

Abstract
Organ development requires well-established intercellular communication to coordinate cell proliferations and differentiations. MicroRNAs (miRNAs) are small, non-coding RNAs that can broadly regulate gene expression and play a critical role in the organ development. In this study, we found that miRNAs could pass through gap junctions between native cochlear supporting cells to play a role in the cochlear development. Connexin26 (Cx26) and Cx30 are predominant isoforms and co-express in the cochlea. Cx26 deficiency but not Cx30 deficiency can cause cochlear developmental disorders. We found that associated with Cx26 deletion induced the cochlear developmental disorders, deletion of Cx26 but not Cx30 disrupted miRNA intercellular transfer in the cochlea, although inner ear gap junctions still retained permeability after deletion of Cx26. Moreover, we found that deletion of Cx26 but not Cx30 reduced miR-96 expression in the cochlea during postnatal development. The reduction is associated with the cochlear tunnel developmental disorder in Cx26 knockout (KO) mice. These data reveal that Cx26-mediated intercellular communication is required for cochlear development and that deficiency of Cx26 can impair miRNA-mediated intercellular genetic communication in the cochlea, which may lead to cochlear developmental disorders and eventually congenital deafness as previously reported.
Tissue homeostasis and organ development rely on the well-orchestrated integration of intercellular communication and gene regulation to synchronize and coordinate cell proliferation and differentiation1. Gap junctions are intercellular channels and represent the only selective intercellular conduit that possesses a large pore size (1.0–1.5 nm), allowing direct exchange of ions and small molecules between cells2. It has been reported that small regulatory RNAs, such as siRNAs and miRNAs, can also pass through gap junctions3,4,5,6,7,8,9,10, which provides a novel mechanism for intercellular genetic communication11. In particular, miRNAs are single-stranded RNAs consisting of ~21 nucleotides and can broadly modulate gene expression by affecting the translation of mRNAs to proteins and mRNA target decay12,13,14,15. To date, approximately 300 conserved miRNA families and thousands of additional poorly conserved miRNAs have been identified in mammals. Approximately two thirds of all human protein-coding genes are conserved targets of miRNAs13,14. Thus, miRNAs provide a widespread mechanism for post-transcriptional control of gene expression and are important for the organ development.
Gap junctions have a crucial role in hearing. Connexin26 (Cx26, GJB2) mutations cause most cases of hereditary genetic deafness, responsible for >50% of nonsyndromic hearing loss16,17. Recently, we found that Cx26 deficiency can cause cochlear developmental disorders leading to congenital deafness18,19. However, the underling mechanism for developmental disorders remains unclear. In this study, we found that miRNAs can pass through gap junctions in the cochlea. Cx26 and Cx30 are predominant connexin isoforms in the cochlea20,21. Associated with Cx26 deficiency induced cochlear developmental disorders, Cx26 deficiency but not Cx30 deletion disrupted miRNA-mediated intercellular genetic communication in the cochlea.
Results
Gap junction and Cx26 and Cx30 expression in the cochlea
The organ of Corti has hair cells and supporting cells (Fig. 1a). The auditory sensory hair cells have no gap junctional coupling and connexin expression (Fig. 1f and also see ref. 20, 21). Gap junctions and connexin expression only existed in supporting cells (Fig. 1c–f). The organ of Corti contains four types of supporting cells, i.e., Deiters cells (DC), pillar cells (PC), Hensen cells (HC), and Claudius cells (CC) (see Supplementary Fig. S1). All of them had Cx26 and Cx30 expression and were well-coupled (Fig. 1 and also see ref. 20,21).
Figure 1. Cochlear structure and co-expression of Cx26 and Cx30 in the cochlea.

(a) Schematic drawing of the cochlear structure in the cross-section. PC: Pillar cell, DC: Deiters cell, HC: Hensen cell, CC: Claudius cell. (b–e) Immunofluorescent staining for Cx26 (green) and Cx30 (red) in the cochlea. A white arrow in panel (b) indicates the cochlear tunnel. TM: tectorial membrane. (f) A high-magnitude image in the organ of Corti. Outer hair cells were visualized by prestin labeling (red). Scale bars: 25 μm in (b–e), 10 μm in (f).
