

Abstract
Melanoma continues to cause more deaths than any other skin cancer, necessitating the development of new avenues of treatment. One promising new opportunity comes in the form of mechanism-based therapeutic targets. We recently reported the overexpression and delocalization of the class III histone deacetylase SIRT1 in melanoma, and demonstrated that its small molecule inhibition via Tenovin-1 decreased cell growth and viability of melanoma cells, possibly by a p53 mediated induction of p21. Here, we support our data using additional SIRT inhibitors, viz. Sirtinol and Ex-527, which suggests possible benefits of concomitantly inhibiting more than one Sirtuin for an effective cancer management strategy. This “Extra View” paper also includes a discussion of our results in the context of similar recent and concurrent studies. Furthermore, we expand upon our findings in an analysis of new research that may link the cellular localization and growth effects of SIRT1 with the PI3K signaling pathway.
Keywords: cellular localization, melanoma, PI3K, SIRT1, SIRT1 inhibitors
Introduction
Melanoma causes more deaths than any other skin cancer, and its incidence in the US has been on the rise for the past 40 years. Despite recent advances in treatment, an estimated 9,710 people will die of melanoma in 2014, necessitating the development of more effective melanoma therapies.1 Until recently, the standard of care involved surgical excision, chemotherapy, or immunotherapy, with poor response rates and a 10-year survival rate of less than 10%.2 In the past few years, new treatments have become available specifically targeting BRAF mutations, which are found in 27–68% of melanomas.3 Despite promising initial response rates to these treatments, clinical trials show that progression-free survival of patients is limited to 3.6–5.6 months, with subsequent relapse in tumor progression.4 In the search for a potential molecular therapeutic target, we recently suggested a possible role for the histone deacetylase SIRT1 in melanoma.5
SIRT1 is a member of the class III histone deacetylases known as the Sirtuins, which depend on nicotinamide adenine dinucleotide (NAD+) as a substrate. SIRT1 has been shown to act through its non-histone protein targets to play a role in a diverse assortment of biological processes, including metabolism, neurodegeneration, and immune function, among others.6 SIRT1 also plays a role in many different types of cancer, although its function as a tumor suppressor or promoter is unclear, with contradictory reports throughout the scientific literature. The role of SIRT1 in melanoma is beginning to be unraveled only recently as 3 separate studies published in 2014, including our own suggested a role for SIRT1 in this deadly neoplasm.5,7,8 In addition to the finding that SIRT1 inhibition has effects on p53 protein levels in wild-type p53 melanoma cells, another recent paper by our lab found that several other pathways may be affected by SIRT1 inhibition, including a possible role in cell cycle regulation.9
In our recent paper, we showed that SIRT1 is overexpressed in melanoma cell lines and human tissue samples. In melanoma cells, chemical inhibition of SIRT1 with the small molecule inhibitor Tenovin-1 led to a decrease in cellular proliferation and viability which was accompanied by increased protein levels of p53 and p21.5 In a few additional experiments we attempted to replicate our findings with other SIRT inhibitors. It is possible that SIRT2 or SIRT3 could be partially responsible for the effects we saw in our previous paper, as they have been shown to play a role in cell cycle regulation and apoptosis.10,11 Tenovin-1 is not a specific inhibitor of SIRT1, as it also inhibits other Sirtuins with an IC50 value of 21 μM for SIRT1, 10 μM for SIRT2, and 67 μM for SIRT3.12 In our previous study, we used doses of Tenovin-1 up to 25 μM, which would affect SIRT1 and/or SIRT2. In additional experiments, we sought to confirm our results through the use of 2 additional Sirtuin inhibitors: Sirtinol and Ex-527. Sirtinol shows more specificity for SIRT2 inhibition than SIRT1, with IC50's of 38 μM and 131 μM, respectively.13 For our experiments, we chose our highest dose of Sirtinol as 50 μM, which would allow us to test SIRT2 inhibition. Ex-527 on the other hand, is much more specific for SIRT1, with IC50's of 98 nM, 19.6 μM, and 48.7 μM for SIRT1, SIRT2, and SIRT3.14 We chose a top dose that would possibly inhibit all 3 SIRTs, 50 μM. Thus, through treatment with these additional 2 inhibitors, we should have a better idea of whether inhibiting more than one Sirtuin at a time will have a greater impact on melanoma cell growth and viability, or if using SIRT1 or SIRT2 inhibition individually would be a better treatment option.
