

Abstract
Many biologically significant processes, such as cell differentiation and cell cycle progression, gene transcription and DNA replication, chromosome stability and epigenetic silencing etc. depend on the crucial interactions between cellular proteins and DNA. Chromatin immunoprecipitation (ChIP) is an important experimental technique for studying interactions between specific proteins and DNA in the cell and determining their localization on a specific genomic locus. In recent years, the combination of ChIP with second generation DNA-sequencing technology (ChIP-seq) allows precise genomic functional assay. This review addresses the important applications of ChIP-seq with an emphasis on its role in genome-wide mapping of transcription factor binding sites, the revelation of underlying molecular mechanisms of differential gene regulation that are governed by specific transcription factors, and the identification of epigenetic marks. Furthermore, we also describe the ChIP-seq data analysis workflow and a perspective for the exciting potential advancement of ChIP-seq technology in the future.
Keywords: chromatin, high throughput, immunoprecipitation, sequencing
Abbreviations
- AR
androgen receptor
- BWA
Burrows-Wheeler aligner
- C. elegans
Caenorhabditis elegans
- ChIP
chromatin immunoprecipitation
- CRs
chromatin regulators
- ChIP-seq
ChIP sequencing
- DNA
deoxyribonucleic acid
- DNase
deoxyribonuclease
- EMSA
electrophoresis mobility shift assay
- ENCODE
encyclopedia of DNA elements
- FDR
false discovery rate
- GR
glucocorticoid receptor
- HDAC
histone deacetylase
- HEK
human embryonic kidney
- HSPCs
haematopoietic stem progenitor cells
- HM
histone modification
- HTChIP
high throughput ChIP
- IL-β
interleukin β
- IFN
interferon
- JNK
c-Jun NH2-terminal kinase
- K
lysine
- MACS
model-based analysis of ChIP-seq
- MEME
multiple Em for motif elicitation
- modENCODE
Model Organism ENCyclopedia Of DNA Elements
- NF-κB
nuclear factor κB
- PCR
polymerase chain reaction
- RNA
ribonucleic acid
- R-ChIP
robotic ChIP
- SNP
single nucleotide polymorphism
- SOAP
short oligonucleotide alignment program
- SPP
ChIP-seq processing pipeline
- STAT
signal transducers and activators of transcription
- SUMO
small ubiquitin-like modifier
- TFs
transcription factors
- TFBS
transcription factor binding sites
- TSS
transcription start site
- UCSC
University of California Santa Cruz
- UV
ultraviolet
The biological significance of protein interactions with DNA is critical for many vital cellular functions, such as DNA-replication, recombination, repair, gene expression, cell differentiation, cell cycle progression, chromosome stability, and epigenetic silencing etc. In eukaryotic cells, genetic elements are maintained in dynamic chromatin structures. Chromatin is a complex of DNA and proteins in the nucleus of a cell. The primary protein components of chromatin are histones, including histone H1, 2A, 2B, 3 and 4. Histones and other regulatory proteins bind to the DNA and maintain its 3-dimensional structure. Chromatin functions to package DNA into a smaller volume to fit into the cell. It strengthens the DNA to allow mitosis and prevents DNA damage.
A variety of phenotypic changes important in normal development and in diseases are temporally and spatially controlled by chromatin-coordinated gene expression.1 Due to their critical influence on cellular phenotype, the DNA-protein interactions have been intensely investigated using a variety of biochemical and genomic approaches. The techniques traditionally used are electrophoresis gel mobility shift (EMSA) and DNase I footprinting assays. However, these in vitro methods are not within the cellular context, thereby, their limited utility has sparked the development of other approaches to analyze DNA-protein interactions. Chromatin immunoprecipitation (ChIP) has become a very popular technique for identifying regions of a genome associated with specific proteins within their native chromatin context. ChIP helps to detect the DNA-protein interactions that take place in living cells by capturing proteins at the sites of their binding to DNA, thus avoiding some of the shortfalls associated with EMSA or DNase I footprinting assays. In vivo locations of binding sites of various transcription factors, histones, and other proteins have been determined using the ChIP technique.2-7
The original ChIP technique was developed by Gilmour and Lis while studying the association of RNA polymerase II with transcribed and poised genes in Escherichia coli and Drosophila.8-10 They used UV irradiation to covalently cross-link proteins in contact with neighboring DNA in intact living cells. The UV cross-link was further replaced with the formaldehyde cross-link approach by Solomon and Varshavsky.11
As shown in Figure 1, the ChIP technique usually involves fixing intact cells with formaldehyde, a reversible protein-DNA cross-linking agent that serves to fix or preserve the protein-DNA interactions occurring in the cell. The cells are then lysed and chromatin fragments are isolated from the nuclei by sonication or nuclease digestion. This is followed by the selective immunoprecipitation of protein–DNA complexes by utilizing specific protein antibodies and their conjugated beads. The crosslinks are then reversed, and the immunoprecipitated DNA is analyzed using various methods.
