

Abstract
Spindle poles are defined by centrosomes; therefore, an abnormal number or defective structural organization of centrosomes can lead to loss of spindle bipolarity and genetic integrity. Previously, we showed that Tpr (translocated promoter region), a component of the nuclear pore complex (NPC), interacts with Mad1 and dynein to promote proper chromosome segregation during mitosis. Tpr also associates with p53 to induce autophagy. Here, we report that Tpr depletion induces mitotic catastrophe and enhances the rate of tetraploidy and polyploidy. Mechanistically, Tpr interacts, via its central domain, with Aurora A but not Aurora B kinase. In Tpr-depleted cells, the expression levels, centrosomal localization and phosphorylation of Aurora A were all reduced. Surprisingly, an Aurora A inhibitor, Alisertib (MLN8237), also disrupted centrosomal localization of Tpr and induced mitotic catastrophe and cell death in a time- and dose-dependent manner. Strikingly, over-expression of Aurora A disrupted Tpr centrosomal localization only in cells with supernumerary centrosomes but not in bipolar cells. Our results highlight the mutual regulation between Tpr and Aurora A and further confirm the importance of nucleoporin function in spindle pole organization, bipolar spindle assembly, and mitosis; functions that are beyond the conventional nucleocytoplasmic transport and NPC structural roles of nucleoporins. Furthermore, the central coiled-coil domain of Tpr binds to and sequesters extra Aurora A to safeguard bipolarity. This Tpr domain merits further investigation for its ability to inhibit Aurora kinase and as a potential therapeutic agent in cancer treatment.
Keywords: alisertib, AURKA, centrosome, centriole, tpr
Introduction
The centrosome consists of a pair of centrioles surrounded by pericentriolar material (PCM). It serves as a microtubule-organizing center and controls vital cellular processes, including cell division and polarity. Numerical and/or structural abnormalities of centrosomes are associated with carcinogenesis, aneuploidy, cell cycle arrest, chromosomal instability and transformation, apoptotic cell death, and ciliopathies and brain disease.1 Normally, centrioles duplicate only once per cell cycle during interphase, through a process that relies on several specific centrosomal proteins with a sequential recruitment order.2 After nuclear envelope breakdown, and during mitosis, the size and microtubule (MT)-nucleating capacity of centrosomes increase dramatically as a result of PCM recruitment. More than 2 hundred proteins are involved in centrosome assembly, organization, and function while the PCM consists of γ-tubulin and a number of other proteins, many of which display coiled-coiled domains.3,4 However, the molecular details of how these coiled-coil proteins function in centrosome homeostasis are largely unknown.
The nuclear pore complex (NPC) consists of hundreds of copies of 30 different proteins called nucleoporins and functions as the mediator of exchange between the nucleus and the cytoplasm in eukaryotic cells.5,6 It is also involved in coordinating other nuclear processes, such as transcription regulation, chromatin silencing and DNA damage repair.7,8 Despite much progress, we still do not possess a complete high-resolution structure for the NPC. Furthermore, different NPC variants are suspected in different tissues and organisms and these have yet to be elucidated.7
The NPCs of vertebrate cells disassemble during prophase and the nuclear pore proteins (nucleoporins) are delivered to the mitotic cytoplasm. Accumulating evidence indicates that NPCs have active mitotic functions.9 Recently, we and others have shown that nucleoporins participate in spindle assembly, kinetochore organization, the spindle assembly checkpoint, centrosome homeostasis, and cytokinesis, all of which are processes that control chromosome segregation and are important for maintenance of genome integrity.8
The nucleoporin, Tpr (translocated promoter region), was initially identified as an oncogenic activator of the met and trk proto-oncogenes.10-12 Tpr is a component of the NPC that localizes to intranuclear filaments or nuclear baskets.13,14 Mammalian Tpr is a 267 kDa protein.15 It contains a ~1600-residue α-helical coiled-coil multiple heptad repeat or leucine zipper motif N-terminal domain, and a highly acidic, unstructured 800 amino acid carboxy terminus contains a nuclear localization sequence (NLS).8,11,16
We have previously shown that Tpr and checkpoint protein complexes (Mad1-Mad2) are transported poleward by dynein/dynactin complex molecular motors and that depleting Tpr causes a chromosome lagging phenotype.10 In addition, Tpr is suggested to spatially regulate Mad1/Mad2 proteostasis through the SUMO-isopeptidases, SENP1 and SENP2.17 Tpr-Mad1 might also provide a store of inhibitory signals that limits the speed of subsequent mitosis.18,19
However, Tpr is normally undetectable at kinetochores and is mainly localized on mitotic spindles and in spindle pole regions in mammalian cells.10,17 Moreover, proteomic studies in human4 and Drosophila20 identified Tpr peptides in centrosomes. The mechanism of Tpr recruitment to spindle poles/centrosomes and the functions of Tpr on centrosomes are largely unknown. Here, we report the unexpected centrosomal roles of coiled-coil protein, Tpr, and Aurora A kinase. Our findings identify a novel regulator of centrosome maturation, and also highlight a therapeutic strategy for killing tumor cells that over-express Aurora A kinase and that have high levels of Aurora A phosphorylation via Tpr. Our data suggest that a reduction in the level of normal Tpr at the NPC may lead to chromosomal instability21 and contribute considerably to the mechanism of tumorigenesis. Thus Tpr is a novel target for therapeutic intervention during tumor progression.
Results
Tpr depletion leads to mitotic catastrophe and enhances polyploidy cell formation
Previously, we observed chromosome lagging during cell cycle progression in HeLa cells stably expressing H2B-GFP and transfected with siRNA against Tpr.10 To determine whether or not lagged chromosomes caused by Tpr depletion would return to the main chromatids in late anaphase and telophase, we first monitored knock-down efficiencies by comparing immunoblots of lysates obtained from Tpr-specific siRNA-transfected cells with lysates from scrambled siRNA-transfected controls. We observed an approximate 90–95% decrease in Tpr levels in knock-down cells compared with controls (Fig. 1A). We next assessed the subcellular localization of chromosomes (using DAPI to stain DNA) during telophase and cytokinesis after Tpr depletion. Consistent with previous reports,10,22 more than 50% of Tpr-depleted cells in mitosis contained errors in chromosome segregation, and 34% of these cells had lagging chromosomes, whereas these defects were not found in control siRNA cells (n=200 mitotic cells) (Fig. 1B). Specifically, we found that Tpr depletion-induced lagging chromosomes remained lagging even after cytokinesis (Plk1 was used as a cytokinesis marker) and that the percentage of multinucleated and micronucleated cells increased dramatically. A marked increase in polyploid cells (approximately 21%, n=200 mitotic cells) was also observed (Fig. 1C), whereas only approximately 2% of control cells showed polyploidy. These observations suggested that lagging chromosomes caused by Tpr depletion enhanced polyploid cell formation and that modulation of Tpr might cause irreversible mitotic catastrophe.
Figure 1.