Results and Discussion
To assess the impact of Sirtinol (SIRT2 inhibition) and Ex-527 (SIRT1, 2, and 3 inhibitions) on melanoma cell growth and viability, we performed a trypan blue exclusion assay. A375, Hs294T, and G361 melanoma cells were treated for 48 hours with 0, 10, 25, or 50 μM Sirtinol or Ex-527 prior to collection and analysis. We found that both Sirtinol and Ex-527 caused a dose-dependent decrease in cell growth across the treatment levels tested, with Sirtinol showing a slightly greater decrease than Ex-527 (Fig. 1A). Sirtinol-treated samples also showed a significant dose-dependent decrease in cell viability, whereas Ex-527-treated samples showed little to no change across treatment levels, with a statistically significant decrease across all 3 cell lines at the 50 μM concentration (Fig. 1B). The effects of Sirtinol treatment on cell viability were confirmed by staining treated G361 cells with Annexin V and Propidium Iodide (PI) and analyzing via flow cytometry (Fig. 2A). The results showed a noticeable, dose-dependent increase in cells positive for both Annexin V and PI across all treatment levels, indicating an increase in cellular apoptosis under treatment with Sirtinol. This suggests that inhibiting SIRT2 alone may have better effectiveness on growth reduction and decreasing viability than inhibition of the 3 Sirtuins together (1–3). However, the results found in our previous paper show a marked decrease in cell growth using low levels of Tenovin-1, which suggests that targeting SIRT1 and 2 in combination may be a better approach, although Sirtinol is better at inhibiting cell viability than either of the other 2 inhibitors. Interestingly, the 25 μM dose of Ex-527 should be inhibiting those same Sirtuins, but the effects on cell viability and growth were not as pronounced. This may be due to a different mechanism of action, and needs to be looked into further.
Figure 1.

Melanoma cell growth and viability after treatment with Sirtinol and Ex-527. A375, Hs294T, and G361 cells (all p53WT) were treated for 48 hours with either Sirtinol or Ex-527 at concentrations of 0, 10, 25, or 50 μM prior to analysis. All treatment levels contained an equal amount (0.5%) of the DMSO used as a diluent. All data are expressed as mean +/- SE of 3 experiments. Statistical analysis was performed using a 2-tailed Student's t-test (ns = no significance, *P < 0 .05, **P < 0 .01, ***P < 0 .001). Effects of Sirtinol and Ex-527 on melanoma cell growth (A) and viability (B). Cells were collected using 0.25% trypsin/2.21 mM EDTA (Cellgro, VA), pelleted via centrifugation, then rinsed with phosphate buffered saline (PBS) (pH 7.4). Cells were then stained with Trypan Blue Dye (Gibco, CA) and analyzed for total and viable cell counts via automated cell counter. Cell growth is displayed as the total number of cells in each treatment group relative to the untreated control. Cell viability is shown as the number of viable cells relative to the total number of cells at each treatment level. (C) Effects of Sirtinol and Ex-527 on melanoma cell viability and metabolic activity (MTT assay). After treatment, cells were rinsed with PBS (pH 7.4), then incubated at 37°C in the dark with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT reagent) (Sigma-Aldrich, MO) diluted to 0.5 mg/mL in the appropriate cell culture medium. After 2 hours, the media was removed and the resulting formazan crystals were solubilized in DMSO for 10 minutes prior to analysis. Results are displayed as a mean of the optical density at 540 nm for each treatment level normalized to the untreated control.
Figure 2.