Figure 1.
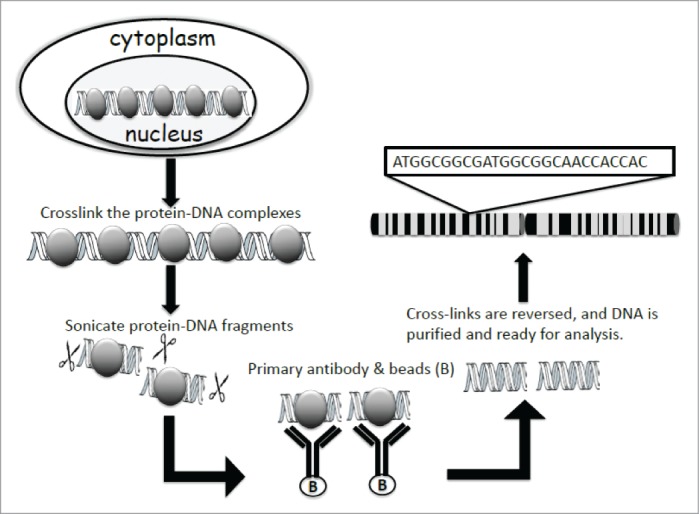
Principle of ChIP assay. The protein-DNA complexes are crosslinked in the nucleus, so the protein of interest and its chromatin binding site can be fixed. After lysing the cells, the protein-DNA complexes are sonicated into 200-1000bp fragments, and immunoprecipitated by probing with specific antibody. The crosslinks of the protein-DNA complexes are then reversed, and DNA is further purified and subjected to further analysis.
Standard PCR methods are most commonly used to identify the sequence identities of the precipitated DNA fragments. ChIP can also be combined with other routinely used molecular biology techniques: real time PCR, Southern blot analysis, and Western blot analysis.12-14 PCR methods can only be used to identify known target genes for a given protein: existing knowledge of the target gene is needed in order to design promoter-specific primers or probes. This limitation led to the emergence of new techniques designed to identify unknown gene sequences that interact with a given protein. A ChIP assay can be combined with genomic micro-array (ChIP-chip) techniques, sequencing (ChIP-seq), or cloning strategies, which allow for genome-wide analysis of protein-DNA interactions.2-4 While ChIP cloning is effective for isolating individual target genes, ChIP-Chip and ChIP-seq provide a more global approach.
From the original ChIP assay to the advanced ChIP-chip to today's most advanced ChIP-seq technique, ChIP techniques have rapidly evolved into an extremely powerful analysis of endogenous protein/DNA interactions with a variety of important applications that far exceed what could have been anticipated when the traditional ChIP assay was first invented.15
In this review, we will mainly focus on reviewing the representative applications of the most advanced ChIP-seq technique and further provide the workflow of its data analysis, and provide perspective regarding the exciting potential advances of ChIP-seq in the future.
Chip-Seq
The ChIP-seq method has become the most popular ChIP variation choice due to its ability to rapidly decode millions of DNA fragments simultaneously with high efficiency and relatively low cost. The ChIP-seq technique involves reverse cross-linking of immunoprecipitated DNA, fragmentation, and analysis by massively parallel DNA sequencing.16,17 ChIP-seq can be used to survey interactions accurately between protein, DNA, and RNA, enabling the interpretation of regulation events central to many biological processes and disease states. Since ChIP-seq provides the actual DNA sequences of the precipitated fragments, the data obtained is of higher resolution. Furthermore, as compared to ChIP-Chip, ChIP-seq is not influenced by the annealing efficiency of genomic DNA fragments to the immobilized oligonucleotides on the microarray tile, which is often weak and variable. Thus, the information provided by ChIP-seq is more accurate and quantitative than ChIP-Chip.18
Application of ChIP-seq is rapidly revolutionizing different areas of science. Below, we will give several major examples of the important roles that the ChIP-seq approach has played in discovering transcription factor binding sites, the study of transcription factor-mediated different gene regulation, and the identification of genome-wide histone marks.
