. Daley JM, Sung P. 53BP1, BRCA1, and the choice between recombination and end joining at DNA double-strand breaks. Mol Cell Biol 2014; 34:1380-8; PMID:24469398; http://dx.doi.org/ 10.1128/MCB.01639-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kakarougkas A, Jeggo PA. DNA DSB repair pathway choice: an orchestrated handover mechanism. Br J Radiol 2014; 87:20130685; PMID:24363387; http://dx.doi.org/ 10.1259/bjr.20130685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rothkamm K, Kruger I, Thompson LH, Lobrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol 2003; 23:5706-15; PMID:12897142; http://dx.doi.org/ 10.1128/MCB.23.16.5706-5715.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hinz JM, Yamada NA, Salazar EP, Tebbs RS, Thompson LH. Influence of double-strand-break repair pathways on radiosensitivity throughout the cell cycle in CHO cells. DNA Repair (Amst) 2005; 4:782-92; PMID:15951249; http://dx.doi.org/ 10.1016/j.dnarep.2005.03.005 [DOI] [PubMed] [Google Scholar]
- 13. Mao Z, Bozzella M, Seluanov A, Gorbunova V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 2008; 7:2902-6; PMID:18769152; http://dx.doi.org/ 10.4161/cc.7.18.6679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hartlerode AJ, Scully R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem J 2009; 423:157-68; PMID:19772495; http://dx.doi.org/ 10.1042/BJ20090942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell 2012; 47:497-510; PMID:22920291; http://dx.doi.org/ 10.1016/j.molcel.2012.07.029 [DOI] [PubMed] [Google Scholar]
- 16. Polo SE, Jackson SP. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev 2011; 25:409-33; PMID:21363960; http://dx.doi.org/ 10.1101/gad.2021311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Venkitaraman AR. Modifying chromatin architecture during the response to DNA breakage. Crit Rev Biochem Mol Biol 2010; 45:2-13; PMID:19874211; http://dx.doi.org/ 10.3109/10409230903325446 [DOI] [PubMed] [Google Scholar]
- 18. Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 2013; 14:197-210; ; http://dx.doi.org/ 10.1038/nrm3546 [DOI] [PubMed] [Google Scholar]
- 19. Polo SE. Reshaping chromatin after DNA damage: the choreography of histone proteins. J Mol Biol 2014; PMID:24887097; http://dx.doi.org/10.1016/j.jmb.2014.05.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rashi-Elkeles S, Warnatz HJ, Elkon R, Kupershtein A, Chobod Y, Paz A, Amstislavskiy V, Sultan M, Safer H, Nietfeld W, et al. Parallel profiling of the transcriptome, cistrome, and epigenome in the cellular response to ionizing radiation. Sci Signaling 2014; 7:rs3; PMID:24825921; http://dx.doi.org/ 10.1126/scisignal.2005032 [DOI] [PubMed] [Google Scholar]
- 21. Storici F, Bebenek K, Kunkel TA, Gordenin DA, Resnick MA. RNA-templated DNA repair. Nature 2007; 447:338-41; PMID:17429354; http://dx.doi.org/ 10.1038/nature05720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shen Y, Nandi P, Taylor MB, Stuckey S, Bhadsavle HP, Weiss B, Storici F. RNA-driven genetic changes in bacteria and in human cells. Mutat Res 2011; 717:91-8; PMID:21515292; http://dx.doi.org/ 10.1016/j.mrfmmm.2011.03.016 [DOI] [PubMed] [Google Scholar]
- 23. Keskin H, Shen Y, Huang F, Patel M, Yang T, Ashley K, Mazin AV, Storici F. Transcript-RNA-templated DNA recombination and repair. Nature 2014; 515:436-9; PMID:25186730; http://dx.doi.org/ 10.1038/nature13682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Melo CA, Drost J, Wijchers PJ, van de Werken H, de Wit E, Oude Vrielink JA, Elkon R, Melo SA, Leveille N, Kalluri R, et al. eRNAs are required for p53-dependent enhancer activity and gene transcription. Mol Cell 2013; 49:524-35; PMID:23273978; http://dx.doi.org/ 10.1016/j.molcel.2012.11.021 [DOI] [PubMed] [Google Scholar]