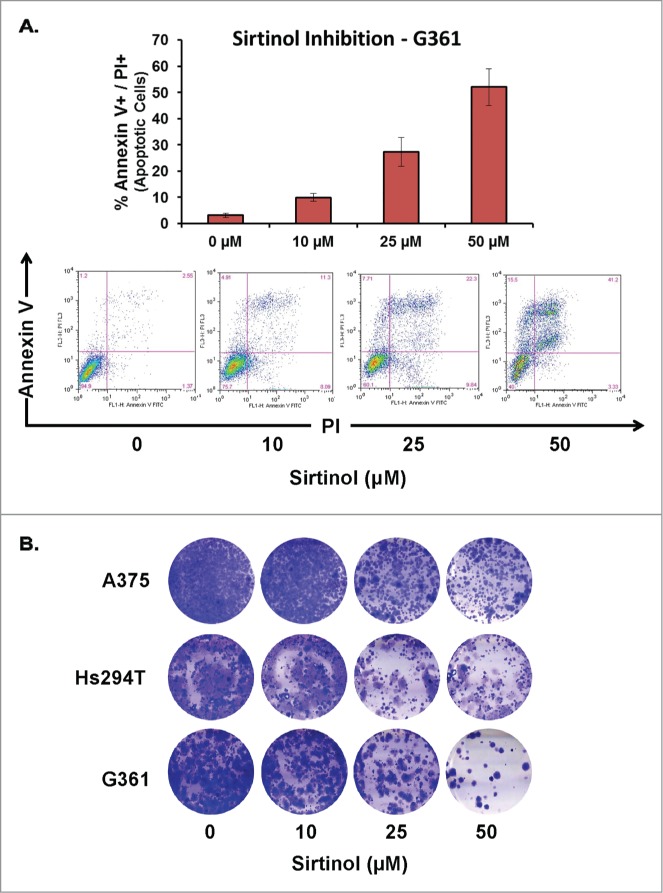
Apoptosis and clonogenic survival of melanoma cells after treatment with Sirtinol. A375, Hs294T, and G361 cells (all p53WT) were treated for 48 hours with Sirtinol at concentrations of 0, 10, 25, or 50 μM prior to analysis. All treatment levels contained an equal amount (0.5%) of the DMSO used as a diluent. (A) Effect of Sirtinol on melanoma cell apoptosis as analyzed via Annexin V/Propidium Iodide (PI) binding assay. Following treatment, the cells were washed with PBS and stained with FITC conjugated Annexin V antibody and PI (BD Biosciences, CA) according to the vendor's protocol. The cells were then analyzed on a FACScan benchtop cytometer at the UWCCC Flow cytometry facility and analyzed by FlowJo software (Treestar, OR). Representative 2-dimensional dot plots of Annexin V-FITC and PI fluorescence are displayed along with quantification of the total Annexin V+/PI+ cells at each treatment level. (B) Effect of Sirtinol on the clonogenic survival of melanoma cells. After treatment, 3000 cells from each treatment level were re-plated in a 6-well tissue culture plate. Cells were allowed to grow under normal tissue culture conditions for 10–14 d, changing media every 3 d prior to fixation and staining with 0.5% crystal violet solution. Cells were stained at 4°C for 1 hour, rinsed with PBS (pH 7.4), then air dried and photographed.
To confirm the effects of Sirtinol and Ex-527 on cell growth and viability, an MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay was performed. All 3 cell lines showed a dose-dependent decrease in OD540 after treatment with each inhibitor, indicating a decrease in cellular growth, viability, and/or metabolism. This decrease was similar in Sirtinol and Ex-527 treated cells, despite the greater effects of Sirtinol on cell growth and viability determined by trypan blue exclusion assay. This could indicate that Ex-527 has a greater impact on cellular metabolic rates than Sirtinol in these cell lines. Overall, we see that both SIRT inhibitors impact cell growth, viability, and/or metabolism, confirming the results of the trypan blue exclusion assay (Fig. 1C). However, as with cellular growth, Tenovin-1 greatly outperformed both Sirtinol and Ex-527 in inhibition of metabolic activity.
Finally, to determine whether the effects of the treatment were long-lasting, we performed a clonogenic survival assay using increasing levels of Sirtinol. For this assay, cells were treated with Sirtinol for 48 hours, then replated at low density and allowed to grow for 10–14 d prior to staining with crystal violet. As shown in Figure 2B, Sirtinol treatment resulted in a dose-dependent decrease in clonogenic survival for all 3 cell lines tested, indicating a lasting inhibition of cellular growth capabilities in treated cells. Although the results we previously published using Tenovin-1 show greater effects, these results have similar trends, indicating that different SIRT inhibitors have the same effect, and that it is likely that the cellular effects may be due to more than one SIRT isoform. It is also possible, although unlikely, that there are off-target or non-specific effects of the drugs and that other signaling pathways may be involved in the outcomes of our results. However, to date none of these have been found or indicated for any of the inhibitors that we tested.