ChIP-seq in the discovery of transcription factor binding sites and network
In 2007, Robertson G et al. first developed the ChIP-seq method, and used it to identify mammalian DNA sequences bound by transcription factors in vivo.19 They used ChIP-seq to map the signal transducers and activators of transcription 1 (STAT1) targets in interferon γ (IFNγ)-stimulated and unstimulated human cervical cancer HeLa S3 cells. By using ChIP-seq, they identified 41,582 and 11,004 putative STAT1-binding regions in stimulated and unstimulated cells, respectively. Of the 34 loci known to contain STAT1 interferon-responsive binding sites, ChIP-seq found 24 (71%). ChIP-seq targets were enriched in sequences similar to known STAT1 binding motifs. This report demonstrated the high coverage and accuracy of the ChIP-seq approach.
Recently, Cheung's group generated extensive global binding maps of androgen receptor (AR) and commonly over-expressed transcriptional corepressors including histone deacetylase 1 (HDAC1), HDAC2, HDAC3, etc., in prostate cancer cells.20,21 Their results surprisingly revealed that HDACs are directly involved in androgen-regulated transcription and wired into an AR-centric transcriptional network via a spectrum of distal enhancers and/or proximal promoters. Moreover, they show that these corepressors function to mediate repression of AR-induced gene transcription that promotes epithelial differentiation and inhibits metastasis. The AR transcriptional network derived from ChIP-seq studies by Cheung's group is critical to the strategic manipulation of AR activity for the targeted eradication of prostate cancer cells, further confirming the essential role that ChIP-seq plays in the discovery of transcriptional networks.
In addition to mammalian systems, in Caenorhabditis elegans (C. elegans), regulation of gene expression by sequence-specific transcription factors is central to developmental programs and depends on the binding of transcription factors with target sites in the genome. As part of the Model Organism ENCyclopedia Of DNA Elements (modENCODE) Consortium, Reinke's group has used ChIP-seq to determine the genome-wide binding sites of 22 transcription factors at diverse developmental stages.22 For each factor, they determined both coding and non-coding candidate gene targets. They revealed that the typical binding sites of almost all factors are within a few hundred nucleotides of the transcript start site. Based on the data they acquired, they further built a regulatory network among the 22 factors to determine their functional relationships to each other and found that some factors appear to act preferentially as regulators while others as target genes. Ultimately, the comprehensive mapping of transcription factor binding sites generated by Reinke's group will identify features of transcriptional networks that regulate C. elegans developmental processes.
ChIP-seq in the discovery of the underlying mechanisms of transcription factor-mediated differential gene regulation
In addition to identification of transcription factor binding sites, ChIP-seq can also be applied to identify distinct mechanisms involved in differential gene regulation.
Using the important transcription factor nuclear factor κB (NF-κB) as an example, we recently used ChIP-seq to study the role of lysine methylation of the p65 subunit of the NF-κB in differential gene regulation.23 We analyzed the effects of the mutants of lysine (K) 37 and 218/221 of p65 in response to IL-1β in 293 cells. ChIP-seq analysis showed that the K218/221 mutation greatly reduces the affinity of p65 for many promoters while the K37 mutation does not. Structural modeling showed that the newly introduced methyl groups of K218/221 interact directly with DNA to increase the affinity of p65 for specific κB sites. Thus, using ChIP-seq, we proved that K218/221 and K37 mutations have dramatically different effects because methylations of these residues affect different genes by distinct mechanisms. Since NF-κB plays an important role in cancer and inflammatory diseases, this important publication pointed out a critical mechanism that cells may use to differentially regulate NF-κB-dependent genes in different physiological or disease states.