Blockage of miRNA intercellular transfer by gap junctional blockers
The intercellular transfer of miRNAs between cochlear supporting cells could be blocked by gap junctional blockers. Fig. 3 shows that application of 50 μM 18α-glycyrrhetinic acid (18-AGA) or 0.1 mM carbenoxolone (CBX) gap junctional blockers blocked miR-F diffusion between cells. The injected miR-F nucleotides were restricted within the injected cell and did not diffuse into the adjacently-contacted cells. Gap junction blocker blocked not only miR-F diffusion but also dye ethidium bromide (EB) diffusion between cells (Fig. 3d).
Figure 3. Blockage of miRNA and EB diffusion between the cochlear supporting cells by gap junctional blockers.
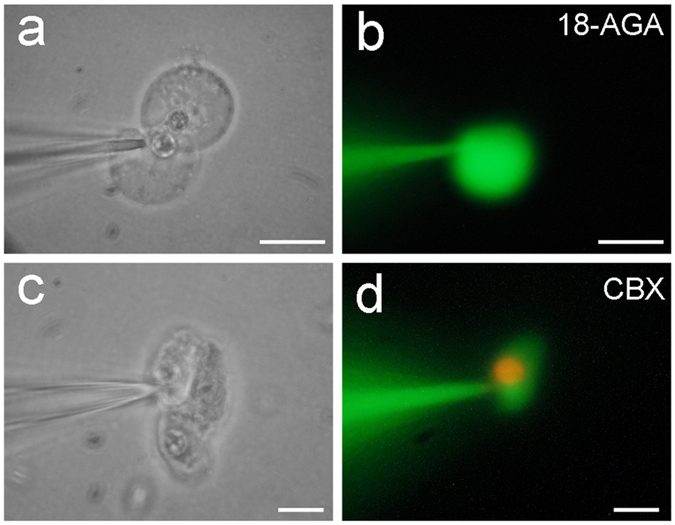
(a,b) Blockage of intercellular diffusion of miR-F between Hensen cells by application of 50 μM 18-AGA. (c,d) Intercellular difussion of miR-F and EB between Claudius cells was blocked by application of 0.1 mM CBX. All images were captured after injection for 30 min. Scale bars: 10 μm.
Cochlear development disorders and disruption of miRNA intercellular transfer in Cx26 KO mice
As previously reported18,22, deletion of Cx26 could induce cochlear developmental disorders (Fig. 4). The tectorial membrane was attached to the inner sulcus cells and the cochlear tunnel was filled (Fig. 4e). Deletion of Cx26 also disrupted intercellular transfer of miRNAs in the cochlea (Fig. 5). The injected miR-F was restricted to the injected cell (Fig. 5c–f). In all 12 injections, no intercellular diffusion of miR-F was visible. However, deletion of Cx26 did not completely disrupt inner ear gap junctions, which still retained permeability to dye EB (Fig. 5g,h). Input capacitance (Cin) recording also indicated that cochlear supporting cells in Cx26 KO mice still retained good gap junctional coupling. Cin in the recording Hensen cells in Fig. 5e,f and Claudius cells in Fig. 5g,h was ~75 pF and 19.3 pF, respectively, showing that they were well-coupled.
Figure 4. Cx26 deletion in the cochlear sensory epithelium and developmental disorders in Cx26 KO mice.

(a,b) Immunofluorescent staining for Cx26 in the cochlear sensory epithelium of WT mice. (c,d) Immunofluorescent staining for Cx26 (green) and Cx30 (red) of the cochlear sensory epithelium in the Cx26 KO mice. No Cx26 labeling is visible but Cx30 labeling remains. (e,f) Cochlear developmental disorders in Cx26 KO mice. The tectorial-membrane attaches to the inner sulcus cells (indicated by arrow heads) and the cochlear tunnel is filled (indicated by a black arrow). Outer hair cells (OHCs) are visualized by prestin labeling (red). Immunofluorescent staining for Cx26 (green) is negative. Scale bars: 100 μm in (a–d), 25 μm in (e,f).
Figure 5. Disruption of the intercellular transfer of miRNAs between cochlear supporting cells in Cx26 KO mice.