- 25. Wang Y, Huang JW, Calses P, Kemp CJ, Taniguchi T. MiR-96 downregulates REV1 and RAD51 to promote cellular sensitivity to cisplatin and PARP inhibition. Cancer Res 2012; 72:4037-46; PMID:22761336; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-0103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hu H, Du L, Nagabayashi G, Seeger RC, Gatti RA. ATM is down-regulated by N-Myc-regulated microRNA-421. Proc Natl Acad Sci U S A 2010; 107:1506-11; PMID:20080624; http://dx.doi.org/ 10.1073/pnas.0907763107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lal A, Pan Y, Navarro F, Dykxhoorn DM, Moreau L, Meire E, Bentwich Z, Lieberman J, Chowdhury D. miR-24-mediated downregulation of H2AX suppresses DNA repair in terminally differentiated blood cells. Nat Struct Mol Biol 2009; 16:492-8; PMID:19377482; http://dx.doi.org/ 10.1038/nsmb.1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moskwa P, Buffa FM, Pan Y, Panchakshari R, Gottipati P, Muschel RJ, Beech J, Kulshrestha R, Abdelmohsen K, Weinstock DM, et al. miR-182-mediated downregulation of BRCA1 impacts DNA repair and sensitivity to PARP inhibitors. Mol Cell 2011; 41:210-20; PMID:21195000; http://dx.doi.org/ 10.1016/j.molcel.2010.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Batista PJ, Chang HY. Long noncoding RNAs: cellular address codes in development and disease. Cell 2013; 152:1298-307; PMID:23498938; http://dx.doi.org/ 10.1016/j.cell.2013.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. d'Adda di Fagagna F. A direct role for small non-coding RNAs in DNA damage response. Trends Cell Biol 2014; 24:171-8; PMID:24156824; http://dx.doi.org/ 10.1016/j.tcb.2013.09.008 [DOI] [PubMed] [Google Scholar]
- 31. Wei W, Ba Z, Gao M, Wu Y, Ma Y, Amiard S, White CI, Rendtlew Danielsen JM, Yang YG, Qi Y. A role for small RNAs in DNA double-strand break repair. Cell 2012; 149:101-12; PMID:22445173; http://dx.doi.org/ 10.1016/j.cell.2012.03.002 [DOI] [PubMed] [Google Scholar]
- 32. Francia S, Michelini F, Saxena A, Tang D, de Hoon M, Anelli V, Mione M, Carninci P, d'Adda di Fagagna F. Site-specific DICER and DROSHA RNA products control the DNA-damage response. Nature 2012; 488:231-5; PMID:22722852; http://dx.doi.org/ 10.1038/nature11179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Maute RL, Schneider C, Sumazin P, Holmes A, Califano A, Basso K, Dalla-Favera R. tRNA-derived microRNA modulates proliferation and the DNA damage response and is down-regulated in B cell lymphoma. Proc Natl Acad Sci U S A 2013; 110:1404-9; PMID:23297232; http://dx.doi.org/ 10.1073/pnas.1206761110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ruby JG, Jan CH, Bartel DP. Intronic microRNA precursors that bypass Drosha processing. Nature 2007; 448:83-6; PMID:17589500; http://dx.doi.org/ 10.1038/nature05983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bogerd HP, Karnowski HW, Cai X, Shin J, Pohlers M, Cullen BR. A mammalian herpesvirus uses noncanonical expression and processing mechanisms to generate viral MicroRNAs. Mol Cell 2010; 37:135-42; PMID:20129062; http://dx.doi.org/ 10.1016/j.molcel.2009.