Moreover, Paakinaho V et al. recently used ChIP-seq to analyze how SUMOylation of the glucocorticoid receptor (GR) influences the activity of endogenous GR target genes and the receptor chromatin binding in isogenic HEK293 cells expressing wild-type GR (wtGR) or SUMOylation-defective GR.24 They showed that SUMOylation modulates the chromatin occupancy of GR on several loci associated with cellular growth in a fashion that parallels differentially regulated gene expression between the 2 cell lines. Their data indicate that, instead of simply repressing GR activity, SUMOylation sophisticatedly regulates the GR activity in a target locus selective fashion that controls GR-dependent genes that influence cell growth.
The above examples show how ChIP-seq, when used in combination with site mutation of the post-translation modifications of a given transcription factor, could help unravel the fundamental mechanism of transcription factor-governed differential gene regulation.
ChIP-seq in the discovery of histone marks
ChIP-seq has been a powerful tool in the studies of histone modification status in cells. Mikkelsen et al. generated genome-wide maps of the chromatin state in pluripotent and lineage-committed cells by using ChIP-seq assay.25 They examined how chromatin states change as cells move from immature to adult states in a genome-wide manner. Findings from this work suggested that tri-methylation (me3) on H3K4 and H3K27 effectively discriminates genes that are expressed, poised for expression, or stably repressed, and therefore reflect cell state and lineage potential. This example vividly proves the importance of ChIP-seq analysis in the identification of H3K4me3 and H3K27me3 as facilitators of gene annotation.
In addition to histone methylation, researchers also found histone acetylation can serve as a histone mark in annotating the status of gene transcription. By using the ChIP-seq approach, Park et al. did a genome-wide analysis of H4K5 acetylation (H4K5ac) status, and revealed the association of H4K5ac with fear memory in brains of mice. They suggested that H4K5ac in the hippocampus is prevalent throughout the genome and is a characteristic mark of actively transcribed genes. Enrichment of the H4K5ac at consensus transcription factor binding sites (TFBS) in the promoter and proximal to the transcription start site (TSS) confer the gene active transcription state.26
Furthermore, histone phosphorylation has also been identified as a mark of active gene transcription. Tiwari et al. showed by genome-wide ChIP-seq analysis that c-Jun NH2-terminal kinase (JNK) binds to a large set of active promoters during the differentiation of stem cells into neurons.27 They identified histone H3 Ser10 (H3S10) as a substrate for JNK, and further proved that activation of JNK signaling in post-mitotic neurons increases phosphorylation of H3S10 at JNK-bound promoters and promotes the expression of target genes that are related to stem cell differentiation. These results established an interesting link between JNK, H3S10, and promoters of a novel class of target genes during stem cell differentiation into neuron.
Additionally, Vaquero-Sedas's group analyzed the epigenetic status of telomeres using ChIP-seq data in Arabidopsis.28 These experiments revealed that Arabidopsis telomeres have a higher density of histone H3 than that of centromeres, which reflects their short nucleosomal organization. Furthermore, they further confirmed that Arabidopsis telomeres have lower levels of heterochromatic marks, higher levels of some euchromatic marks, and similar or lower levels of other euchromatic marks than centromeres.
The above examples strongly support the indispensably important role of the ChIP-seq approach in the field of chromosome and histone mark research.
ChIP-seq data analysis workflow
Given the popular use of the ChIP-seq technique, it is worth further discussing both data analysis and work flow of this technique.
The main analytical steps of ChIP-seq data analysis workflow are quality control, mapping, peak detection, motif analysis, annotation, and visualization.
As primary quality control on the data, raw reads with poor quality bases and with adaptors or other contaminants are filtered. Then, remaining reads are mapped to the reference genome using a short-read mapping program such as BWA, Bowtie, or SOAP.29-31 As secondary quality control step, reads mapped to a unique genomic position are kept, and reads mapped to the same genomic position are filtered as they suggest potential PCR or optical duplicates. The percentage of uniquely mapped reads is expected to be over 50% for a good quality library.32
In a high quality ChIP-seq experiment, mapped reads are clustered (enriched) around the genomic locations where ChIPed protein binds. Peak detection algorithms are used to identify either enriched regions compared to a control sample (e.g. nonspecific antibody) or differential binding of a protein between two or more biological conditions. Depending on the underlying statistical model, a significance metric (e.g. p-value, q-value) is assigned to each putative peak and the False Discovery Rate (FDR) determined to correct for multiple comparisons. Selection of the peak detection algorithm is the key to meaningful interpretation of ChIP-seq data. MACS and SPP are well-established and commonly accepted peak detection algorithms.33,34 A detailed comparison of peak detection algorithms and practical guidelines is already published.35,36 As the last analytical step, enriched regions identified by the peak detection algorithm are assembled and evaluated for motif discovery using a motif analysis program, such as MEME-ChIP or peak-motifs.37,38 Motif analysis is not only used to discover new binding motifs, but also to validate known motifs.