Additional data from a simultaneous publication8 to our previous study has corroborated our results as well as those included here. In this paper, Ohanna et al. showed that SIRT1 activity levels are overexpressed across several melanoma cell lines. In addition, SIRT1 siRNA-treated melanoma cells showed increased levels of p53, as well as a senescent phenotype indicated by G0/G1 cell cycle arrest and positive senescence-associated-β-galactosidase (SA-β-Gal) staining. They found that treatment with Sirtinol or Ex-527 showed similar SA-β-Gal staining results to those obtained with siRNA treatment, suggesting that the growth inhibition seen here is most likely associated with cellular senescence rather than a temporary growth arrest. However, it is important to note that the work done by Ohanna et al. was in different cell types, at different concentrations, and for a longer time period (96 hrs) than our work.
Altogether, our previous work, the data shown here, and the findings of Ohanna et al. suggest that all 3 of these chemical inhibitors of Sirtuins are effective in the inhibition of melanoma cell growth regardless of which of the Sirtuins is being more strongly inhibited. Cell viability is most greatly impacted by Sirtinol, followed by Tenovin-1, and is least impacted by Ex-527 at these treatment levels. This could indicate a possible greater role for SIRT2 in melanoma cells, however, the dosage used in these treatments could also be playing a role. In addition, the inhibition of the different Sirtuins could be working synergistically in these treatments, and their individual impacts cannot be definitively known without separate inhibition of each.
Whether or not separate inhibition of SIRT1 and SIRT2 is possible at the signaling level remains to be seen. It is possible that SIRT2 activity could be impacted by the activity levels of SIRT1, making it impossible to individually inhibit the 2 Sirtuins. This possibility is based on the known interactions between SIRT1, SIRT2, and the histone acetylase p300 (Fig. 3). SIRT1 has been shown to target p300 for deacetylation, rendering it inactive.15 Therefore, inhibition of SIRT1 would lead to more p300 in the active form. p300 has been shown to acetylate SIRT2, interfering with its catalytic activity.16 Thus, inhibition of SIRT1 could lead to SIRT2 inhibition through the activity of p300. It is possible that the normal cellular distribution of these proteins minimizes the effects of these potential interactions, with SIRT1 primarily in the nucleus and SIRT2 primarily in the cytoplasm, necessitating a nuclear-cytoplasmic shuttling of p300 to achieve SIRT2 inhibition as a downstream effect of SIRT1 inhibition. However, as we showed in our recent paper, in 65% of the melanoma tissues tested, there was a shift in the localization of SIRT1 to the cytoplasm.5 Whether this cytoplasmic localization could make such interactions as this one between SIRT1, p300, and SIRT2 more likely remains to be seen. This dynamic interaction could be a new area to explore for possible combinational targeted therapies.
Figure 3.
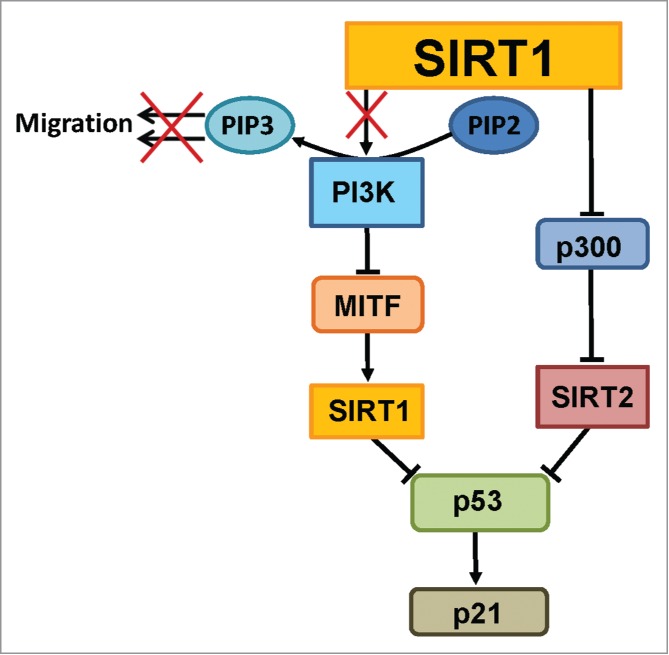
Importance of SIRT1 in regulating melanoma signaling pathways. SIRT1 is involved in important melanoma-related pathways. Kunimoto et al. have shown that blocking SIRT (indicated by the “X”) results in decreased phosphorylation of PIP3 by PI3K at the cell membrane, leading to decreased cell migration.7 In our proposed mechanism, PI3K regulates SIRT1 levels through the intermediate MITF protein. PI3K has been found to negatively regulate MITF,20 which has separately been shown to positively regulate SIRT1.8 Any experiments (including ours here), need to note that when inhibiting SIRT1, the results may not be specific solely due to a p300-mediated inhibition of SIRT2 that happens naturally in the cell. This may be mitigated or compounded by intracellular localization. SIRT1 and SIRT2 both negatively regulate tumor protein p53, as discussed in our previous paper.5 p53 has been shown in turn to positively regulate p21.