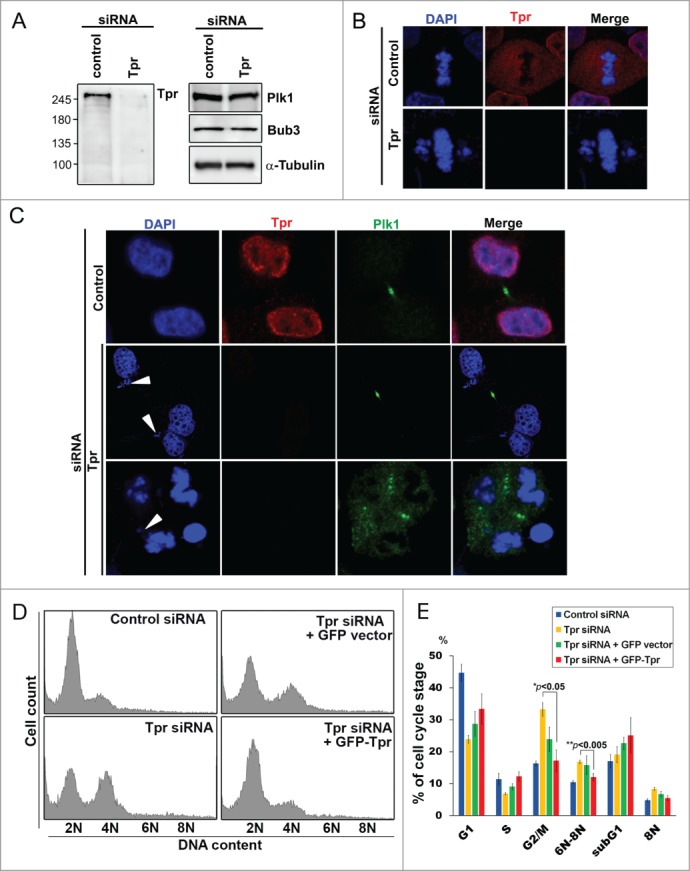
Tpr depletion induces mitotic catastrophe and enhances polyploidy. (A) HeLa cells were transfected with control siRNA or siRNA specific for Tpr (Tpr RNAi). Seventy-two hours post transfection, lysates of Tpr RNAi or control siRNA cells were analyzed by immunoblotting with the antibodies specific for Tpr (mouse anti-Tpr, sc-101294, from Santa Cruz Biotechnology); Plk1 (mouse anti-Plk1, sc-17783, from Santa Cruz Biotechnology) and Bub3 (mouse anti-Bub3, 611731, from BD Transduction Laboratories). The same membrane was stripped and re-probed with anti-α-tubulin [mouse α-tubulin (DM1A)T9026 from Sigma-Aldrich] (as loading control). Numbers indicate molecular mass markers in kilodaltons. (B) Confocal images of mitotic HeLa cells transfected with control or Tpr siRNA and analyzed 72 h post-transfection. Cells were analyzed by immunofluorescence using antibodies against Tpr (rabbit anti-Tpr, sc-67116, from Santa Cruz Biotechnology). Goat anti-mouse Alexa Fluor-488 or rabbit Rhodamine were used as secondary antibodies. DNA was counterstained using DAPI. (C) HeLa cells transfected with control or Tpr siRNA and analyzed 72 h post-transfection. Confocal images of multi-nuclei and polyploid cells that were often found after Tpr depletion, but not in control cells. Cells were stained and were analyzed by immunofluorescence using antibodies against Plk1 (mouse anti-Plk1, sc-17783, from Santa Cruz Biotechnology) (a telophase/cytokinesis marker) and Tpr (rabbit anti-Tpr, sc-67116, from Santa Cruz Biotechnology). Goat anti-mouse Alexa Fluor-488 or rabbit Rhodamine were used as secondary antibodies. DNA was counterstained using DAPI. White arrow heads indicate lagging chromosomes. (D) Ninety-six hours post-transfection with control or Tpr siRNAs, the cell-cycle profiles of HeLa cells were examined by flow cytometry. A control siRNA profile is shown (top, left). (E) Percentage of cells in cell-cycle phases after flow cytometry analysis. Asterisks indicate significant p values (*p < 0.05 or **p < 0.005).
Next, to further characterize the effects of Tpr depletion on cell cycle distribution, Tpr siRNA-treated and control cells were incubated for 4 days and their DNA content was then analyzed by flow cytometry. We noted that mock siRNA transfection did not affect the cell cycle of HeLa cells compared with untreated HeLa cells (Fig. 1D and E). Conversely, Tpr depletion caused a significant increase in the proportion of G2/M cells and reduced the number of G1 cells when compared with control cells (Fig. 1E). We also observed a 1.5-fold increase in 6N-8N cells (polyploidy, **p < 0.005) in Tpr-depleted cells compared with controls (Fig. 1E, G2/M is indicated as *p < 0.05). Moreover, to determine whether the observed Tpr depletion mitotic catastrophe phenotypes are different manifestations of the same defect or whether mitotic roles of Tpr can be uncoupled, we employed a rescue strategy by over-expressing siRNA-resistant GFP-Tpr (full-length) in Tpr knockdown cells. The mitotic catastrophe phenotype was partially rescued (Fig. 1E, rescue of 6N-8N cells is indicated as **p < 0.005). These data suggest that Tpr depletion induced mitotic catastrophe, impaired chromosome alignment and caused G2/M phase arrested and polyploidy formation, indicating a crucial role for Tpr in faithful cell cycle progression.
Tpr interacts with aurora a during cell division
Recently, we found that Tpr depletion increased nuclear accumulation of the tumor suppressor p53 and facilitated autophagy.23 To examine whether Tpr depletion affected the behavior of p53 during mitosis, we analyzed mitotic chromosome morphology and centrosome localization in synchronously growing cells that received either Tpr-specific siRNAs or control siRNA. In Tpr-depleted cells, chromosome lagging and “bending” defects were often found and, more importantly, p53 was mislocalized from centrosomes, as assessed by p53 immunostaining (Fig. S1). This indicated that knock-down of Tpr also impaired the centrosomal localization of p53 and enhanced polyploidy. It is nevertheless reasonable to speculate that there may be other proteins involved in regulating the centrosomal role of Tpr-p53 during mitosis to prevent irreversible mitotic catastrophe and polyploidy formation.
Our next focus was to identify the molecular mechanism underlying p53 mislocalization at the spindle pole regions and polyploidy formation after Tpr reduction. p53 regulates several mitotic kinases at centrosomes. One of these is Aurora A kinase, which has similar physiological and functional characteristics at spindle poles compared with those of Tpr. To assess whether Tpr was associated with Aurora A in mitosis, HeLa cells were synchronized by a double thymidine block, and immunoprecipitations were performed. Endogenous Aurora A but not Aurora B was present in the anti-Tpr immunoprecipitates, indicating that Tpr binds Aurora A in vivo (Fig. 2A). Conversely, using anti-Aurora A antibodies, we immunoprecipitated Tpr, but not other nucleoporins, such as Nup62 (Fig. 2B). Consistent with the immunoprecipitation data, we found that Tpr co-localized transiently with Aurora A at the spindle pole region (Fig. 2C), from metaphase to anaphase. To examine more closely the binding of Tpr to Aurora A, and to biochemically map the region of Tpr that interacts with Aurora A, we expressed 3 cDNA fragments covering the entire length of Tpr [named Tpr-N (1–774 aa), Tpr-M (775–1700 aa), and Tpr-C (1701–2351 aa)]10 in a cell-free reticulocyte translation system. Then, we performed Ni-NTA pulldown assays. Only the in vitro translated Tpr-M (775–1700 aa) fragment exhibited binding activity to recombinant His-Aurora A (Fig. 2D). These data suggest that a population of Aurora A directly interacted and co-localized with the large internal coiled-coil domain in Tpr-M in a transient manner at the metaphase–anaphase transition during mitosis.
Figure 2.