Alignment results can be visualized as ChIP-seq signal coverage tracks in the University of California Santa Cruz (UCSC) genome browser (genome.ucsc.edu).39 This browser is a web- based application that provides extensive genomic annotation including genes (e.g. refseq, Ensembl), SNPs, evolutionary conservation, sequence properties, and patterns (e.g., CpG islands, repeats), as well as tracks for regulatory elements (e.g., transcription factor binding sites, methylation) from the ENCODE consortium. This visualization allows interpretation of peaks in the context of functionally relevant genomic regions (e.g., genes, gene promoters, transcription start sites).
Perspective
ChIP-seq identifies sites of transcription factor binding, mechanisms of differential gene regulation, as well as the distribution of histone marks. Consortia such as the ENCyclopedia Of DNA Elements (ENCODE) have produced large datasets using manual protocols. Initially, DNA sequencing capacity and cost were major barriers to large scale ChIP-seq, but sequencing capacity has increased rapidly and the immunoprecipitation step has now emerged as being rate-limiting. The tedious and often variable results from one practitioner to another demand an automated ChIP protocol that could improve data reproducibility and efficiency per experiment.
ChIP-seq – attempts to be automatic
In order to scale up the number of ChIP reactions, Wu et al. first introduced a high throughput, low consumption, automated microfluidic device for ChIP named high throughput ChIP (HTChIP) or the microfluidic ChIP assay.40 HTChIP is an extremely effective tool that uses a microfluidic device to simultaneously process 16 distinct biological samples. Thus using the microfluidic device, complex protocols with multiple components can be streamlined in a systematic manner. The immunoprecipitation efficiency observed in HTChIP performed in a microfluidic environment is much higher than that of conventional ChIP assay.
More excitingly, Gasper et al. recently reported a fully automated robotic ChIP (R-ChIP) technique.41 This pipeline is fully automated and does not require manual intervention at any step. It utilizes a multipurpose programmable liquid handling robotic platform (Tecan Freedom EVO 200) that allows up to 96 reactions without compromising yield or quality. Thus R-ChIP not only increases ChIP throughput, uniformity, and quality, but also reduces human time and error.
ChIP-seq and database
ChIP-seq knowledge is being rapidly added to public repositories leading to an emergence of several databases for evaluation of gene expression and protein-protein networks. Chacon et al recently built a BloodChIP database (http://www.med.unsw.edu.au/CRCWeb.nsf/page/ BloodChIP) that has the genome-wide binding profiles of 7 key haematopoietic transcription factors (TFs) in human stem/progenitor cells.42 BloodChIP compares binding profiles of TFs integrated with chromatin marks and expression data with binding profiles in normally differentiated and leukemic cells. It thus facilitates understanding of the dynamic changes of the transcriptional network as haematopoietic stem progenitor cells (HSPCs) differentiate or transform into malignant cells. Data from BloodChIP can be obtained as TF-gene and protein-protein networks and it evaluates the association of genes with cellular processes and tissue expression.
Moreover, Wang et al. constructed the chromatin regulator (CR) Cistrome database, available online at http://compbio.tongji.edu.cn/cr and http://cistrome.org/cr/.43 CR Cistrome is a ChIP-seq database for the CRs and histone modification (HM) linkages in human and mouse. This database is a collection of all publicly available ChIP-seq data on CRs in human and mouse. It can be categorized into four cohorts: the reader, writer, eraser, and remodeler cohorts. This database also provides ChIP-seq analysis data for the targeted HM readers, writers, and erasers, and elucidates the relationships between them in the form of schematics. This database is of great importance for the development and hypotheses test regarding transcriptional and epigenetic regulation.