(a,b) Intercellular transfer of miR-F in the mouse cochlear sensory epithelium. The injection site (indicated by a red asterisk in panel (a)) locates at the Hensen cell region in the cochlear sensory epithelium. (c,d) Disruption of miR-F intercellular transfer in the cochlear sensory epithelium in Cx26 KO mice. A red asterisk in panel c indicates the injection site, where locates at the Hensen cell region. (e,f) Disruption of intercellular transfer of miRNA between cochlear supporting cells in Cx26 KO mice in the isolated cell preparation. The injected miR-F is limited in the injected Hensen cell. Cin is ~75 pF, indicating that these cells are well-coupled by gap junctions. (g,h) Disruption of intercellular diffusion of miR-F but not EB between cochlear supporting cells. The pipette was filled with a mixture of miR-F and EB. An arrow indicates that a neighboring Claudius cell only has red EB labeling but no miR-F labeling. Cin is 19.3 pF, indicating that two cells are coupled. All images were captured after injection for 30 min. Scale bars: 25 μm in (a–d), 10 μm in (e–h).
Normal cochlear development and miRNA intercellular transfer in Cx30 KO mice
Cx30 is co-expressed with Cx26 in the cochlea (Fig. 1, and also see ref. 20, 21). However, deletion of co-expressed Cx30 displayed normal cochlear development (Fig. 6a–b). Intercellular transfer of miR-F also appeared normal in Cx30 KO mice and intercellular diffusion of miR-F among supporting cells was visible (Fig. 6c–f).
Figure 6. Normal cochlear development and miRNA intercellular transfer in the cochlea in Cx30 KO mice.

(a–b) Normal cochlear development in the Cx30 KO mice. A white arrow in panel (a) indicates the open cochlear tunnel. Panel (b) shows immunofluorescent staining for Cx30. No Cx30 labeling is visible. (c–d) Intercellular transfer of miR-F between cochlear supporting cells in Cx30 KO mice. Cin = 43 pF. (e–f) Intercellular transfer of miR-F and EB between a pair of Hensen cells in Cx30 KO mice. Cin = 35.5 pF. All images were captured after injection for 30 min. Scale bars: 25 μm (a–b), 10 μm in (c–f).
Reduction of miRNA expression in Cx26 KO mice during cochlear postnatal development
MicroRNA-96 is critical for cochlear development23. In mouse postnatal development, the cochlear tunnel starts to open at postnatal day 5 (P5) and fully opens at P10 (Fig. 7a). We found that prior to the cochlear tunnel opening, the expression of miR-96 in the cochlea was increased at P3 (Fig. 7b). Then, the expression decreased and reached a steady state at P10. However, the expression of miR-96 in Cx26 KO mice was not increased at P3 and remained at lower level during the postnatal period (Fig. 7b). On the other hand, the expression of miR-96 in the cochlea in Cx30 KO mice, which displayed normal cochlear development (Fig. 6a), was similar to WT mice, increasing at P3 and then reducing afterward during the postnatal development (Fig. 7b). There was no significant difference in miR-96 expression between Cx30 KO mice and WT mice (P = 0.43, one-way ANOVA).
Figure 7. Cochlear tunnel development and expression of miR-96 in the cochlea during cochlear postnatal development.