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cheloufi S, Dos Santos CO, Chong MM, Hannon GJ. A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature 2010; 465:584-9; PMID:20424607; http://dx.doi.org/ 10.1038/nature09092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Deem AK, Li X, Tyler JK. Epigenetic regulation of genomic integrity. Chromosoma 2012; 121:131-51; PMID:22249206; http://dx.doi.org/ 10.1007/s00412-011-0358-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shi L, Oberdoerffer P. Chromatin dynamics in DNA double-strand break repair. Biochim Biophys Acta 2012; 1819:811-9; PMID:22285574; http://dx.doi.org/ 10.1016/j.bbagrm.2012.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Greenberg RA. Histone tails: directing the chromatin response to DNA damage. FEBS letters 2011; 585:2883-90; PMID:21621538; http://dx.doi.org/ 10.1016/j.febslet.2011.05.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Luijsterburg MS, van Attikum H. Chromatin and the DNA damage response: the cancer connection. Mol Oncol 2011; 5:349-67; PMID:21782533; http://dx.doi.org/ 10.1016/j.molonc.2011.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Black JC, Van Rechem C, Whetstine JR. Histone lysine methylation dynamics: establishment, regulation, and biological impact. Mol Cell 2012; 48:491-507; PMID:23200123; http://dx.doi.org/ 10.1016/j.molcel.2012.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu H, Galka M, Mori E, Liu X, Lin YF, Wei R, Pittock P, Voss C, Dhami G, Li X, et al. A method for systematic mapping of protein lysine methylation identifies functions for HP1beta in DNA damage response. Mol Cell 2013; 50:723-35; PMID:23707759; http://dx.doi.org/ 10.1016/j.molcel.2013.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004; 119:941-53; PMID:15620353; http://dx.doi.org/ 10.1016/j.cell.2004.12.012 [DOI] [PubMed] [Google Scholar]
- 44. Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006; 439:811-6; PMID:16362057; http://dx.doi.org/ 10.1038/nature04433 [DOI] [PubMed] [Google Scholar]
- 45. Shi Y, Whetstine JR. Dynamic regulation of histone lysine methylation by demethylases. Mol Cell 2007; 25:1-14; PMID:17218267; http://dx.doi.org/ 10.1016/j.molcel.2006.12.010 [DOI] [PubMed] [Google Scholar]
- 46. Chen Z, Zang J, Whetstine J, Hong X, Davrazou F, Kutateladze TG, Simpson M, Mao Q, Pan CH, Dai S, et al. Structural insights into histone demethylation by JMJD2 family members. Cell 2006; 125:691-702; PMID:16677698; http://dx.doi.org/ 10.1016/j.cell.2006.04.024 [DOI] [PubMed] [Google Scholar]
- 47. Shi YG, Tsukada Y. The discovery of histone demethylases. Cold Spring Harbor Perspect Biol 2013; 5; PMID:24003214; http://dx.doi.org/10.1101/cshperspect.a017947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wissmann M, Yin N, Muller JM, Greschik H, Fodor BD, Jenuwein T, Vogler C, Schneider R, Gunther T, Buettner R, et al. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat Cell Biol 2007; 9:347-53; PMID:17277772; http://dx.doi.org/ 10.1038/ncb1546 [DOI] [PubMed] [Google Scholar]
- 49. Shin S, Janknecht R. Activation of androgen receptor by histone demethylases JMJD2A and JMJD2D. Biochem Biophys Res Commun 2007; 359:742-6; PMID:17555712; http://dx.doi.org/ 10.1016/j.bbrc.2007.05.179 [DOI] [PubMed] [Google Scholar]
- 50. Zhang D, Yoon HG, Wong J. JMJD2A is a novel N-CoR-interacting protein and is involved in repression of the human transcription factor achaete scute-like homologue 2 (ASCL2/Hash2). Mol Cell Biol 2005; 25:6404-14; PMID:16024779; http://dx.doi.org/ 10.1128/MCB.25.15.6404-6414.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mallette FA, Richard S. JMJD2A promotes cellular transformation by blocking cellular senescence through transcriptional repression of the tumor suppressor CHD5. Cell Rep 2012; 2:1233-43; PMID:23168260; http://dx.doi.org/ 10.1016/j.celrep.2012.09.033 [DOI] [PubMed] [Google Scholar]