Due to the invaluable information added to our existing knowledge, considerable progress has been made in our understanding of chromatin structure, nuclear events involved in transcription, transcription regulatory networks, and histone modifications. With all the recent advances in ChIP-seq variations, it would be reasonable to envision that more exciting advances in ChIP-seq will be made in the years to come, and this effort will undoubtedly pave the way to the future of ChIP-seq in diagnostic applications.
Funding Statement
The research is supported by grants 23-862-07TL (to TL) and 036433730102 (to TL).
Acknowledgments
We thank Ms. Lisa King at the Department of Pharmacology and Toxicology at Indiana University School of Medicine for her generous and professional help with revising this review.
References
- 1. Nowak DE, Tian B, Brasier AR. Two-step cross-linking method for identification of NF-κB gene network by chromatin immunoprecipitation. Biotechniques 2005; 39:715-25; PMID:16315372; http://dx.doi.org/ 10.1007/978-1-61779-376-9_7 [DOI] [PubMed] [Google Scholar]
- 2. Weinmann SA, Bartley MS, Zhang T, Zhang QM, Farnham JP. Use of chromatin immunoprecipitation to clone novel E2F target promoters. Mol. Cell Biol 2001; 21:6820-32; PMID:11564866; http://dx.doi.org/ 10.1128/MCB.21.20.6820-6832.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Weinmann SA, Farnham JP. Identification of unknown target genes of human transcription factors using chromatin immunoprecipitation. Methods 2002; 26:37-47; PMID:12054903; http://dx.doi.org/ 10.1016/S1046-2023(02)00006-3 [DOI] [PubMed] [Google Scholar]
- 4. Weinmann SA, Yan SP, Oberley JM, Huang HT, Farnham JP. Isolating human transcription factor targets by coupling chromatin immunoprecipitation and CpG island microarray analysis. Genes Dev 2002; 16:235-44; PMID:11799066; http://dx.doi.org/ 10.1101/gad.943102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Spencer AV, Sun MJ, Li L, Davie RJ. Chromatin immunoprecipitation: a tool for studying histone acetylation and transcription factor binding. Methods 2003; 31:67-5; PMID:12893175; http://dx.doi.org/ 10.1016/S1046-2023(03)00089-6 [DOI] [PubMed] [Google Scholar]
- 6. Yan Y, Kluz T, Zhang P, Chen BH, Costa M. Analysis of specific lysine histone H3 and H4 acetylation and methylation status in clones of cells with a gene silenced by nickel exposure. Toxicol Appl Pharmacol 2003; 190:272-7; PMID:12902198; http://dx.doi.org/ 10.1016/S0041-008X(03)00169-8 [DOI] [PubMed] [Google Scholar]
- 7. Ren B, Robert F, Wyrick JJ, Aparicio O, Jennings GE, Simon I, Zeitlinger J, Schreiber J. Genome-wide location and function of DNA binding proteins. Science 2004; 290:2306-9; PMID:11125145; http://dx.doi.org/ 10.1126/science.290.5500.2306 [DOI] [PubMed] [Google Scholar]
- 8. Gilmour DS, Lis JT. Detecting protein-DNA interactions in vivo: Distribution of RNA polymerase on specific bacterial genes. Proc Natl Acad Sci 1984; 81:4275-9; PMID:6379641; http://dx.doi.org/ 10.1073/pnas.81.14.4275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gilmour DS, Lis JT. In vivo interactions of RNA polymerase II with genes of Drosophila melanogaster. Mol Cell Biol 1985; 5:2009-18; PMID:3018544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gilmour DS, Lis JT. RNA polymerase II interacts with the promoter region of the noninduced hsp70 gene in Drosophila melanogaster cells. Mol Cell Biol 1986; 6:3984-9; PMID:3099167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Solomon MJ, Larsen PL, Varshavsky A. Mapping protein-DNA interactions in vivo with formaldehyde: Evidence that histone H4 is retained on a highly transcribed gene. Cell 1988; 53:937-47; PMID:2454748; http://dx.doi.org/ 10.1016/S0092-8674(88)90469-2 [DOI] [PubMed] [Google Scholar]