As discussed in our earlier study, the high incidence of cytoplasmic SIRT1 in melanoma tissues was not unexpected, as Byles et al. have shown a shift to cytoplasmic localization in breast, lung, and prostate cancer cell lines, as well as in prostate cancer tissues.17 In addition, they found that the overall increase in SIRT1 levels in cancer cells relative to normal cells was largely due to this increase in cytoplasmic SIRT1 levels. When they looked into the cause for the increased levels of cytoplasmic SIRT1, they found that the stability of SIRT1 in the cytoplasm could be reduced through the inhibition of insulin-like growth factor-1R (IGF-1R) or phosphoinositide 3-kinase (PI3K) activity, whereas nuclear SIRT1 stability was unaffected. Furthermore, inhibition of PI3K decreased prostate cancer cell growth and proliferation at a significantly higher level when SIRT1 was overexpressed in the cytoplasm than when SIRT1 siRNA was used in conjunction with the PI3K inhibitor in cancer cells. Following this trend, there was little effect in normal prostate cells where SIRT1 is primarily nuclear. However, Tanno et al. found that PI3K inhibition had the opposite effect on SIRT1 cellular localization in the mouse myoblast cell line C2C12.18 This study was the first to show cytoplasmic localization of SIRT1, but found that PI3K inhibition by LY2940024 resulted in dose-dependent exclusion of SIRT1 from the nucleus, possibly due to certain nuclear export and nuclear localization signals within the SIRT1 protein. Alternatively, they found that SIRT1 localization varies depending on the tissue type and degree of differentiation. Thus, the PI3K signaling cascade likely plays a role in the regulation of cytoplasmic SIRT1 and its role in prostate cancer growth and proliferation, yet it is most likely dependent upon the tissue type, differentiation status, and/or species.
While the effect of PI3K in melanoma cell growth and proliferation has not yet been extensively investigated, a recent paper by Kunimoto et al. discussed the possible link between PI3K and cytoplasmic SIRT1 in a mechanism regulating the migration of melanoma cells.7 PI3K is known to play a role in the formation of the growth factor-stimulated membrane extensions at the leading edge of migrating cells known as lamellipodia. In response to serum or platelet-derived growth factor (PDGF) stimulation, PI3K phosphorylates phosphatidylinositol-4,4-bisphosphate (PIP2) to form phosphatidylinositol-3,4,5-triphosphate (PIP3) at the cell membrane, and subsequently activates and recruits Akt to the cell membrane.19 Kunimoto et al. showed that nicotinamide (NAM), a Sirtuin inhibitor, reduces in vitro wound healing and transwell migration, and prevents lamellipodium extension in melanoma cells in response to growth factor treatment (Fig. 3). They showed that NAM inhibits the increase of PIP3 at the cell membrane and the accumulation of phospho-Akt in response to PDGF treatment. This suggests that SIRT1, which was shown to be almost exclusively cytoplasmic in these cell lines, positively regulates PI3K-dependent melanoma cell migration. When combined with the data from Byles et al. and Tanno et al., this suggests that a regulatory feedback mechanism could exist between cytoplasmic SIRT1 and PI3K.