- 52. Kim TD, Oh S, Shin S, Janknecht R. Regulation of tumor suppressor p53 and HCT116 cell physiology by histone demethylase JMJD2D/KDM4D. PLoS One 2012; 7:e34618; PMID:22514644; http://dx.doi.org/ 10.1371/journal.pone.0034618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cloos PA, Christensen J, Agger K, Helin K. Erasing the methyl mark: histone demethylases at the center of cellular differentiation and disease. Genes Dev 2008; 22:1115-40; PMID:18451103; http://dx.doi.org/ 10.1101/gad.1652908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Black JC, Allen A, Van Rechem C, Forbes E, Longworth M, Tschop K, Rinehart C, Quiton J, Walsh R, Smallwood A, et al. Conserved antagonism between JMJD2A/KDM4A and HP1gamma during cell cycle progression. Mol Cell 2010; 40:736-48; PMID:21145482; http://dx.doi.org/ 10.1016/j.molcel.2010.11.008 [DOI] [PubMed] [Google Scholar]
- 55. Mallette FA, Mattiroli F, Cui G, Young LC, Hendzel MJ, Mer G, Sixma TK, Richard S. RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. Embo J 2012; 31:1865-78; PMID:22373579; http://dx.doi.org/ 10.1038/emboj.2012.47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Palomera-Sanchez Z, Bucio-Mendez A, Valadez-Graham V, Reynaud E, Zurita M. Drosophila p53 is required to increase the levels of the dKDM4B demethylase after UV induced DNA damage to demethylate histone H3-lysine 9. J Biol Chem 2010; PMID:20675387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Young LC, McDonald DW, Hendzel MJ. Kdm4b histone demethylase is a DNA damage response protein and confers a survival advantage following gamma-irradiation. J Biol Chem 2013; 288:21376-88; PMID:23744078; http://dx.doi.org/ 10.1074/jbc.M113.491514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Khoury-Haddad H, Guttmann-Raviv N, Ipenberg I, Huggins D, Jeyasekharan AD, Ayoub N. PARP1-dependent recruitment of KDM4D histone demethylase to DNA damage sites promotes double-strand break repair. Proc Natl Acad Sci U S A 2014; 111:E728-37; PMID:24550317; http://dx.doi.org/ 10.1073/pnas.1317585111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M, et al. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell 2006; 125:467-81; PMID:16603238; http://dx.doi.org/ 10.1016/j.cell.2006.03.028 [DOI] [PubMed] [Google Scholar]
- 60. Loh YH, Zhang W, Chen X, George J, Ng HH. Jmjd1a and Jmjd2c histone H3 Lys 9 demethylases regulate self-renewal in embryonic stem cells. Genes Dev 2007; 21:2545-57; PMID:17938240; http://dx.doi.org/ 10.1101/gad.1588207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pedersen MT, Helin K. Histone demethylases in development and disease. Trends Cell Biol 2010; 20:662-71; PMID:20863703; http://dx.doi.org/ 10.1016/j.tcb.2010.08.011 [DOI] [PubMed] [Google Scholar]
- 62. Zoabi M, Nadar-Ponniah PT, Khoury-Haddad H, Usaj M, Budowski-Tal I, Haran T, Henn A, Mandel-Gutfreund Y, Ayoub N. RNA-dependent chromatin localization of KDM4D lysine demethylase promotes H3K9me3 demethylation. Nucleic Acids Res 2014; 42:13026-38; PMID:25378304; http://dx.doi.org/ 10.1093/nar/gku1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol 2012; 13:411-24; PMID:22713970; http://dx.doi.org/ 10.1038/nrm3376 [DOI] [PubMed] [Google Scholar]
- 64. Sajish M, Zhou Q, Kishi S, Valdez DM, Jr, Kapoor M, Guo M, Lee S, Kim S, Yang XL, Schimmel P. Trp-tRNA synthetase bridges DNA-PKcs to PARP-1 to link IFN-gamma and p53 signaling. Nat Chem Biol 2012; 8:547-54; PMID:22504299; http://dx.doi.org/ 10.1038/nchembio.937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ahel I, Ahel D, Matsusaka T, Clark AJ, Pines J, Boulton SJ, West SC. Poly(ADP-ribose)-binding zinc finger motifs in DNA repair/checkpoint proteins. Nature 2008; 451:81-5; PMID:18172500; http://dx.doi.org/ 10.1038/nature06420 [DOI] [PubMed] [Google Scholar]