- 12. Singal R, van Wert MJ, Ferdinand L. Methylation of alpha-type embryonic globin gene alpha pi represses transcription in primary erythroid cells. Blood 2002; 100:4217-22; PMID:12393573; http://dx.doi.org/ 10.1182/blood-2002-02-0457 [DOI] [PubMed] [Google Scholar]
- 13. Orlando V, Strutt H, Paro R. Analysis of chromatin structure by in vivo formaldehyde cross-linking. Methods 1997; 11:205-14; PMID:8993033; http://dx.doi.org/ 10.1006/meth.1996.0407 [DOI] [PubMed] [Google Scholar]
- 14. Wells J, Farnham JP. Characterizing transcription factor binding sites using formaldehyde crosslinking and immunoprecipitation. Methods 2002; 26:48-56; PMID:12054904; http://dx.doi.org/ 10.1016/S1046-2023(02)00007-5 [DOI] [PubMed] [Google Scholar]
- 15. Buck JM, Lieb DJ. ChIP-chip: considerations for the design, analysis, and application of genome-wide chromatin immuno-precipitation experiments. Genomics 2004; 83:349-60; PMID:14986705; http://dx.doi.org/ 10.1016/j.ygeno.2003.11.004 [DOI] [PubMed] [Google Scholar]
- 16. Mardis ER. ChIP-seq: Welcome to the new frontier. Nat Methods 2007; 4:613-4; PMID:17664943; http://dx.doi.org/ 10.1038/nmeth0807-613 [DOI] [PubMed] [Google Scholar]
- 17. Mardis ER. Next-generation DNA sequencing methods. Annu. Rev Genomics Hum Genet 2008; 9:387-402; PMID:18576944; http://dx.doi.org/ 10.1146/annurev.genom.9.081307.164359 [DOI] [PubMed] [Google Scholar]
- 18. Carey MF, Peterson CL, Smale ST. Chromatin Immunoprecipitation (ChIP). Cold Spring Harb Protoc 2009; 9:pdb.prot5279; PMID:20147264; http://dx.doi.org/ 10.1101/pdb.prot5279 [DOI] [PubMed] [Google Scholar]
- 19. Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods 2007; 4:651-7; PMID:17558387; http://dx.doi.org/ 10.1038/nmeth1068 [DOI] [PubMed] [Google Scholar]
- 20. Chng KR, Chang CW, Tan SK, Yang C, Hong SZ, Sng NY, Cheung E. A transcriptional repressor co-regulatory network governing androgen response in prostate cancers. EMBO J 2012; 31:2810-3; PMID:22531786; http://dx.doi.org/ 10.1038/emboj.2012.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chng KR, Cheung E. Sequencing the transcriptional network of androgen receptor in prostate cancer. Cancer Lett 2013; 340:254-60; PMID:23196061; http://dx.doi.org/ 10.1016/j.canlet.2012.11.009 [DOI] [PubMed] [Google Scholar]
- 22. Niu W, Lu ZJ, Zhong M, Sarov M, Murray JI, Brdlik CM, Janette J, Chen C, Alves P, Preston E, et al. Diverse transcription factor binding features revealed by genome-wide ChIP-seq in C. elegans. Genome Res 2011; 21:245-54; PMID:21177963; http://dx.doi.org/ 10.1101/gr.114587.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lu T, Yang M, Huang DB, Wei H, Ozer GH, Ghosh G, Stark GR. Role of lysine methylation of NF-κB in differential gene regulation. Proc Natl Acad Sci U S A. 2013; 110:13510-5; PMID:23904479; http://dx.doi.org/ 10.1073/pnas.1311770110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Paakinaho V, Kaikkonen S, Makkonen H, Benes V, Palvimo JJ. SUMOylation regulates the chromatin occupancy and anti-proliferative gene programs of glucocorticoid receptor. Nucleic Acids Res 2014; 42:1575-92; PMID:24194604; http://dx.doi.org/ 10.1093/nar/gkt1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007; 448:553-60; PMID:17603471; http://dx.doi.org/ 10.1038/nature06008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Park CS, Rehrauer H, Mansuy IM. Genome-wide analysis of H4K5 acetylation associated with fear memory in mice. BMC Genomics 2013; 14:539; PMID:23927422; http://dx.doi.org/ 10.1186/1471-2164-14-539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tiwari VK, Stadler MB, Wirbelauer C, Paro R, Schübeler D, Beisel C. A chromatin-modifying function of JNK during stem cell differentiation. Nat Genet 2011; 44:94-100; PMID:22179133; http://dx.doi.org/ 10.1038/ng.1036 [DOI] [PubMed] [Google Scholar]