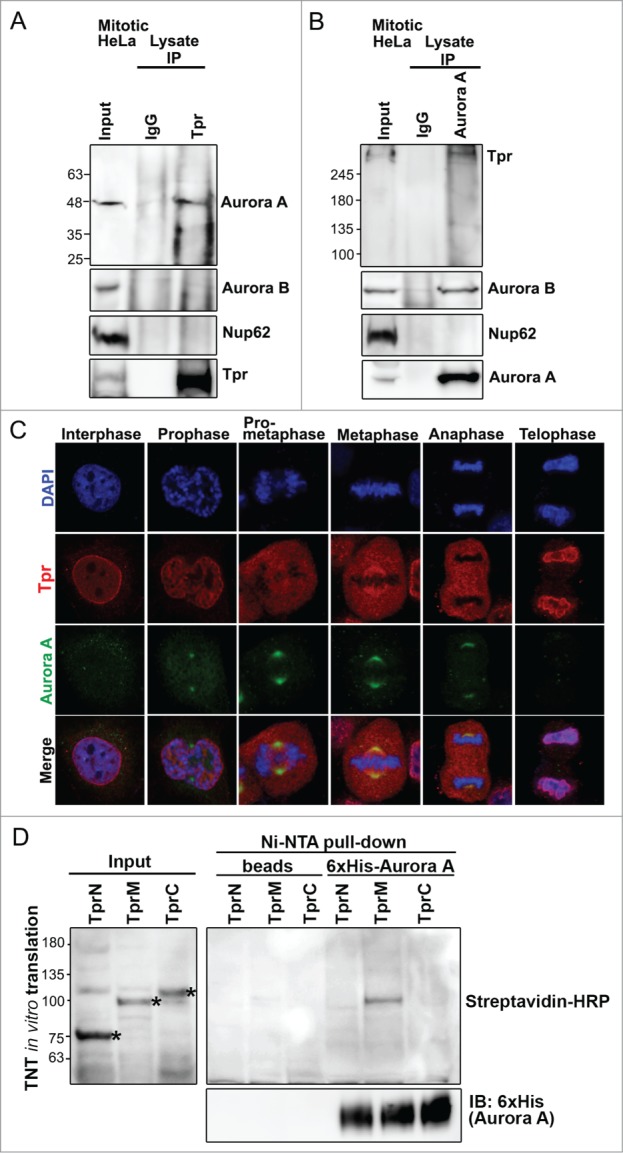
Tpr interacts with Aurora A during mitosis. (A) Immunoprecipitation of mitotic HeLa cell extracts incubated with anti-Tpr (mouse anti-Tpr, sc-101294 from Santa Cruz Biotechnology) or non-specific rabbit antibodies (IgG) (#2729; Cell Signaling Technology) were analyzed by immunoblotting with anti-Aurora-A (IAK1 610939 from BD Transduction Laboratories), anti-Aurora-B (ab2254, from Abcam), anti-Nup62 (m414, MMS-120R from COVANCE) and anti-Tpr (sc-101294 from Santa Cruz Biotechnology) antibodies. Numbers indicate molecular mass markers in kilodaltons. (B) Immunoprecipitates of mitotic HeLa cell extracts incubated with anti-Aurora A (IAK1 610939 from BD Transduction Laboratories) or non-specific rabbit antibodies (IgG) (#2729; Cell Signaling Technology) were analyzed by immunoblotting with the indicated antibodies (refer to Fig. 2A). (C) Co-immunostaining of Tpr and Aurora A in the cell cycle. Confocal images of HeLa cells at different mitotic stages, stained with anti-Aurora A (green) antibody and anti-Tpr (red; this antibody gives strong spindle signals in metaphase). Goat anti-mouse Alexa Fluor-488 or rabbit Rhodamine were used as secondary antibodies. Chromatin was stained with DAPI (blue). Data correspond to the sum of 3 independent experiments. (D) Aurora A is directly pulled-down by the Tpr-M domain in vitro. Tpr fragments were expressed in vitro, affinity-purified together with 6xHis-Aurora A, and separated by SDS-PAGE. Tpr fragments were prepared using the TNT Quick-Coupled Transcription/translation system (Promega) together with TranscendTM Biotin-Lysyl-tRNA (Promega). Asterisks indicate Tpr fragment (N, M and C) respectively. Streptavidin horseradish peroxidase (HRP) (1:4,000) was used for detection. Tpr fragments were untagged. Numbers indicate molecular mass markers in kilodaltons.
Tpr depletion alters aurora a centrosome localization, reduces aurora a phosphorylation and induces chromosome lagging or other segregation defects
The above observations led us to surmise that Tpr and Aurora A contribute to spindle pole/centrosome assembly during mitosis. Therefore, we confirmed the Tpr mitotic topography with respect to the spindle pole/centrosomal apparatus. We used specific antibodies against Tpr and γ-tubulin (centrosome marker) to examine their localizations at different stages of the cell cycle. Confocal microscopy of HeLa cells indicated a co-localization of Tpr and γ-tubulin in centrosomes (Fig. S2). Next, we determined whether increased levels of Tpr or Tpr fragments could alter Aurora A spindle or centrosomal localization. Full-length Tpr tagged with GFP or Tpr-N, Tpr-M or Tpr-C fragments or GFP vector alone were transiently transfected; however, in all cases Aurora A was still located on the centrosome and no obvious mitotic abnormalities were found (Fig. S3). The above results prompted us to test the consequences of Tpr depletion on Aurora A and p-Aurora A (Aurora A phosphorylation). Notably, we found that levels of Aurora A and p-Aurora A protein were significantly reduced in Tpr-depleted samples (Fig. 3A). Furthermore, compared with the staining pattern in control siRNA cells, down-regulation of Tpr also induced Aurora A and p-Aurora A to diffuse from the centrosome region and caused a marked ~25% increase in chromosome segregation defects (Fig. 3B and C; n=200 mitotic cells) and supernumerary centrosomes (co-staining with γ-tubulin as centrosome marker) (Fig. 3D and E). Meanwhile, we also observed that in approximately 8% of Tpr-depleted cells, Aurora A was still localized on the centrosome. However, in these cells another interesting chromosome defect phenotype of relatively long chromatin fibers was observed (Fig. S4, n = 200 mitotic cells).
Figure 3 (See previous page).
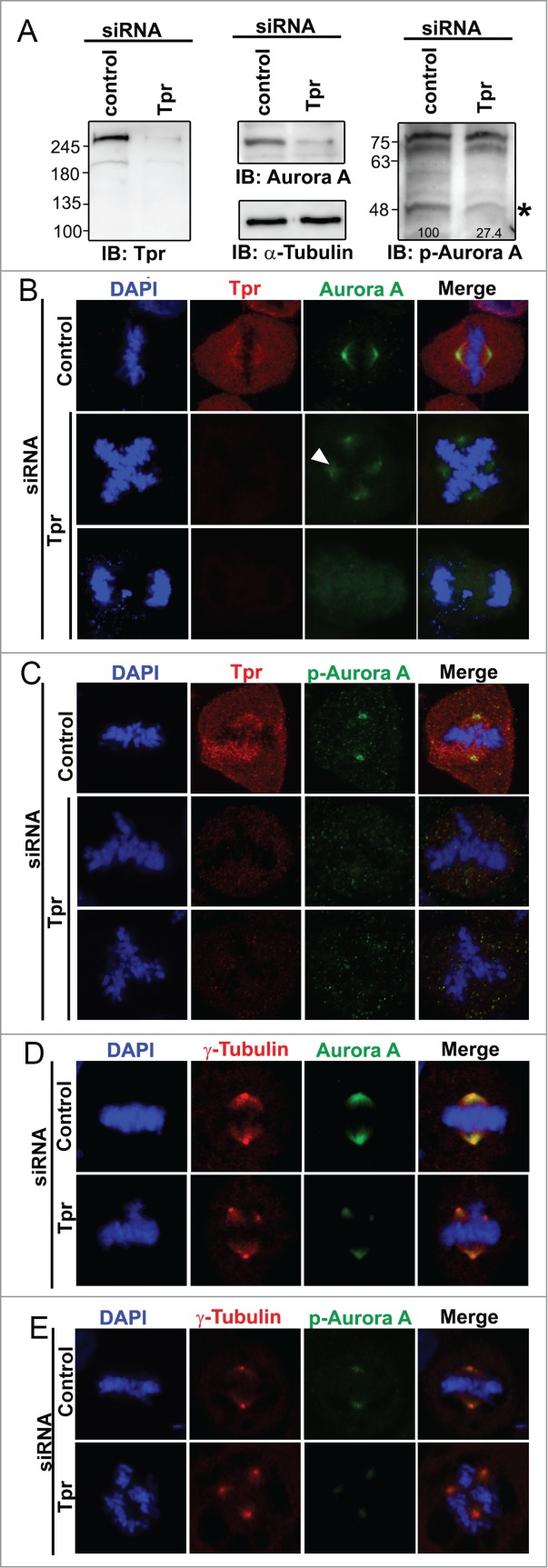
Tpr depletion alters Aurora A centrosome localization, reduces Aurora A phosphorylation and induces chromosome lagging or other segregation defects HeLa cells were transfected with control siRNA or Tpr siRNAs. Seventy-two hours after transfection, lysates were analyzed by immunoblotting with the indicated antibodies (see Fig. 2A or materials and methods for antibody details). The same membrane was stripped and re-probed with anti-α-tubulin (as loading control). Two non-specific bands (~70 KDA) are included to demonstrate equal loading as internal control (Right panel). Numbers indicate molecular mass markers in kilodaltons. (B and C) Confocal images of mitotic HeLa cells transfected with control or Tpr siRNA and analyzed 72 h post-transfection. Cells were analyzed by immunofluorescence using antibodies against (B) anti-Aurora A (IAK1 610939 from BD Transduction Laboratories) and Tpr (rabbit anti-Tpr, sc-67116, from Santa Cruz Biotechnology); (C) anti-Phospho-Aurora A (Thr 288) (#3079, from Cell Signaling Technology) and Tpr (mouse anti-Tpr, sc-101294, from Santa Cruz Biotechnology). Goat anti-mouse Alexa Fluor-488 or rabbit Rhodamine were used as secondary antibodies. DNA was counterstained using DAPI. D-E) Confocal images of mitotic HeLa cells transfected with control or Tpr siRNA and analyzed 72 h post-transfection. Cells were analyzed by immunofluorescence using antibodies against (D) anti-Aurora A and γ-tubulin (T6557 from Sigma-Aldrich); (E) anti- Aurora A phosphorylation (pT288) and γ-tubulin (T6557 from Sigma-Aldrich). Goat anti-mouse Alexa Fluor-488 or rabbit Rhodamine were used as secondary antibodies. DNA was counterstained using DAPI.