- 66. Pleschke JM, Kleczkowska HE, Strohm M, Althaus FR. Poly(ADP-ribose) binds to specific domains in DNA damage checkpoint proteins. J Biol Chem 2000; 275:40974-80; PMID:11016934; http://dx.doi.org/ 10.1074/jbc.M006520200 [DOI] [PubMed] [Google Scholar]
- 67. Gagne JP, Isabelle M, Lo KS, Bourassa S, Hendzel MJ, Dawson VL, Dawson TM, Poirier GG. Proteome-wide identification of poly(ADP-ribose) binding proteins and poly(ADP-ribose)-associated protein complexes. Nucleic Acids Res 2008; 36:6959-76; PMID:18981049; http://dx.doi.org/ 10.1093/nar/gkn771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zaja R, Mikoc A, Barkauskaite E, Ahel I. Molecular insights into poly(ADP-ribose) recognition and processing. Biomolecules 2012; 3:1-17; PMID:24970154; http://dx.doi.org/ 10.3390/biom3010001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ahel D, Horejsi Z, Wiechens N, Polo SE, Garcia-Wilson E, Ahel I, Flynn H, Skehel M, West SC, Jackson SP, et al. Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science 2009; 325:1240-3; PMID:19661379; http://dx.doi.org/ 10.1126/science.1177321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chou DM, Adamson B, Dephoure NE, Tan X, Nottke AC, Hurov KE, Gygi SP, Colaiacovo MP, Elledge SJ. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc Natl Acad Sci U S A 2010; 107:18475-80; PMID:20937877; http://dx.doi.org/ 10.1073/pnas.1012946107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Li GY, McCulloch RD, Fenton AL, Cheung M, Meng L, Ikura M, Koch CA. Structure and identification of ADP-ribose recognition motifs of APLF and role in the DNA damage response. Proc Natl Acad Sci U S A 2010; 107:9129-34; PMID:20439749; http://dx.doi.org/ 10.1073/pnas.1000556107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Okano S, Lan L, Caldecott KW, Mori T, Yasui A. Spatial and temporal cellular responses to single-strand breaks in human cells. Mol Cell Biol 2003; 23:3974-81; PMID:12748298; http://dx.doi.org/ 10.1128/MCB.23.11.3974-3981.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Haince JF, McDonald D, Rodrigue A, Dery U, Masson JY, Hendzel MJ, Poirier GG. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J Biol Chem 2008; 283:1197-208; PMID:18025084; http://dx.doi.org/ 10.1074/jbc.M706734200 [DOI] [PubMed] [Google Scholar]
- 74. Zhang F, Chen Y, Li M, Yu X. The oligonucleotide/oligosaccharide-binding fold motif is a poly(ADP-ribose)-binding domain that mediates DNA damage response. Proc Natl Acad Sci U S A 2014; 111:7278-83; PMID:24799691; http://dx.doi.org/ 10.1073/pnas.1318367111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Li M, Yu X. Function of BRCA1 in the DNA damage response is mediated by ADP-ribosylation. Cancer Cell 2013; 23:693-704; PMID:23680151; http://dx.doi.org/ 10.1016/j.ccr.2013.03.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jha DK, Strahl BD. An RNA polymerase II-coupled function for histone H3K36 methylation in checkpoint activation and DSB repair. Nat Commun 2014; 5:3965; PMID:24910128; http://dx.doi.org/ 10.1038/ncomms4965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Seward DJ, Cubberley G, Kim S, Schonewald M, Zhang L, Tripet B, Bentley DL. Demethylation of trimethylated histone H3 Lys4 in vivo by JARID1 JmjC proteins. Nat Struct Mol Biol 2007; 14:240-2; PMID:17310255; http://dx.doi.org/ 10.1038/nsmb1200 [DOI] [PubMed] [Google Scholar]