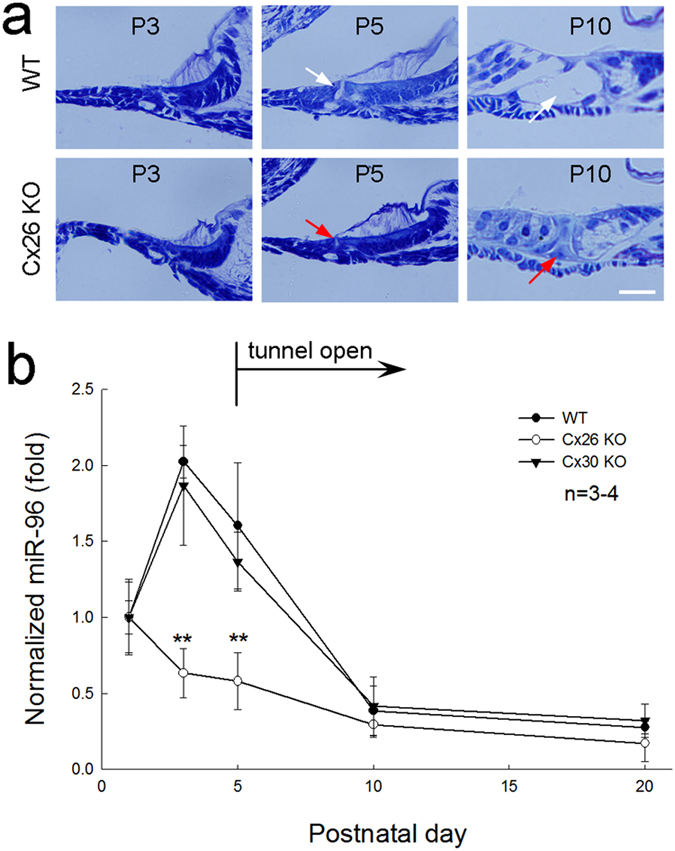
(a) Postnatal development of the organ of Corti in wild-type (WT) and Cx26 KO mice. The cochlear tunnel occurs at postnatal day 5 (P5) in WT mice (indicated by white arrows) but is filled in the Cx26 KO mice (indicated by red arrows). Scale bars: 20 μm. (b) Dynamic changes of miR-96 expression in the Cx26 KO, Cx30 KO, and WT mouse cochlea during postnatal development. WT littermates were used as control. The expression levels were normalized to that at P1 for comparison of dynamic changes. **P < 0.001, one-way ANOVA with a Bonferroni correction.
Discussion
In this study, we found that miRNAs could pass through gap junctions between native cochlear supporting cells (Figs 2, 5a,b and Supplementary Fig. S2). Deletion of Cx26 disrupted cochlear development and miRNA intercellular transfer in the cochlea (Figs. 4 and 5). However, the inner ear gap junctions still remainted permeability to cationic dye EB after Cx26 deletion (Fig 5g,h). Deletion of Cx26 also reduced miR-96 expression in the cochlea during postnatal development and the reduction is associated with over-development of the cochlear tunnel (Fig. 7). However, consistent with the normal cochlear development in Cx30 KO mice, deletion of Cx30 did not affect intercellular transfer of miRNA and miR-96 expression in the cochlea (Figs 6 and 7b). We previously reported that miRNAs can pass through gap junctional channels and regulate gene expression in neighboring cells to achieve intercellular genetic communication11. Our new data further demonstrate that this gap junction-mediated miRNA intercellular communication may have an important role in the cochlear development.
Figure 2. Time-lapse recording of intercellular transfer of miR-F between cochlear supporting cells.

(a–d) Diffusion of miR-F between Hensen cells. The miR-F nucleotides were injected into a Hensen cell by the patch pipette. Arrows indicate a neighboring cell with miR-F labeling. (e–h) Intercellular diffusion of miR-F and EB between Claudius cells. The patch pipette was filled with a mixture of miR-F and EB. EB labeling is mainly visible at cell nuclei because of EB binding to DNAs. Scale bars: 10 μm.
Deletion of Cx26 can result in filling of the cochlear tunnel and attachment of the tectorial membrane to inner sulcus cells leading to loss of the under-tectorial-membrane space (Figs 4e,7a, and also see ref. 17, 18, 19,22). Currently, the underlying mechanism remains unclear. In this experiment, we found that prior to the cochlear tunnel opening, miR-96 expression had a rapid increase at P3 (Fig. 7). This peak was missed in Cx26 KO mice but not in Cx30 KO mice (Fig. 7b). We hypothesize that this up-regulation of miRNA expression may be associated with arresting of cochlear supporting cell differentiation leading to formation of the cochlear tunnel. Deletion of Cx26 impaired intercellular transfer of miRNAs between cochlear supporting cells (Fig. 5), which may lead to over-proliferation and differentiation of cochlear supporting cells inducing over-development of the cochlear tunnel and loss of the under-tectorial-membrane space. This concept is further supported by the fact that deletion of Cx26 disrupted miRNA permeability but not EB permeability in the cochlea (Fig. 5). This implies that cochlear gap junctions in Cx26 KO mice still retain permeability to ions and other small molecules, since co-expressed Cx30 expression remains (Fig. 4d, and also see ref. 24). Thus, some of gap junctional functions in the cochlea, such as K+-recycling, in Cx26 KO mice may still remain normal. These data also further support our previous reports that Cx26 deficiency impairs cochlear developmental disorders and active cochlear amplification rather than K+-recycling resulting in hearing loss17,18,19,24,25.