Aurora a inhibitor, alisertib (MLN8237), disrupts Tpr spindle pole localization and induces chromosome defects and multi-nuclei formation
We sought to evaluate Alisertib for its ability to alter Tpr protein levels and function. Consistent with recent reports,24-26 we found that exposure of HeLa cells to Alisertib (10–1000 nM) induced G2/M cell cycle arrest and cell death (sub-G1 phase) in a dose- and time-dependent manner (Fig. 4A and B). In addition, cells treated at 100nM concentration displayed mitotic spindle abnormalities and chromosome misalignment, phenotypes that were previously described to be associated with Aurora A inhibition in HCT116 colorectal carcinoma cells.24 At a higher concentration (1 μM), cells often underwent a repeated round of DNA synthesis, suggesting that Alisertib could also induce multinucleated cells and failure of cytokinesis. Hence, we decided to observe the effect of Alisertib on Tpr nuclear rim morphology in HeLa cells (Fig. 4C). Following treatment with Alisertib, we observed a concentration-dependent reduction in Aurora A T288 phosphorylation, validating that Alisertib was effective at inhibiting Aurora A kinase activity, while levels of total Tpr signal were also reduced (Fig. 4D–E). Notably, we found that Tpr was not bound to the spindle or spindle poles, in contrast to control cells27 (Fig. 4F). These data demonstrated that Alisertib increased chromosomal segregation defects and caused unusually large nuclei and multinucleated cells (Fig. 4D–F). Strikingly, we found that Alisertib also caused Tpr to diffuse from spindle pole regions. These data also indicated that Tpr reduction induced mitotic and centrosomal aberration and enhanced chromosomal instability, which may lead to increased rates of mutation or chromosomal translocation during early phases of tumor development.
Figure 4.

Aurora A inhibitor, MLN8237, promotes polyploidy and alters cell-cycle progression and induces multi-nuclei formation by disrupted Tpr spindle pole localization. (A) HeLa cells were treated with MLN8237 for 24 and 48 h and cell-cycle progression was analyzed by flow cytometry. MLN8237 treatment (as indicated with various concentrations) induces G2–M arrest and polyploidy. (B) Percentage of G1, G2/M, S, and Sub-G1 cells were calculated based on the results shown in (A). Asterisks indicates significant p values (*p < 0.05 or **p < 0.005). (C) Schedule of fixing or collecting mitotic HeLa cells after MLN8237 treatment. (D-F) Confocal images of MLN8237-treated HeLa cells, stained with pT288 (Aurora phosphorylation) (green) and Tpr (red) at the indicated concentrations. (D) 24 h and (E) 48 h after treatment. (F) White arrows indicate Tpr spindle pole localization disruption in the spindle pole/centrosomal areas in the observed cells. (Refer to materials and methods for antibody details). White arrow heads indicate Tpr diffusing from spindle poles/centrosome regions and white arrows indicate normal centrosome area.
Overexpression of aurora a disrupted tpr spindle pole/centrosomal localization and enhanced multiple centrosome formation
Over-expression of Aurora A has been shown to induce genomic instability and tumorigenesis, while Aurora A is up-regulated in several human cancers and is correlated with poor prognosis.28 To test whether overexpression of Aurora A has any effect on the stability of endogenous Tpr, HeLa cells were transfected with Flag-tagged Aurora A and Tpr stability was investigated. The expression of endogenous Tpr or other nuclear pore proteins (nuclear pore marker, m414) was not affected by the over-expression of Aurora A, confirming the western blot analysis data (Fig. 5A). Consistently, we found that ~25% of transfected cells had multipolar centrosomes (n = 200 mitotic cells). Next, we examined the subcellular distribution of Tpr in mitotic cells over-expressing Aurora A. Double staining with Tpr or m414 showed that over-expressed Aurora A had little, if any, effect on nuclear rim staining, which retained its usual punctuate distribution (data not shown). We noticed, however, that Tpr was localized to the spindle pole/centrosome in non-Aurora A transfected mitotic cells. Interestingly, closer observation showed that Tpr localization was abolished from spindle pole regions, and chromosome separation defects were found only in mitotic cells over-expressing Aurora A and that these cells often contained supernumerary centrosomes (Fig. 5B); co-staining with γ-tubulin as centrosome marker (Fig. 5C). These data indicate that overexpressed Aurora A altered Tpr spindle pole/centrosomal localization.
Figure 5.

Overexpression of Aurora A-enhanced multiple centrosome formation by disrupting Tpr spindle pole/centrosomal localization. Bipolarity was restored by co-transfection of the Tpr-M domain. (A) HeLa cells transfected with FLAG-vector or FLAG-Aurora A expression plasmid were analyzed by immunoblotting for FLAG (mouse anti-FLAG, F1804 from Sigma-Aldrich), Aurora A (IAK1 610939 from BD Transduction Laboratories), Tpr (mouse anti-Tpr, sc-101294, from Santa Cruz Biotechnology) and nuclear pore marker, m414 (MMS-120R from COVANCE). Numbers indicate molecular mass markers in kilodaltons. (B and C) Confocal images of FLAG-Aurora A expressing HeLa cells, stained with FLAG or pT288 (for Aurora phosphorylation, green) and Tpr (red) (B); Anti-Phospho-Aurora A (Thr 288) and γ-tubulin (C). Goat anti-mouse Alexa Fluor-488 or rabbit Rhodamine were used as secondary antibodies. DNA was counterstained using DAPI. White arrow heads indicate normal bipolar cells and white arrows indicate Tpr diffused from centrosome areas only in cells with multiple centrosomes. (D) Bipolarity of FLAG-Aurora A expressing cells was restored by co-transfection of GFP-Tpr-M. Confocal images of HeLa cells co-transfected with GFP or GFP-Tpr-M and FLAG-vector or FLAG-Aurora A, stained with FLAG (red) or GFP (green).
Aurora-a overexpression-induced supernumerary centrosomes were rescued by co-transfection of the tpr-M domain
Because we determined that Aurora A interacts with the Tpr-M domain (Fig. 2), we considered that Tpr might be a target of Aurora A kinase activity. If this hypothesis is correct, over-expression of GFP-Tpr-M (775–1700 aa) would mimic the Aurora A reduction phenotype through sequestration of endogenous Aurora A, and might partially rescue the supernumerary centrosome phenotype induced by overexpression of Aurora A. Therefore, we next examined the effect of expressing Tpr-M in HeLa cells to challenge our hypothesis further. Consistent with our prediction, all co-transfected GFP-M and FLAG-Aurora A cells were bipolar (n = 42 cells). In addition, we found that cells co-transfected with Tpr-M and Aurora A had much stronger staining at the spindle pole (Fig. 5D). A highly plausible interpretation of these results is that Tpr-M binds to and sequesters endogenous and exogenous Aurora A. This action is analogous to that of the Aurora A inhibitor.
Discussion
Nucleoporins, once thought to be exclusively structural components of the nuclear pore with roles only in nuclear transportation, are emerging as regulators of diverse cellular functions. We and others have demonstrated that a number of nuclear pore proteins (Nups) play active roles during mitosis.9,10,29 Recently, we showed that several nucleoporins are involved in mitotic spindle, kinetochore and centrosome homeostasis during mitosis.30-40
In the present study, we demonstrate a novel centrosomal role for the coiled-coil nucleoporin, Tpr, which regulates Aurora A both spatially and temporally. We propose that Tpr plays an important role in coordinating the centrosome localization of Aurora A and the Aurora A phosphorylation process during mitosis (Fig. 6).
Figure 6.

Speculative working model for the mutual regulation between Tpr and Aurora A in mitotic progression and centrosome homeostasis. We propose that the coiled-coil Tpr-M domain binds to and sequesters endogenous and exogenous Aurora A and thereby causes a reduction in Aurora A phosphorylation. This action is analogous to that of the Aurora A inhibitor, Alisertib. This domain merits further investigation as a therapeutic agent for those cancers in which Aurora kinaseA is over-expressed.