- 78. Christensen J, Agger K, Cloos PA, Pasini D, Rose S, Sennels L, Rappsilber J, Hansen KH, Salcini AE, Helin K. RBP2 belongs to a family of demethylases, specific for tri-and dimethylated lysine 4 on histone 3. Cell 2007; 128:1063-76; PMID:17320161; http://dx.doi.org/ 10.1016/j.cell.2007.02.003 [DOI] [PubMed] [Google Scholar]
- 79. Yamane K, Tateishi K, Klose RJ, Fang J, Fabrizio LA, Erdjument-Bromage H, Taylor-Papadimitriou J, Tempst P, Zhang Y. PLU-1 is an H3K4 demethylase involved in transcriptional repression and breast cancer cell proliferation. Mol Cell 2007; 25:801-12; PMID:17363312; http://dx.doi.org/ 10.1016/j.molcel.2007.03.001 [DOI] [PubMed] [Google Scholar]
- 80. Li X, Liu L, Yang S, Song N, Zhou X, Gao J, Yu N, Shan L, Wang Q, Liang J, et al. Histone demethylase KDM5B is a key regulator of genome stability. Proc Natl Acad Sci U S A 2014; 111:7096-101; PMID:24778210; http://dx.doi.org/ 10.1073/pnas.1324036111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Watanabe S, Watanabe K, Akimov V, Bartkova J, Blagoev B, Lukas J, Bartek J. JMJD1C demethylates MDC1 to regulate the RNF8 and BRCA1-mediated chromatin response to DNA breaks. Nat Struct Mol Biol 2013; 20:1425-33; PMID:24240613; http://dx.doi.org/ 10.1038/nsmb.2702 [DOI] [PubMed] [Google Scholar]
- 82. Mosammaparast N, Kim H, Laurent B, Zhao Y, Lim HJ, Majid MC, Dango S, Luo Y, Hempel K, Sowa ME, et al. The histone demethylase LSD1/KDM1A promotes the DNA damage response. J Cell Biol 2013; 203:457-70; PMID:24217620; http://dx.doi.org/ 10.1083/jcb.201302092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ayoub N, Jeyasekharan AD, Bernal JA, Venkitaraman AR. HP1-beta mobilization promotes chromatin changes that initiate the DNA damage response. Nature 2008; 453:682-6; PMID:18438399; http://dx.doi.org/ 10.1038/nature06875 [DOI] [PubMed] [Google Scholar]
- 84. Sun Y, Jiang X, Xu Y, Ayrapetov MK, Moreau LA, Whetstine JR, Price BD. Histone H3 methylation links DNA damage detection to activation of the tumour suppressor Tip60. Nat Cell Biol 2009; 11:1376-82; PMID:19783983; http://dx.doi.org/ 10.1038/ncb1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Trojer P, Zhang J, Yonezawa M, Schmidt A, Zheng H, Jenuwein T, Reinberg D. Dynamic histone H1 isotype 4 methylation and demethylation by histone lysine methyltransferase G9a/KMT1C and the Jumonji domain-containing JMJD2/KDM4 proteins. J Biol Chem 2009; 284:8395-405; PMID:19144645; http://dx.doi.org/ 10.1074/jbc.M807818200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Jack AP, Bussemer S, Hahn M, Punzeler S, Snyder M, Wells M, Csankovszki G, Solovei I, Schotta G, Hake SB. H3K56me3 is a novel, conserved heterochromatic mark that largely but not completely overlaps with H3K9me3 in both regulation and localization. PLoS One 2013; 8:e51765; PMID:23451023; http://dx.doi.org/ 10.1371/journal.pone.0051765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Falk M, Lukasova E, Gabrielova B, Ondrej V, Kozubek S. Chromatin dynamics during DSB repair. Biochim Biophys Acta 2007; 1773:1534-45; PMID:17850903; http://dx.doi.org/ 10.1016/j.bbamcr.2007.07.002 [DOI] [PubMed] [Google Scholar]
- 88. Ayrapetov MK, Gursoy-Yuzugullu O, Xu C, Xu Y, Price BD. DNA double-strand breaks promote methylation of histone H3 on lysine 9 and transient formation of repressive chromatin. Proc Natl Acad Sci U S A 2014; 111:9169-74; PMID:24927542; http://dx.doi.org/ 10.1073/pnas.1403565111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ayoub N, Jeyasekharan AD, Venkitaraman AR. Mobilization and recruitment of HP1: a bimodal response to DNA breakage. Cell Cycle 2009; 8:2945-50; PMID:19657222 [PubMed] [Google Scholar]