In the experiment, we found that deletion of Cx30 did not affect miRNA permeability in the cochlea (Fig. 6) and had no influence on miR-96 expression (Fig. 7b). This is consistent with our previous report that inner ear gap junctions have strong charge-selectivity and that Cx26 is mainly responsible for anionic permeability in the cochlea26. MicroRNAs are anionic at physiological pH. It has been found that Cx30 channels are impermeable to anionic molecules27,28 and miRNAs11. Thus, deletion of Cx30 could have little effect on permeability to miRNAs in the cochlea (Fig. 6). These data also suggest that Cx26 may have a critical role not only in intercellular signaling in the cochlea26 but also in intercellular genetic communication and development in the cochlea. This may be a reason why Cx26 rather than Cx30 deletion can induce cochlear developmental disorders.
Cx30 deficiency can also induce hearing loss17,29,30,31. However, hearing loss mainly results from endocochlear potential (EP) reduction17,19,29 rather than from cochlear developmental disorders as shown in Cx26 deficiency.
MicroRNAs provide a widespread mechanism for post-transcriptional control of gene expression. Gene expression can be regulated by many factors at many stages, such as enhancer and promoter, transcription factors, and mRNA polyA polymerization. However, none of these regulatory factors is intercellular-exchangeable through gap junctions except small non-coding RNAs such as miRNA. Gap junctions extensively exist in almost all cell types and organs. Recently, it has been found that miRNAs can be exchanged between tumor cells in a gap junction-dependent manner7,8,9. Thus, gap junction mediated intercellular genetic communication can play an important role in organ development and may also be important for tumor genesis or inhibition.
Methods
Cx26 KO and Cx30 KO mice and genotyping
Cx26 KO mice were generated by crossing Cx26loxP/loxP mice (European Mouse Mutant Archive, EM00245) with the Pax2-Cre mouse line (the Mutation Mouse Regional Center, Chapel Hill, NC)18. The Cx26 floxed allele was detected on tail genomic DNA by PCR amplification using the following primers: Cx26F: 5′-CTT TCC AAT GCT GGT GGAGTG-3′ and Cx26R: 5′-ACA GAA ATG TGT TGG TGA TGG-3′18. Cx26loxP/loxP and wild-type (WT) mice generated 400 and 300 bps bands, respectively. For the Pax2-Cre transgene, the following primers were used: CreF: 5′-GCC TGC ATT ACC GGT CGA TGC AAC GA- 3′ and CreR: 5′-GTG GCA GAT GGC GCG GCA ACA CCA TT- 3′. The band size was 700 bps. Cx30 KO mice19,29 were also purchased from EMMA (EM000323). Primer pairs for detecting Cx30 KO were Cx30 KO-1 (LACZ e Neo): 5′-GGT ACC TTC TAC TAA TTA GCT TGG -3′; Cx30 KO2 (LACZ e Neo): 5′-AGG TGG TAC CCA TTG TAG AGG AAG -3′; Cx30 KO-3 (LACZ e Neo) 5′-AGC GAG TAA CAA CCC GTC GGA TTC -3′. The bands of Cx30 KO and WT mice were 460 and 544 bps, respectively.
The experimental procedures were approved by the University of Kentucky′s Animal Care & Use Committee and conducted according to the standards of the NIH Guidelines for the Care and Use of Laboratory Animals.
Cochlear cell isolation and intracellular injection
Adult mice (30–60 day old) were decapitated and the temporal bone was removed. As we previously reported32,33,34,35, the otic capsule was opened and the cochlea was isolated by micro-dissection in a standard extracellular solution (142 NaCl, 5.37 KCl, 1.47 MgCl2, 2 CaCl2, 10 HEPES in mM, 300 mOsm, pH 7.2). The sensory epithelium was micro-dissected by a sharpened needle. The isolated sensory epithelium was dissociated by trypsin (1 mg/ml) for 3–5 min32,33,34,35. The dissociated cells were then transferred to a dish for recording. The cochlear supporting cells and hair cells can be unambiguously identified under microscope by their own morphological shapes (Fig. S1, and also see ref. 26,32,33). The dissociated supporting cells also retained good gap junctional coupling33,34,35.