Previously, we have shown that Tpr associates with the molecular motors, dynein and dynactin, both of which orchestrate the spindle checkpoint proteins, Mad1 and Mad2, during cell division.10 Over-expression of Tpr enhanced multinucleated cell formation,10 while in Tpr-depleted cells, the levels of p53 and p21 proteins were enhanced. Tpr downregulation also increased p53 nuclear accumulation and facilitated autophagy.23 Here we report an additional mitotic spindle pole/centrosome role for Tpr during the metaphase-to-anaphase transition. Our data also indicate the potential of Tpr as an anticancer agent via its inhibition of Aurora A kinase.
In this study, we first showed that down-regulation of Tpr caused lagging chromosomes and tetraploidy and polyploidy. We also investigated the effect of Tpr depletion on cell cycle progression. Tpr depletion enhanced G2 arrest and 6N/8N in HeLa cells (Fig. 1). Recent studies have suggested that mitotic catastrophe is a type of cell death that results from abnormal mitosis, and usually culminates in the formation of large aneuploid cells with multinucleation.41 Typical features of irreversible mitotic catastrophe include cell ‘blebbing,’ formation of micronuclei, and chromosome fragmentation.42 Our observations indicate that lagging chromosomes caused by Tpr depletion enhanced polyploid cell formation causing irreversible mitotic catastrophe. However, Tpr depletion also affects sumoylation,43 which plays an important role in polyploidy formation.44,45 We could not rule out the possibility that other pathways might also interact with Tpr in polyploidy formation. Further studies are necessary to clarify any other molecular events that are critical for Tpr function in cell cycle progression.
Recently, we and others found that in Tpr-depleted cells, the levels of p53 and p21 proteins were enhanced.23,43 Furthermore, Tpr depletion increased p53 nuclear accumulation and facilitated autophagy.23 Here we found that Tpr depletion also altered p53 centrosomal localization during mitosis (Fig. S1). Notably, Aurora A interacts with and phosphorylates p53 at Ser215 and Ser315 and down-regulates its transactivation activity and protein stability.46,47 Aurora A inhibition in cells lacking or with mutant p53 induced polyploidization48 and Aurora A malfunction also led to tetraploid and polyploid cells.49 Moreover, Aurora A inhibition also induced autophagy.50
The Aurora kinases comprise a family of serine/threonine kinases that play an essential role in cell-cycle progression, most notably during G2 and M phases. Three human homologs, Aurora kinase A, B, and C, have been characterized and maintain discrete functions within the cell cycle. Aurora A shares substantial structural and sequence similarity with Aurora B, even though these proteins have distinct mitotic functions and distinct subcellular localizations. These differences in both function and localization are attributed in part to the association of each kinase with a unique group of cofactor proteins.51,52 Aurora A accumulates at the centrosome/spindle pole regions throughout mitosis and is also localized along spindle MTs. These physiological and functional characteristics are similar to those of Tpr; therefore, we next elucidated the relationship between Tpr and Aurora during mitosis. We found that Aurora A directly interacted and co-localized with the large internal coiled-coil domain in Tpr-M at the metaphase–anaphase transition (Fig. 2).
Consistent with this finding, we also observed a correlation between the levels of Aurora A phosphorylation and Tpr depletion. We found that levels of Aurora A and p-Aurora A protein were reduced after Tpr downregulation. We also found that Tpr depletion caused Aurora A and p-Aurora A to diffuse from the spindle pole region and to cause a marked increase in chromosome segregation defects (Fig. 3).
Aurora kinases have central roles in mitosis and are frequently over-expressed in many types of tumor. They are believed to be important anti-cancer drug targets. A number of small-molecule Aurora kinase inhibitors have been developed over recent years and more than 20 compounds are currently under development or in clinical trials.25,51-53 Phosphorylation at Thr-288 within the activation loop (A-loop) of Aurora A is necessary for its kinase activity.54 Recent studies show that MLN8237 (also called Alisertib) selectively inhibits Aurora A and it is undergoing clinical trials for anti-cancer therapies.26,28 Polyploidy induced by Aurora kinase inhibitors is associated with compromised p53-dependent post-mitotic checkpoint function.55 Consistent with this, we found that Alisertib or Tpr inhibition causes the development of extra centrosomes, chromosome segregation defects that increase over time, cell cycle arrest, and enhanced and multinucleated cell formation (Fig. 4). Remarkably, we showed that Alisertib also caused Tpr to diffuse from spindle pole regions. These data also indicate that Tpr reduction prompts mitotic and centrosomal aberration, increasing chromosomal instability. This potentially increases the risk of mutation or chromosomal translocation during early phases of cancer development.
Finally, we also show that the Tpr M domain binds to and sequesters endogenous and exogenous Aurora A and causes a reduction in the level and phosphorylation of Aurora A. This action is analogous to that of the Aurora A inhibitor (Fig. 5). We suggest focusing on the Tpr M (775-1700 aa) fragment as a pharmaceutical target that might function in a similar way to the Aurora A specific inhibitor, Alisertib. Our preliminary data indicate that Tpr-M, when simply overexpressed, did not induce any visible chromosomal instability.
Is there any clinical evidence relating to Tpr down-regulation and Aurora A overexpression? Colorectal cancer (CRC) is a prime example of a tumor exhibiting chromosomal instability, which is thought to contribute to carcinogenesis, cancer progression and therapy resistance.21,56 Over-expression of Aurora A is often detected in human CRC cancer cells.57 Remarkably, when 6 different human colorectal cancer tumors were examined, Tpr expression levels were found to be decreased by 4- to 5-fold in all patients.7,58
It is obviously naive to think one can simply over-express Tpr to inhibit Aurora A and Aurora A phosphorylation without causing significant side effects because Tpr has other important roles in nucleocytoplasmic transport, nuclear pore structure and spindle-check point regulation. Moreover, we found that Tpr depletion increased p53 nuclear accumulation and facilitated autophagy.23 Excitingly, p53 inhibited both Mad2 and Aurora A in mitosis.59 Understanding Tpr-Mad1/2 or Tpr-Aurora A together with mitotic p53 may lead to improvements in the rational development of targeted cancer therapies. Our data also raise the interesting idea that Tpr itself may function as a suppressor in carcinogenesis. Our work points to Tpr as a potential inhibitor of Aurora kinase, and is of great interest for its therapeutic potential (Fig. 6).
Materials and Methods
Plasmids
The full-length Aurora A coding region was PCR-amplified from pCMV-SPORT6-Aurora A (a kind gift from Dr. Golemis, Fox Chase Cancer Center, USA) and subcloned into pCMV-3xFLAG and pET28a. The full-length Tpr, and the 3 plasmids encoding Tpr fragments (N, M and C) were described previously.10 All constructs were confirmed by DNA sequencing.
Mammalian Cell Culture, Transfections, RNA Interference and Drug Treatments
HeLa cells were obtained from the American Type Culture Collection (ATCC), and were propagated in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (Life Technologies) and 50 U/ml penicillin-streptomycin (Nacalai Tesque). Cells were cultured in 5% CO2 in a humidified incubator at 37°C. HeLa cells were synchronized in S phase by the double thymidine block using 2 mM thymidine.10
For inhibitor treatment, cells were treated with Alisertib (Selleck Chemicals) at the indicated final concentration. For transfection, cells were plated onto 12-well tissue culture plates at a density of ~105 cells per well. The next day, cells were transfected with DNA constructs (GFP-Tpr-fragment or FLAG-Aurora A plasmids) using Lipofectamine 2000 (Life Technologies) following the manufacturers' protocol. Generally, 2 μg of DNA was used for every transfection. After 6 h, the media was changed to fresh growth media as indicated above. For siRNA transfections, siRNA duplexes targeting Tpr (sc-45343) and control siRNA (sc-37007) were purchased from Santa Cruz Biotechnology. HeLa cells were plated onto 12- or 6-well tissue culture plates at a density of 105 cells per well. The following day, cells were transfected with 250 pmol siRNA using Lipofectamine 2000, following the manufacturer's protocol (Life Technologies). HeLa cells were imaged 72 h after transfection. If necessary, transfection efficiency was monitored with Block-iT (Invitrogen).