- 28. Vaquero-Sedas MI, Luo C, Vega-Palas MA. Analysis of the epigenetic status of telomeres by using ChIP-seq data. Nucleic Acids Res 2012; 40:e163; PMID:22855559; http://dx.doi.org/ 10.1093/nar/gks730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25:1754-60; PMID:19451168; http://dx.doi.org/ 10.1093/bioinformatics/btp698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009; 10: R25; PMID:19261174; http://dx.doi.org/ 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li R, Li Y, Kristiansen K, Wang J. SOAP: short oligonucleotide alignment program. Bioinformatics 2008; 24:713-4; PMID:18227114; http://dx.doi.org/ 10.1093/bioinformatics/btn025 [DOI] [PubMed] [Google Scholar]
- 32. Shin H, Liu T, Duan X, Zhang Y, Liu XS. Computational methodology for ChIP-seq analysis. Quant Biol 2013; 1:54-70: http://dx.doi.org/ 10.1007/s40484-013-0006-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang Y, Liu T, Meyer C, Eeckhoute J, Johnson D, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based Analysis of ChIP-seq (MACS). Genome Biol 2008; 9:R137; PMID:18798982; http://dx.doi.org/ 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kharchenko PV, Tolstorukov MY, Park PJ. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat Biotechnol 2008; 26:1351-59; PMID:19029915; http://dx.doi.org/ 10.1038/nbt.1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wilbanks EG, Facciotti MT. Evaluation of Algorithm Performance in ChIP-seq Peak Detection. PLoS ONE 2010; 5:e1147; PMID:20628599; http://dx.doi.org/ 10.1371/journal.pone.0011471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bailey T, Krajewski P, Ladunga I, Lefebvre C, Li Q, Liu T, Madrigal P, Taslim C, Zhang J. Practical Guidelines for the Comprehensive Analysis of ChIP-seq Data. PLoS Comput Biol 2013; 9:e1003326; PMID:24244136; http://dx.doi.org/ 10.1371/journal.pcbi.1003326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Machanick P, Bailey TL. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 2011; 27:1696-7; PMID:21486936; http://dx.doi.org/ 10.1093/bioinformatics/btr189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thomas-Chollier M, Herrmann C, Defrance M, Sand O, Thieffry D, van Helden J. RSAT peak- motifs: motif analysis in full-size ChIP-seq data- sets. Nucleic Acids Res 2012; 40:e31; PMID:22156162; http://dx.doi.org/ 10.1093/nar/gkr1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res 2002; 12:996-1006; PMID:12045153; http://dx.doi.org/ 10.1101/gr.229102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wu AR, Kawahara T, Rapicavoli NA, van Riggelen J, Shroff EH, Xu L, Felsher DW, Chang HY, Quake SR. High throughput automated chromatin immunoprecipitation as a platform for drug screening and antibody validation. Lab Chip 2012; 12:2190-8; PMID:22566096; http://dx.doi.org/ 10.1039/c2lc21290k [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gasper WC, Marinov GK, Pauli-Behn F, Scott MT, Newberry K, DeSalvo G, Ou S, Myers RM, Vielmetter J, Wold BJ. Fully automated high-throughput chromatin immunoprecipitation for ChIP-seq: Identifying ChIP-quality p300 monoclonal antibodies. Sci Rep 2014; 4:5152; PMID:24919486; http://dx.doi.org/ 10.1038/srep05152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chacon D, Beck D, Perera D, Wong JW, Pimanda JE. BloodChIP: a database of comparative genome-wide transcription factor binding profiles in human blood cells. Nucleic Acids Res 2014; 42:D172-7; PMID:24185696; http://dx.doi.org/ 10.1093/nar/gkt1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang Q, Huang J, Sun H, Liu J, Wang J, Wang Q, Qin Q, Mei S, Zhao C, Yang X, et al. CR Cistrome: a ChIP-seq database for chromatin regulators and histone modification linkages in human and mouse. Nucleic Acids Res 2014; 42:D450-8; PMID:24253304; http://dx.doi.org/ 10.1093/nar/gkt1151 [DOI] [PMC free article] [PubMed] [Google Scholar]