To assess intercellular permeation of gap junctions to miRNAs, a fluorescence-tagged miRNA (miR-F), which is constructed by a 25 nt miRNA (5′-CCT CTT ACC TCA GTT ACA ATT TATA-3′) labeled with carboxyfluorescein on its 3′ end (Gene Tools, Inc. OR), was used. This miR-F was proven to not be hybridized or degraded and also had no fluorescent tag removal in the cytoplasm36,37.
For dye injection to assess intercellular diffusion, a group or pair of cochlear supporting cells was selected. Intracellular injection was performed by patch clamp recording under the whole-cell configuration26. The patch pipette was 1.5–2 μm in tip diameter and filled with the normal intracellular solution (KCl 140, EGTA 5, and HEPES 10 in mM, pH 7.2 and 300 mOsm) with 100 μM miR-F. The holding voltage was set at −40 mV. Gap junctional coupling between cells was continuously monitored by input capacitance (Cin), which was recorded online at 1–3 Hz and calculated from the transient charge elicited by small (−10 mV) test pulses at the holding potential33,34. The diffusion was captured with a CCD camera under a fluorescence microscope (Nickon, TE300) as we previously reported26.
In some cases, cationic dye ethidium bromide (EB, 0.1 mM) was also used and mixed with miR-F for injection. EB can distinctly identify the transjunctional-diffused cells and clearly demonstrate transjunctional transport, because it can bind to DNAs labeling cell nuclei showing bright fluorescence.
Immunofluorescent staining
The immunofluorescent staining was performed as previously reported21,38. The cochlear section or culture cells were fixed with 4% paraformaldehyde for 30 min and washed out with PBS. After 30 min of incubation in a blocking solution (10% goat serum and 1% BSA in PBS) with 0.1% Triton X-100, the cochlear section or culture cells were incubated with monoclonal mouse anti-Cx26 (1: 400, Cat#33–5800, Invitrogen) in the blocking solution at 4 oC overnight. For double immunofluorescent staining for Cx26 and Cx30, polyclonal rabbit anti-Cx30 (1:400, Cat#71–2200, Invitrogen), or polyclonal goat anti-prestin (1:50, Cat# sc-22694, Santa Cruz Biotech Inc, CA) was used. After being washed with PBS, the section or cells were incubated with corresponding Alexa Fluor 488- or 568-conjugated goat anti-mouse IgG and Alexa Fluor 568-conjugated goat anti-rabbit IgG (1:500, Molecular Probes) in the blocking solution at room temperature (23 oC) for 1 hr. In some cases, following the 2nd antibody incubation, the section or cells were stained by 4′, 6-diamidino-2-phenylindole (DAPI, 0.1 mg/ml, D1306; Molecular Probes) for ~15–20 min to visualize cell nuclei. After completely washing out with PBS, the section or cells were mounted with a fluorescence mounting medium (H-1000, Vector Lab, CA) and observed under a fluorescence microscope (Nickon, T2000) or a confocal microscope (Leica TCS SP2). The fluorescent image was saved in the TIFF format and assembled in Photoshop (Adobe Systems, CA) for presentation.
miRNA extraction and quantitative PCR measurement
The cochlear sensory epithelia were freshly isolated as described above and miRNAs were extracted by mirVana miRNA Isolation Kit (AM1560, Ambion, USA) following manufacturer’s instructions. The purity and quantity of miRNA was determined by a NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies, Inc., Rockland, DE). Then, miRNAs were converted to cDNA using TaqMan® MicroRNA Reverse Transcription Kit (#4366596, Applied Biosystems, CA, USA) with corresponding mouse-specific miRNA reverse transcription templates according to manufacturer’s instructions and measured by use of MyiQ real-time PCR detection system (Bio-Rad Laboratories) with TaqMan® MicroRNA Assay (Applied Biosystems, CA, USA). An internal standard U6 snRNA (#001973, Applied Biosystems, CA) was used as an internal control. The relative quantity of miRNA expression was calculated from the standard curve39 and normalized to the amount of the internal standard U6 snRNA.
Data analysis
Data were expressed as mean ± s.e.m. and plotted by SigmaPlot (SPSS Inc. Chicago, IL). The statistical analyses were performed by SPSS v18.0 (SPSS Inc. Chicago, IL) using one-way ANOVA with a Bonferroni correction.