Antibodies, Immunocytochemistry and Confocal Microscopy
Anti-Tpr rabbit polyclonal antibody for confocal microscopy was a kind gift from Dr. Larry Gerace (The Scripps Research Institute). Anti-Tpr (sc-101294) antibody (for immunoblotting) and anti-Plk1 (sc-17783) and anti-Tpr (sc-67116) antibodies (for immunoprecipitation) were from Santa Cruz Biotechnology. Anti-GFP antibody (A-6455) was from Life Technologies. Anti-Aurora B (ab2254) antibody was from Abcam. Anti-phospho-Aurora A (Thr288) (#3079S) antibody was from Cell Signaling. Anti-α-tubulin (DM1A) and anti-FLAG (F3165) antibodies were from Sigma-Aldrich. Anti-Bub3 (611731) and anti-Aurora A (IAK1 610939) antibodies were from BD Transduction Laboratories. Anti-mAb414 (MMS-120R) antibody was from COVANCE. Anti-6xHis antibody was from Qiagen. Anti-p53 (DO-7) antibody was from Dako. Secondary antibodies (Alexa Fluor or Rhodamine) were from Molecular Probes (Life Technologies).
For microscopy, synchronized HeLa cells were washed in phosphate-buffered saline (PBS) and fixed for 10 min in 4% paraformaldehyde in PBS. Cells were then washed 3 times with PBS and permeabilized with 0.3% Triton X-100 in PBS for 10 min at room temperature. Coverslips were incubated with indicated primary antibody for 2 h. Coverslips were washed 3 times and incubated with Alexa Fluor-conjugated secondary antibody (Life Technologies) for 2 h. After three washes, samples were then mounted onto coverslips using Pro-Long Gold Antifade reagent (Life Technologies) and were examined on a LSM5 EXCITER confocal microscope with 4 laser beams (Carl Zeiss Microscopy), and all images were acquired using a plan-Apochromat from Carl Zeiss Microscopy at × 63 with a 1.4-N.A. objective equipped with ZEN Imaging software (Zeiss).10
Immunoprecipitation
The immunoprecipitation procedures were described previously.10,30,31 Briefly, mitotic HeLa cells were harvested, washed with PBS, spun at 400 × g for 10 min and lysed in 1 ml of cold lysis buffer [50 mM Tris-HCl (pH 7.2), 250 mM NaCl, 0.1% Nonidet P-40, 2 mM EDTA, 10% glycerol] containing 1 × protease inhibitor mixture (Roche-Diagnostics) and 1 mM phenylmethylsulfonyl fluoride. The lysates were centrifuged for 30 min at 4°C at 14 000 × g. The resulting supernatants were pre-cleared with 50 μl protein A/G bead slurry (Santa Cruz Biotechnology), mixed with 10 μl of various antibodies as specified and incubated for 2 h at 4°C with rocking. The beads were then washed 5 times with 500 μl of lysis buffer. After the last wash, 50 μl 1 × sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) blue loading buffer (New England Biolabs) was added to the bead pellet before loading, the samples were separated by PAGE and electro-blotted to PVDF membranes (GE Healthcare). The membrane was probed with indicated antibodies. Signals were detected with an enhanced chemiluminescence system (GE Healthcare) and quantified using an LAS-4000 image analyzer (Fujifilm) according to the manufacturer's specifications.
Cell Cycle Profile Analysis
The FACS procedures used were described previously.10,30,31 Briefly, HeLa cells transfected with siRNA were trypsinized, washed twice with PBS and fixed in 70% ethanol at −20°C overnight. The fixed cells were resuspended in PBS containing 50 μg/ml RNase A (Nacalai Tesque) and 50 μg/ml PI (Sigma-Aldrich). Cellular DNA content was analyzed using a FACSCanto II (BD Biosciences) with FACS Diva software (BD Biosciences).
Expression of Recombinant His-Aurora A Proteins
Protein expression and affinity chromatography were performed as previously described.30,31 Briefly, to express 6 × His-tagged Aurora A in E. coli BL21(DE3) Codon Plus (Agilent Technologies), cells were grown at 37°C to an absorbance at 600 nm (A600) of 0.6 and induced with 0.5 mM isopropyl-β-D-thiogalactopyranoside (IPTG) at 18°C overnight. The cells were harvested by centrifugation and lysed in buffer containing 50 mM Tris-HCl (pH 7.7), 150 mM KCl, 0.1% Triton-X100 and Complete EDTA-free protease inhibitor mixture tablets (Roche). The cells were lysed using a cell sonicator (SMT), and the lysate was clarified by centrifugation at 15 000 × g for 60 min. Aurora A proteins containing the 6 × His tag were purified by nickel-affinity chromatography (Qiagen) and stored at −80°C.
In vitro binding assays
In vitro binding procedures were described previously.10 Briefly, His-tagged Aurora A protein was loaded onto Ni2+-NTA agarose beads (Qiagen) in loading buffer [50 mm Tris-HCl (pH 7.7), 150 mm KCl, 0.1% Triton-X100, 1 × protease inhibitor mixture] for 2 h at 4°C. The beads were then washed 5 times with wash buffer [50 mm Tris-HCl (pH 7.7), 300 mm KCl]. Tpr proteins were expressed using the Promega TNT coupled transcription/translation system according to the manufacturer's protocol or as described previously10. Beads were incubated with in vitro translated Tpr proteins for 2 h at 4°C. The beads were then washed 5 times with wash buffer. After the last wash, 1 × SDS-PAGE blue loading buffer was added to the samples and boiled for 5 min. Proteins were separated by 10% SDS-PAGE, and then electro-blotted onto a PVDF membrane. The membrane was probed with Streptavidin-HRP or anti-6×His antibodies. Signals were detected with an enhanced chemiluminescence system (GE Healthcare) and quantified using an LAS-4000 image analyzer (Fujifilm) according to the manufacturer's specifications.
Statistical analysis
Statistical analyses were performed in Excel. Data are expressed as means ± SD. Comparisons between groups were determined using the unpaired t test. P < 0.05 was considered statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. Golemis for the Aurora-A plasmid.
Funding
This work was supported by Grants-in-Aid for Scientific Research on Innovative Areas, Grants-in-Aid for Challenging Exploratory Research and Grants-in-Aid for Scientific Research (B) from MEXT Japan, and by grants from the Asahi Glass Foundation, the Suzuken Memorial Foundation, the Sumitomo Foundation, the Kowa Life Science Foundation, the Mochida Memorial Foundation, the Sagawa Foundation, the Uehara Memorial Foundation, the Ichiro Kanehara Foundation and the Takeda Science Foundation (to R. W.). This work was also supported by Grants-in-Aid for young scientists (B) (to C.H.).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Nigg EA, Raff JW. Centrioles, centrosomes, and cilia in health and disease. Cell 2009; 139: 663–78; PMID:19914163; http://dx.doi.org/ 10.1016/j.cell.2009.10.036 [DOI] [PubMed] [Google Scholar]
- 2.Bettencourt-Dias M, Glover DM. Centrosome biogenesis and function: centrosomics brings new understanding. Nat Rev Mol Cell Biol 2007; 8: 451–63; PMID:17505520; http://dx.doi.org/ 10.1038/nrm2180 [DOI] [PubMed] [Google Scholar]
- 3.Andersen JS, Wilkinson CJ, Mayor T, Mortensen P, Nigg EA, Mann M. Proteomic characterization of the human centrosome by protein correlation profiling. Nature 2003; 426: 570–4; PMID:14654843; http://dx.doi.org/ 10.1038/nature02166 [DOI] [PubMed] [Google Scholar]