- 90. Ayoub N, Jeyasekharan AD, Bernal JA, Venkitaraman AR. Paving the way for H2AX phosphorylation: chromatin changes in the DNA damage response. Cell Cycle 2009; 8:1494-500; PMID:19377276; http://dx.doi.org/ 10.4161/cc.8.10.8501 [DOI] [PubMed] [Google Scholar]
- 91. Li ML, Yuan G, Greenberg RA. Chromatin yo-yo: expansion and condensation during DNA repair. Trends Cell Biol 2014; 24:616-8; PMID:25305135; http://dx.doi.org/ 10.1016/j.tcb.2014.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Chiolo I, Minoda A, Colmenares SU, Polyzos A, Costes SV, Karpen GH. Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell 2011; 144:732-44; PMID:21353298; http://dx.doi.org/ 10.1016/j.cell.2011.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Adam S, Polo SE. Blurring the line between the DNA damage response and transcription: the importance of chromatin dynamics. Exp Cell Res 2014; 329:148-53; PMID:25062983; http://dx.doi.org/ 10.1016/j.yexcr.2014.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Huang J, Sengupta R, Espejo AB, Lee MG, Dorsey JA, Richter M, Opravil S, Shiekhattar R, Bedford MT, Jenuwein T, et al. p53 is regulated by the lysine demethylase LSD1. Nature 2007; 449:105-8; PMID:17805299; http://dx.doi.org/ 10.1038/nature06092 [DOI] [PubMed] [Google Scholar]
- 95. Huang J, Perez-Burgos L, Placek BJ, Sengupta R, Richter M, Dorsey JA, Kubicek S, Opravil S, Jenuwein T, Berger SL. Repression of p53 activity by Smyd2-mediated methylation. Nature 2006; 444:629-32; PMID:17108971; http://dx.doi.org/ 10.1038/nature05287 [DOI] [PubMed] [Google Scholar]
- 96. Shi X, Kachirskaia I, Yamaguchi H, West LE, Wen H, Wang EW, Dutta S, Appella E, Gozani O. Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol Cell 2007; 27:636-46; PMID:17707234; http://dx.doi.org/ 10.1016/j.molcel.2007.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Black JC, Manning AL, Van Rechem C, Kim J, Ladd B, Cho J, Pineda CM, Murphy N, Daniels DL, Montagna C, et al. KDM4A lysine demethylase induces site-specific copy gain and rereplication of regions amplified in tumors. Cell 2013; 154:541-55; PMID:23871696; http://dx.doi.org/ 10.1016/j.cell.2013.06.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Berry WL, Janknecht R. KDM4/JMJD2 histone demethylases: epigenetic regulators in cancer cells. Cancer Res 2013; 73:2936-42; PMID:23644528; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-4300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Kupershmit I, Khoury-Haddad H, Awwad SW, Guttmann-Raviv N, Ayoub N. KDM4C (GASC1) lysine demethylase is associated with mitotic chromatin and regulates chromosome segregation during mitosis. Nucleic Acids Res 2014; 42:6168-82; PMID:24728997; http://dx.doi.org/ 10.1093/nar/gku253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Frescas D, Guardavaccaro D, Kuchay SM, Kato H, Poleshko A, Basrur V, Elenitoba-Johnson KS, Katz RA, Pagano M. KDM2A represses transcription of centromeric satellite repeats and maintains the heterochromatic state. Cell Cycle 2008; 7:3539-47; PMID:19001877; http://dx.doi.org/ 10.4161/cc.7.22.7062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Cheng Z, Cheung P, Kuo AJ, Yukl ET, Wilmot CM, Gozani O, Patel DJ. A molecular threading mechanism underlies Jumonji lysine demethylase KDM2A regulation of methylated H3K36. Genes Dev 2014; 28:1758-71; PMID:25128496; http://dx.doi.org/ 10.1101/gad.246561.114 [DOI] [PMC free article] [PubMed] [Google Scholar]