Additional Information
How to cite this article: Zhu, Y. et al. Connexin26 gap junction mediates miRNA intercellular genetic communication in the cochlea and is required for inner ear development. Sci. Rep. 5, 15647; doi: 10.1038/srep15647 (2015).
Supplementary Material
Acknowledgments
We are grateful to Dr Garrett Soukup at Creighton University for helpful discussions and comments on the manuscript. This work was supported by NIDCD R01-05989
Footnotes
The authors declare no competing financial interests.
Author Contributions Y.Z., L.Z., L.M. and H.B.Z. designed and performed experiments and analyzed data. H.B.Z. wrote paper.
References
- Mittelbrunn M. & Sánchez-Madrid F. Intercellular communication: diverse structures for exchange of genetic information. Nat. Rev. Mol. Cell Biol. 13, 328–335 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris A. L. Emerging issues of connexin channels: biophysics fills the gap. Q. Rev. Biophys. 34, 325–472 (2001). [DOI] [PubMed] [Google Scholar]
- Valiunas V. et al. Connexin-specific cell-to-cell transfer of short interfering RNA by gap junctions. J. Physiol. 568, 459–468 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brink P. R., Valiunas V., Gordon C., Rosen M. R. & Cohen I. S. Can gap junctions deliver? Biochim. Biophys. Acta. 1818, 2076–2081 (2012). [DOI] [PubMed] [Google Scholar]
- Wolvetang E. J., Pera M. F. & Zuckerman K. S. Gap junction mediated transport of shRNA between human embryonic stem cells. Biochem. Biophys. Res. Commun. 363, 610–615 (2012). [DOI] [PubMed] [Google Scholar]
- Kizana E., Cingolani E. & Marbán E. Non-cell-autonomous effects of vector-expressed regulatory RNAs in mammalian heart cells. Gene Ther. 16, 1163–1168 (2009). [DOI] [PubMed] [Google Scholar]
- Katakowski M., Buller B., Wang X., Rogers T. & Chopp M. Functional microRNA is transferred between glioma cells. Cancer Res. 70, 8259–8263 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory L. A., Ricart R. A., Patel S. A., Lim P. K. & Rameshwar P. MicroRNAs, Gap Junctional Intercellular Communication and Mesenchymal Stem Cells in Breast Cancer Metastasis. Curr. Cancer Ther. Rev. 7, 176–183 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim P. K. et al. Gap junction-mediated import of microRNA from bone marrow stromal cells can elicit cell cycle quiescence in breast cancer cells. Cancer Res. 71, 1550–1560 (2011). [DOI] [PubMed] [Google Scholar]
- van Rooij E., Purcell A. L. & Levin A. A. Developing microRNA therapeutics. Circ. Res. 110, 496–507 (2012). [DOI] [PubMed] [Google Scholar]
- Zhao H. B., Zhu Y., Zong L. & Liang R. Q. Gap junction mediated miRNA intercellular communication. The 36th Association Research in Otolaryngology Annual Meeting. Baltimore, MD, USA. Mt. Royal, NJ: Mira Digital Publish (2013, Feb. 16-20).