While it doesn't confirm the existence of such a feedback loop, the work of Ohanna et al. provides a mechanism through which PI3K could regulate SIRT1 expression in melanoma cells. Microphthalmia-associated transcription factor (MITF) has previously been shown to be a downstream target of the PI3K signaling pathway in melanoma cells, with use of the PI3K inhibitor LY294002 resulting in increased levels of MITF (Fig. 3).20 Ohanna et al. have shown that MITF knockdown reduces SIRT1 levels, and forced overexpression of MITF increases SIRT1 levels, indicating a role for MITF in the regulation of SIRT1. This is, however, the opposite effect that one would expect based on the results of Byles et al., since they showed that PI3K inhibition (which increases MITF expression) reduces cytoplasmic SIRT1 and has no effect on nuclear SIRT1. Since MITF is a melanocyte lineage-specific transcription factor, it is possible that the presence of MITF alters the effects of PI3K inhibition on SIRT1 in melanoma cells relative to the lung, breast, and prostate cells studied by Byles et al. Alternatively, Tanno et al. reported that many SIRT1 antibodies do not detect both nuclear and cytoplasmic SIRT1, leading to many discrepancies in the literature concerning its expression and localization.
In summary, based on recent efforts it can be suggested that Sirtuin inhibition imparts anti-proliferative effects in melanoma cells. However, the level of anti-proliferative effects in response to different inhibitors might be impacted by the relative inhibition levels of SIRT1, SIRT2, and SIRT3, although additional data is required to confirm this possibility. Recent papers from other groups have corroborated our findings regarding SIRT1 in melanoma, and led to interesting possibilities concerning the interactions between SIRT1, PI3K, and MITF. The finding that PI3K is involved in both cell migration and SIRT1 cellular localization allows us to look at our recently published data in a new light. Our recent finding of increased cytoplasmic SIRT1 localization in human melanoma tissues could prove useful in developing new mechanism-based treatments of metastatic melanoma.
Funding Statement
This work was partially supported by funding from the NIH (R01AR059130 and R01CA176748) and the Department of Veterans Affairs (VA Merit Review Award 1I01BX001008). We would like to acknowledge the University of Wisconsin Carbone Cancer Center, Cancer Center Support Grant P30 CA014520.
References
- 1. Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, CA Cancer J Clin 2014; 64:9-29; PMID:24399786 [DOI] [PubMed] [Google Scholar]
- 2. Bhatia S, Tykodi SS, Thompson JA. Treatment of metastatic melanoma: an overview. Oncol (Williston Park) 2009; 23:488-96 [PMC free article] [PubMed] [Google Scholar]
- 3. Chakraborty R, Wieland CN, Comfere NI. Molecular targeted therapies in metastatic melanoma. Pharmgenomics Pers Med 2013; 6:49-56; PMID:23843700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Klinac D, Gray ES, Millward M, Ziman M. Advances in personalized targeted treatment of metastatic melanoma and non-invasive tumor monitoring. Front Oncol 2013; 3:54; PMID:23515890; http://dx.doi.org/ 10.3389/fonc.2013.00054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wilking MJ, Singh C, Nihal M, Zhong W, Ahmad N. SIRT1 deacetylase is overexpressed in human melanoma and its small molecule inhibition imparts anti-proliferative response via p53 activation. Arch Biochem Biophys 2014; http://dx.doi.org/ 10.1016/j.abb.2014.04.001; PMID:24751483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stunkel W, Campbell RM. Sirtuin 1 (SIRT1): the misunderstood HDAC. J Biomol Screen 2011; 16:1153-69; PMID:22086720; http://dx.doi.org/ 10.1177/1087057111422103 [DOI] [PubMed] [Google Scholar]