- 4.Jakobsen L, Vanselow K, Skogs M, Toyoda Y, Lundberg E, Poser I, Falkenby LG, Bennetzen M, Westendorf J, Nigg EA., et al. Novel asymmetrically localizing components of human centrosomes identified by complementary proteomics methods. EMBO J 2011; 30: 1520–35; PMID:21399614; http://dx.doi.org/ 10.1038/emboj.2011.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fernandez-Martinez J, Rout MP. A jumbo problem: mapping the structure and functions of the nuclear pore complex. Curr Opin Cell Biol 2012; 24: 92–9; PMID:22321828; http://dx.doi.org/ 10.1016/j.ceb.2011.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tran EJ, Wente SR. Dynamic nuclear pore complexes: life on the edge. Cell 2006; 125: 1041–53; PMID:16777596; http://dx.doi.org/ 10.1016/j.cell.2006.05.027 [DOI] [PubMed] [Google Scholar]
- 7.Simon DN, Rout MP. Cancer and the nuclear pore complex. Adv Exp Med Biol 2014; 773: 285–307; PMID:24563353; http://dx.doi.org/ 10.1007/978-1-4899-8032-8_13 [DOI] [PubMed] [Google Scholar]
- 8.Funasaka T, Wong RW. The role of nuclear pore complex in tumor microenvironment and metastasis. Cancer Metastasis Rev 2011; 30: 239–51; PMID:21298575; http://dx.doi.org/ 10.1007/s10555-011-9287-y [DOI] [PubMed] [Google Scholar]
- 9.Chatel G, Fahrenkrog B. Nucleoporins: leaving the nuclear pore complex for a successful mitosis. Cell Signal 2011; 23: 1555–62; PMID:21683138; http://dx.doi.org/ 10.1016/j.cellsig.2011.05.023 [DOI] [PubMed] [Google Scholar]
- 10.Nakano H, Funasaka T, Hashizume C, Wong RW. Nucleoporin translocated promoter region (Tpr) associates with dynein complex, preventing chromosome lagging formation during mitosis. J Biol Chem 2010; 285: 10841–9; PMID:20133940; http://dx.doi.org/ 10.1074/jbc.M110.105890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nezi L, Rancati G, De Antoni A, Pasqualato S, Piatti S, Musacchio A. Accumulation of Mad2-Cdc20 complex during spindle checkpoint activation requires binding of open and closed conformers of Mad2 in saccharomyces cerevisiae. J Cell Biol 2006; 174: 39–51; PMID:16818718; http://dx.doi.org/ 10.1083/jcb.200602109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peschard P, Park M. From Tpr-met to met, tumorigenesis and tubes. Oncogene 2007; 26: 1276–85; PMID:17322912; http://dx.doi.org/ 10.1038/sj.onc.1210201 [DOI] [PubMed] [Google Scholar]
- 13.Frosst P, Guan T, Subauste C, Hahn K, Gerace L. Tpr is localized within the nuclear basket of the pore complex and has a role in nuclear protein export. J Cell Biol 2002; 156: 617–30; PMID:11839768; http://dx.doi.org/ 10.1083/jcb.200106046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krull S, Thyberg J, Bjorkroth B, Rackwitz HR, Cordes VC. Nucleoporins as components of the nuclear pore complex core structure and Tpr as the architectural element of the nuclear basket. Mol Biol Cell 2004; 15: 4261–77; PMID:15229283; http://dx.doi.org/ 10.1091/mbc.E04-03-0165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mitchell PJ, Cooper CS. Nucleotide sequence analysis of human tpr cDNA clones. Oncogene 1992; 7: 383–8; PMID:1549355 [PubMed] [Google Scholar]
- 16.Cordes VC, Reidenbach S, Rackwitz HR, Franke WW. Identification of protein p270/tpr as a constitutive component of the nuclear pore complex-attached intranuclear filaments. J Cell Biol 1997; 136: 515–29; PMID:9024684; http://dx.doi.org/ 10.1083/jcb.136.3.515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schweizer N, Ferras C, Kern DM, Logarinho E, Cheeseman IM, Maiato H. Spindle assembly checkpoint robustness requires tpr-mediated regulation of Mad1/Mad2 proteostasis. J Cell Biol 2013; 203: 883–93; PMID:24344181; http://dx.doi.org/ 10.1083/jcb.201309076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodriguez-Bravo V, Maciejowski J, Corona J, Buch HK, Collin P, Kanemaki MT, Shah JV, Jallepalli PV. Nuclear pores protect genome integrity by assembling a premitotic and mad1-dependent anaphase inhibitor. Cell 2014; 156: 1017–31; PMID:24581499; http://dx.doi.org/ 10.1016/j.cell.2014.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buchwalter A, Hetzer MW. Nuclear pores set the speed limit for mitosis. Cell 2014; 156: 868–9; PMID:24581486; http://dx.doi.org/ 10.1016/j.cell.2014.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muller H, Schmidt D, Steinbrink S, Mirgorodskaya E, Lehmann V, Habermann K, Dreher F, Gustavsson N, Kessler T, Lehrach H., et al. Proteomic and functional analysis of the mitotic drosophila centrosome. EMBO J 2010; 29: 3344–57; PMID:20818332; http://dx.doi.org/ 10.1038/emboj.2010.210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pfau SJ, Amon A. Chromosomal instability and aneuploidy in cancer: from yeast to man. EMBO Rep 2012; 13: 515–27; PMID:22614003; http://dx.doi.org/ 10.1038/embor.2012.65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee SH, Sterling H, Burlingame A, McCormick F. Tpr directly binds to Mad1 and Mad2 and is important for the Mad1-Mad2-mediated mitotic spindle checkpoint. Genes Dev 2008; 22: 2926–31; PMID:18981471; http://dx.doi.org/ 10.1101/gad.1677208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Funasaka T, Tsuka E, Wong RW. Regulation of autophagy by nucleoporin tpr. Sci Rep 2012; 2: 878; PMID:23170199; http://dx.doi.org/ 10.1038/srep00878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manfredi MG, Ecsedy JA, Meetze KA, Balani SK, Burenkova O, Chen W, Galvin KM, Hoar KM, Huck JJ, LeRoy PJ., et al. Antitumor activity of MLN8054, an orally active small-molecule inhibitor of aurora a kinase. Proc Natl Acad Sci U S A 2007; 104: 4106–11; PMID:17360485; http://dx.doi.org/ 10.1073/pnas.0608798104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marxer M, Ma HT, Man WY, Poon RY. p53 deficiency enhances mitotic arrest and slippage induced by pharmacological inhibition of aurora kinases. Oncogene 2014; 33: 3550–60; PMID:23955083; http://dx.doi.org/ 10.1038/onc.2013.325 [DOI] [PubMed] [Google Scholar]
- 26.Manfredi MG, Ecsedy JA, Chakravarty A, Silverman L, Zhang M, Hoar KM, Stroud SG, Chen W, Shinde V, Huck JJ., et al. Characterization of alisertib (MLN8237), an investigational small-molecule inhibitor of aurora a kinase using novel in vivo pharmacodynamic assays. Clin Cancer Res 2011; 17: 7614–24; PMID:22016509; http://dx.doi.org/ 10.1158/1078-0432.CCR-11-1536 [DOI] [PubMed] [Google Scholar]
- 27.Kesisova IA, Nakos KC, Tsolou A, Angelis D, Lewis J, Chatzaki A, Agianian B, Giannis A, Koffa MD. Tripolin A, a novel small-molecule inhibitor of aurora a kinase, reveals new regulation of HURP's distribution on microtubules. PLoS One 2013; 8: e58485; PMID:23516487; http://dx.doi.org/ 10.1371/journal.pone.0058485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gautschi O, Heighway J, Mack PC, Purnell PR, Lara PN Jr., Gandara DR. Aurora kinases as anticancer drug targets. Clin Cancer Res 2008; 14: 1639–48; PMID:18347165; http://dx.doi.org/ 10.1158/1078-0432.CCR-07-2179 [DOI] [PubMed] [Google Scholar]
- 29.Raices M, D'Angelo MA. Nuclear pore complex composition: a new regulator of tissue-specific and developmental functions. Nat Rev Mol Cell Biol 2012; 13: 687–99; PMID:23090414; http://dx.doi.org/ 10.1038/nrm3461 [DOI] [PubMed] [Google Scholar]
- 30.Hashizume C, Kobayashi A, Wong RW. Down-modulation of nucleoporin RanBP2/Nup358 impaired chromosomal alignment and induced mitotic catastrophe. Cell Death Dis 2013; 4: e854; PMID:24113188; http://dx.doi.org/ 10.1038/cddis.2013.370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hashizume C, Moyori A, Kobayashi A, Yamakoshi N, Endo A, Wong RW. Nucleoporin Nup62 maintains centrosome homeostasis. Cell Cycle 2013; 12: 3804–16; PMID:24107630; http://dx.doi.org/ 10.4161/cc.26671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakano H, Wang W, Hashizume C, Funasaka T, Sato H, Wong RW. Unexpected role of nucleoporins in coordination of cell cycle progression. Cell Cycle 2011; 10: 425–33; PMID:21270521; http://dx.doi.org/ 10.4161/cc.10.3.14721 [DOI] [PubMed] [Google Scholar]