- Ambros V. The functions of animal microRNAs. Nature 431, 350–355 (2004). [DOI] [PubMed] [Google Scholar]
- Bartel D. P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297 (2004). [DOI] [PubMed] [Google Scholar]
- Bartel D. P. MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L. & Hannon G. J. MicroRNAs: small RNAs with a big role in gene regulation. Nat. Rev. Genet. 5, 522–531 (2004). [DOI] [PubMed] [Google Scholar]
- Castillo F. J. & Castillo I. The DFNB1 subtype of autosomal recessive non-syndromic hearing impairment. Front. Biosci. 17, 3252–3274 (2011). [DOI] [PubMed] [Google Scholar]
- Wingard J. C. & Zhao H. B. Cellular and deafness mechanisms underlying connexin mutation induced hearing loss – A common hereditary deafness. Front. Cell. Neurosci. 9, 202 (2015). doi: 10.3389/fncel.2015.00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang C., Zhu Y., Zong L., Lu G. J. & Zhao H. B. Cell degeneration is not a primary causer for Connexin26 (GJB2) deficiency associated hearing loss. Neurosci. Lett. 528, 36–41 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Chen J., Zhu Y., Liang C. & Zhao H. B. Deafness induced by Connexin26 (GJB2) deficiency is not determined by endocochlear potential (EP) reduction but is associated with cochlear developmental disorders. Biochem. Biophys. Res. Commun. 448, 28–32 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forge A. et al. Gap junctions in the inner ear: comparison of distribution patterns in different vertebrates and assessement of connexin composition in mammals. J. Comp. Neurol. 467, 207–231 (2003). [DOI] [PubMed] [Google Scholar]
- Zhao H. B. & Yu N. Distinct and gradient distributions of connexin26 and connexin30 in the cochlear sensory epithelium of guinea pigs. J. Comp. Neurol. 499, 506–518 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y. et al. Targeted connexin26 ablation arrests postnatal development of the organ of Corti. Biochem. Biophys. Res. Commun. 385, 33–37 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conte I., Banfi S. & Bovolenta P. Non-coding RNAs in the development of sensory organs and related diseases. Cell Mol. Life Sci. 70, 4141–4155 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y. et al. Connexin26 (GJB2) deficiency reduces active cochlear amplification leading to late-onset hearing loss. Neuroscience 284, 719–729 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y. et al. Active cochlear amplification is dependent on supporting cell gap junctions. Nat. Commun. 4, 1786 (2013). doi: 10.1038/ncomms2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H. B. Connexin26 is responsible for anionic molecule permeability in the cochlea for intercellular signaling and metabolic communications. Eur. J. Neurosci. 21, 1859–1868 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manthey D. et al. Intracellular domains of mouse connexin26 and -30 affect diffusional and electrical properties of gap junction channels. J. Membr. Biol. 181, 137–148 (2001). [DOI] [PubMed] [Google Scholar]
- Beltramello M. et al. Permeability and gating properties of human connexins 26 and 30 expressed in HeLa cells. Biochem. Biophys. Res. Commun. 305, 1024–1033 (2003). [DOI] [PubMed] [Google Scholar]
- Teubner B. et al. Connexin30 (Gjb6)-deficiency causes severe hearing impairment and lack of endocochlear potential. Hum. Mol. Genet. 12, 13–21 (2003). [DOI] [PubMed] [Google Scholar]
- Schütz M. et al. The human deafness-associated connexin 30 T5M mutation causes mild hearing loss and reduces biochemical coupling among cochlear non-sensory cells in knock-in mice. Hum. Mol. Genet. 19, 4759–4773 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W. H. et al. A novel missense mutation in the connexin30 causes nonsyndromic hearing loss. PLoS One 6, e21473. doi: 10.1371/journal.pone.0021473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y. & Zhao H. B. ATP-mediated potassium recycling in the cochlear supporting cells. Purinergic Signal. 6, 221–229 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y. & Zhao H. B. ATP activates P2X receptors to mediate gap junctional coupling in the cochlea. Biochem. Biophys. Res. Commun. 426, 528–532 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H. B. & Santos-Sacchi J. Effect of membrane tension on gap junctional conductance of supporting cells in Corti's organ. J. Gen. Physiol. 112, 447–455 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H. B. & Santos-Sacchi J. Voltage gating of gap junctions in cochlear supporting cells: evidence for nonhomotypic channels. J. Membr. Biol. 175, 17–24 (2000). [DOI] [PubMed] [Google Scholar]
- Mudziak R. M. et al. Resistance of morpholino phosphorodiamidate oligomers to enzymatic degradation. Antisense Nucl. Acid. Drug Dev. 6, 267–272 (1996). [DOI] [PubMed] [Google Scholar]
- Summerton J. & Weller D. Morpholino antisense oligmers: design, preparation and properties. Antisense Nucl. Acid. Drug Dev. 7, 187–195 (1997). [DOI] [PubMed] [Google Scholar]
- Liu Y. P. & Zhao H. B. Cellular characterization of Connexin26 and Connnexin30 expression in the cochlear lateral wall. Cell Tissue Res. 333, 395–403 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu N. et al. Prestin up-regulation in chronic salicylate (aspirin) administration: an implication of functional dependence of prestin expression. Cell Mol. Life Sci. 65, 2407–2418 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.