- 7. Kunimoto R, Jimbow K, Tanimura A, Sato M, Horimoto K, Hayashi T, Hisahara S, Sugino T, Hirobe T, Yamashita T, et al. SIRT1 regulates lamellipodium extension and migration of melanoma cells. J Invest Dermatol 2014; 134:1693-700; PMID:24480879; http://dx.doi.org/ 10.1038/jid.2014.50 [DOI] [PubMed] [Google Scholar]
- 8. Ohanna M, Bonet C, Bille K, Allegra M, Davidson I, Bahadoran P, Lacour J-P, Ballotti R, Bertolotto C. SIRT1 promotes proliferation and inhibits the senescence-like phenotype in human melanoma cells. Oncotarget 2014; 5:2085-95; PMID:24742694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Singh CK, George J, Nihal M, Sabat G, Kumar R, Ahmad N. Novel downstream molecular targets of SIRT1 in melanoma: a quantitative proteomics approach. Oncotarget 2014; 5:1987-99; PMID:24743044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Allison SJ, Milner J. SIRT3 is pro-apoptotic and participates in distinct basal apoptotic pathways. Cell Cycle 2007; 6:2669-77; PMID:17957139; http://dx.doi.org/ 10.4161/cc.6.21.4866 [DOI] [PubMed] [Google Scholar]
- 11. Inoue T, Hiratsuka M, Osaki M, Oshimura M. The molecular biology of mammalian SIRT proteins: SIRT2 in cell cycle regulation. Cell Cycle 2007; 6:1011-8; PMID:17457050; http://dx.doi.org/ 10.4161/cc.6.9.4219 [DOI] [PubMed] [Google Scholar]
- 12. Lain S, Hollick JJ, Campbell J, Staples OD, Higgins M, Aoubala M, McCarthy A, Appleyard V, Murray KE, Baker L, et al. Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell 2008; 13:454-63; PMID:18455128; http://dx.doi.org/ 10.1016/j.ccr.2008.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Grozinger CM, Chao ED, Blackwell HE, Moazed D, Schreiber SL. Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening. J Biol Chem 2001; 276:38837-43; PMID:11483616; http://dx.doi.org/ 10.1074/jbc.M106779200 [DOI] [PubMed] [Google Scholar]
- 14. Napper AD, Hixon J, McDonagh T, Keavey K, Pons JF, Barker J, Yau WT, Amouzegh P, Flegg A, Hamelin E, et al. Discovery of indoles as potent and selective inhibitors of the deacetylase SIRT1. J Med Chem 2005; 48:8045-54; PMID:16335928; http://dx.doi.org/ 10.1021/jm050522v [DOI] [PubMed] [Google Scholar]
- 15. Bouras T, Fu M, Sauve AA, Wang F, Quong AA, Perkins ND, Hay RT, Gu W, Pestell RG. SIRT1 deacetylation and repression of p300 involves lysine residues 1020/1024 within the cell cycle regulatory domain 1. J Biol Chem 2005; 280:10264-76; PMID:15632193; http://dx.doi.org/ 10.1074/jbc.M408748200 [DOI] [PubMed] [Google Scholar]
- 16. Han Y, Jin YH, Kim YJ, Kang BY, Choi HJ, Kim DW, Yeo CY, Lee KY. Acetylation of Sirt2 by p300 attenuates its deacetylase activity. Biochem Biophys Res Commun 2008; 375:576-80; PMID:18722353; http://dx.doi.org/ 10.1016/j.bbrc.2008.08.042 [DOI] [PubMed] [Google Scholar]
- 17. Byles V, Chmilewski LK, Wang J, Zhu L, Forman LW, Faller DV, Dai Y. Aberrant cytoplasm localization and protein stability of SIRT1 is regulated by PI3K/IGF-1R signaling in human cancer cells. Int J Biol Sci 2010; 6:599-612; PMID:20941378; http://dx.doi.org/ 10.7150/ijbs.6.599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tanno M, Sakamoto J, Miura T, Shimamoto K, Horio Y. Nucleocytoplasmic Shuttling of the NAD+-dependent Histone Deacetylase SIRT1. J Biol Chem 2007; 282:6823-32; PMID:17197703; http://dx.doi.org/ 10.1074/jbc.M609554200 [DOI] [PubMed] [Google Scholar]
- 19. Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front Oncol 2014; 4:64; PMID:24782981; http://dx.doi.org/ 10.3389/fonc.2014.00064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Khaled M, Larribere L, Bille K, Ortonne J-P, Ballotti R, Bertolotto C. Microphthalmia associated transcription factor is a target of the phosphatidylinositol-3-Kinase pathway. 2003; 121:831-6; PMID:14632202 [DOI] [PubMed] [Google Scholar]