- 33.Wong RW. Interaction between rae1 and cohesin subunit SMC1 is required for proper spindle formation. Cell Cycle 2010; 9: 198–200; PMID:20016259; http://dx.doi.org/ 10.4161/cc.9.1.10431 [DOI] [PubMed] [Google Scholar]
- 34.Wong RW. An update on cohesin function as a 'molecular glue' on chromosomes and spindles. Cell Cycle 2010; 9: 1754–8; PMID:20436296; http://dx.doi.org/ 10.4161/cc.9.9.11806 [DOI] [PubMed] [Google Scholar]
- 35.Wong RW, Blobel G. Cohesin subunit SMC1 associates with mitotic microtubules at the spindle pole. Proc Natl Acad Sci U S A 2008; 105: 15441–5; PMID:18832153; http://dx.doi.org/ 10.1073/pnas.0807660105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wong RW, Blobel G, Coutavas E. Rae1 interaction with NuMA is required for bipolar spindle formation. Proc Natl Acad Sci U S A 2006; 103: 19783–7; PMID:17172455; http://dx.doi.org/ 10.1073/pnas.0609582104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Strambio-De-Castillia C, Niepel M, Rout MP. The nuclear pore complex: bridging nuclear transport and gene regulation. Nat Rev Mol Cell Biol 2010; 11: 490–501; PMID:20571586; http://dx.doi.org/ 10.1038/nrm2928 [DOI] [PubMed] [Google Scholar]
- 38.Funasaka T, Nakano H, Wu Y, Hashizume C, Gu L, Nakamura T, Wang W, Zhou P, Moore MA, Sato H, Wong RW. RNA export factor RAE1 contributes to NUP98-HOXA9-mediated leukemogenesis. Cell Cycle 2011; 10:1456-67; PMID:21467841; http://dx.doi.org/ 10.4161/cc.10.9.15494 [DOI] [PubMed] [Google Scholar]
- 39.Hashizume C, Nakano H, Yoshida K, Wong RW. Characterization of the role of the tumor marker Nup88 in mitosis. Mol Cancer 2010; 9: 119; PMID:20497554; http://dx.doi.org/ 10.1186/1476-4598-9-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hashizume C, Wong RW. Structure and function of nuclear pore complex. Seikagaku 2011; 83: 957–65; PMID:22184889 [PubMed] [Google Scholar]
- 41.Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene 2004; 23: 2825–37; PMID:15077146; http://dx.doi.org/ 10.1038/sj.onc.1207528 [DOI] [PubMed] [Google Scholar]
- 42.Ha GH, Kim HS, Lee CG, Park HY, Kim EJ, Shin HJ, Lee JC, Lee KW, Lee CW. Mitotic catastrophe is the predominant response to histone acetyltransferase depletion. Cell Death Differ 2009; 16: 483–97; PMID:19096391; http://dx.doi.org/ 10.1038/cdd.2008.182 [DOI] [PubMed] [Google Scholar]
- 43.David-Watine B. Silencing nuclear pore protein tpr elicits a senescent-like phenotype in cancer cells. PLoS One 2011; 6: e22423; PMID:21811608; http://dx.doi.org/ 10.1371/journal.pone.0022423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang M, Chiu SY, Hsu W. SUMO-specific protease 2 in Mdm2-mediated regulation of p53. Cell Death Differ 2011; 18: 1005–15; PMID:21183956; http://dx.doi.org/ 10.1038/cdd.2010.168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schimmel J, Eifler K, Sigurethsson JO, Cuijpers SA, Hendriks IA, Verlaan-de Vries M, Kelstrup CD, Francavilla C, Medema RH, Olsen JV., et al. Uncovering SUMOylation dynamics during cell-cycle progression reveals FoxM1 as a key mitotic SUMO target protein. Mol Cell 2014; 53: 1053–66; PMID:24582501; http://dx.doi.org/ 10.1016/j.molcel.2014.02.001 [DOI] [PubMed] [Google Scholar]
- 46.Liu Q, Kaneko S, Yang L, Feldman RI, Nicosia SV, Chen J, Cheng JQ. Aurora-A abrogation of p53 DNA binding and transactivation activity by phosphorylation of serine 215. J Biol Chem 2004; 279: 52175–82; PMID:15469940; http://dx.doi.org/ 10.1074/jbc.M406802200 [DOI] [PubMed] [Google Scholar]
- 47.Katayama H, Sasai K, Kawai H, Yuan ZM, Bondaruk J, Suzuki F, Fujii S, Arlinghaus RB, Czerniak BA, Sen S. Phosphorylation by aurora kinase a induces Mdm2-mediated destabilization and inhibition of p53. Nat Genet 2004; 36: 55–62; PMID:14702041; http://dx.doi.org/ 10.1038/ng1279 [DOI] [PubMed] [Google Scholar]
- 48.Nair JS, Ho AL, Schwartz GK. The induction of polyploidy or apoptosis by the aurora a kinase inhibitor MK8745 is p53-dependent. Cell Cycle 2012; 11: 807–17; PMID:22293494; http://dx.doi.org/ 10.4161/cc.11.4.19323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goldenson B, Crispino JD. The aurora kinases in cell cycle and leukemia. Oncogene 2014; 34:537-45; PMID:24632603; http://dx.doi.org 10.1038/onc.2014.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zou Z, Yuan Z, Zhang Q, Long Z, Chen J, Tang Z, Zhu Y, Chen S, Xu J, Yan M., et al. Aurora kinase A inhibition-induced autophagy triggers drug resistance in breast cancer cells. Autophagy 2012; 8: 1798–810; PMID:23026799; http://dx.doi.org/ 10.4161/auto.22110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Karthigeyan D, Prasad SB, Shandilya J, Agrawal S, Kundu TK. Biology of aurora a kinase: implications in cancer manifestation and therapy. Med Res Rev 2010; 31: 757–93; PMID:20196102; http://dx.doi.org/ 10.4161/auto.22110 [DOI] [PubMed] [Google Scholar]
- 52.Reboutier D, Troadec MB, Cremet JY, Chauvin L, Guen V, Salaun P, Prigent C. Aurora a is involved in central spindle assembly through phosphorylation of ser 19 in P150Glued. J Cell Biol 2013; 201: 65–79; PMID:23547029; http://dx.doi.org/ 10.1083/jcb.201210060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kimura M, Kotani S, Hattori T, Sumi N, Yoshioka T, Todokoro K, Okano Y. Cell cycle-dependent expression and spindle pole localization of a novel human protein kinase, aik, related to aurora of drosophila and yeast Ipl1. J Biol Chem 1997; 272: 13766–71; PMID:9153231; http://dx.doi.org/ 10.1074/jbc.272.21.13766 [DOI] [PubMed] [Google Scholar]
- 54.Bayliss R, Sardon T, Vernos I, Conti E. Structural basis of aurora-a activation by TPX2 at the mitotic spindle. Mol Cell 2003; 12: 851–62; PMID:14580337; http://dx.doi.org/ 10.1016/S1097-2765(03)00392-7 [DOI] [PubMed] [Google Scholar]
- 55.Gizatullin F, Yao Y, Kung V, Harding MW, Loda M, Shapiro GI. The aurora kinase inhibitor VX-680 induces endoreduplication and apoptosis preferentially in cells with compromised p53-dependent postmitotic checkpoint function. Cancer Res 2006; 66: 7668–77; PMID:16885368; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-3353 [DOI] [PubMed] [Google Scholar]
- 56.Ertych N, Stolz A, Stenzinger A, Weichert W, Kaulfuss S, Burfeind P, Aigner A, Wordeman L, Bastians H. Increased microtubule assembly rates influence chromosomal instability in colorectal cancer cells. Nat Cell Biol 2014; 16: 779–91; PMID:24976383; http://dx.doi.org/ 10.1038/ncb2994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bischoff JR, Anderson L, Zhu Y, Mossie K, Ng L, Souza B, Schryver B, Flanagan P, Clairvoyant F, Ginther C., et al. A homologue of drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J 1998; 17: 3052–65; PMID:9606188; http://dx.doi.org/ 10.1093/emboj/17.11.3052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alfonso P, Canamero M, Fernandez-Carbonie F, Nunez A, Casal JI. Proteome analysis of membrane fractions in colorectal carcinomas by using 2D-DIGE saturation labeling. J Proteome Res 2008; 7: 4247–55; PMID:18707159; http://dx.doi.org/ 10.1021/pr800152u [DOI] [PubMed] [Google Scholar]
- 59.de Carcer G, Malumbres M. A centrosomal route for cancer genome instability. Nat Cell Biol 2014; 16: 504–6; PMID:24875738; http://dx.doi.org/ 10.1038/ncb2978 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.