

Abstract
In principle, a decline in base excision repair (BER) efficiency with age should lead to genomic instability and ultimately contribute to the onset of the aging phenotype. Although multiple studies have indicated a negative link between aging and BER, the change of BER efficiency with age in humans has not been systematically analyzed. Here, with foreskin fibroblasts isolated from 19 donors between 20 and 64 y of age, we report a significant decline of BER efficiency with age using a newly developed GFP reactivation assay. We further observed a very strong negative correlation between age and the expression levels of SIRT6, a factor which is known to maintain genomic integrity by improving DNA double strand break (DSB) repair. Our mechanistic study suggests that, similar to the regulatory role that SIRT6 plays in DNA DSB repair, SIRT6 regulates BER in a PARP1-depdendent manner. Moreover, overexpression of SIRT6 rescues the decline of BER in aged fibroblasts. In summary, our results uncovered the regulatory mechanisms of BER by SIRT6, suggesting that SIRT6 reactivation in aging tissues may help delay the process of aging through improving BER.
Keywords: aging, base excision repair, mono-ADP-ribosylation, PARP1, SIRT6, SIRTUIN
Introduction
One of the major hallmarks of aging is increased genomic instability, typically conferred by the accumulation of genetic alterations with age.1,2 Dysfunctional base excision repair (BER) machinery may give rise to mutations and even chromosomal rearrangements, thereby contributing to aging related loss of tissue functionality and other types of diseases associated with aging.
On average, each cell in the human body sustains between 100 to 500 8-hydroxyguanine lesions per day as a result of exposure to either endogenous or exogenous reactive oxygen species (ROS) and alkylating agents.3 The BER pathway has evolved to repair such damage to the DNA bases of the genome.4 The repair machinery of BER is composed of several factors, including: DNA glycosylases such as OGG1, AP endonuclease APE1, Pol β, PARP1, XRCC1, Lig III and many other ancillary factors.5 In brief, in response to base damage in DNA, DNA glycosylases remove the covalent bond connecting the base and the sugar, leaving an abasic site which may be recognized by APE1. APE1 then cleaves the 5′ side of the abasic site, creating a single strand break in DNA. Eventually PARP1 helps recruit XRCC1, Pol β, Lig III and other repair factors to complete the repair process.6
Several pieces of evidence indicate that the efficacy of BER may negatively change with age. The expression level of Pol β in both rat and mouse brain extracts declines with age.7-9 The number of apurinic/apyrimidinic sites (AP sites) generated in response to H2O2 or MMS by glycosylases is higher in young cells than in old cells, indicating a decrease of glycosylase activity in old cells.10 Additionally, the ability of 2 DNA glycosylases, OGG1 and UDG, to translocate from the cytoplasm to the matrix of mitochondria is impaired in the livers of old mice and in presenescent human fibroblasts.11,12 However, due to the lack of a tool for rapid and quantitative analysis of BER efficiency, the relationship between age and BER efficiency, and the age associated regulation of BER, has not been analyzed in human samples.
SIRT6 is one of the 7 mammalian homologs of yeast Sir2, whose overexpression robustly extends lifespan in Saccharomyces cerevisiae.13 SIRT6 overexpression significantly prolongs lifespan in mice,14 strongly suggesting it plays an essential role in aging. Several studies indicate that SIRT6 might execute its anti-aging function by participating in DNA double strand break (DSB) repair.15-20 In response to DNA damage, SIRT6 helps recruit SNF2H, a chromatin remodeler, therefore facilitating the recruitment of 53BP1 and BRCA1 to damage sites.19 SIRT6 promotes classical nonhomologous end joing (NHEJ) by stabilizing DNA-PKcs at damage site.18 It also stimulates alternative NHEJ and homologous recombination (HR) by activating PARP1 through its mono-ADP-ribosylation activity.16,21 In addition, it deacetylates CtIP to promote end resection during HR.15 In presenescent cells, restoration of SIRT6 strongly improves HR in a PARP1 dependent manner.17 Intriguingly, although the molecular mechanisms remain to be elucidated, SIRT6 was first proposed to be involved in BER.22 Knocking out SIRT6 in mice leads to a phenotype of progeria.22 In comparison to wild type cells, SIRT6 knockout cells are more sensitive to MMS, an alkylating agent, and H2O2, an oxidative agent, both of which cause damage to DNA bases. This hypersensitivity, caused by loss of SIRT6, could be rescued by overexpressing the Pol β dRP lyase domain, strongly implicating the involvement of SIRT6 in regulating the BER pathway.
Here, for the first time, we report a GFP plasmid reactivation assay for measuring BER efficiency and systematically examine the BER efficiency in 19 fibroblast cell lines isolated from donors at different ages. We observed a significant decline of BER efficiency with age. The age dependent impairment of BER cannot be explained by the change of the BER factor expression. Instead, SIRT6 expression strongly negatively correlates with age. More importantly, SIRT6 expression positively correlates with BER efficiency, confirming its role in BER pathway. Further mechanistic analysis suggests that SIRT6 regulates BER in a PARP1 dependent manner. Overexpression of SIRT6 in cells from an old donor successfully restores the level of BER efficiency to that of cells from young donors, indicating that SIRT6 may be a potential target for delaying aging through reactivation of the BER pathway in aging cells.
Results
BER efficiency declines with age
To quantitatively and rapidly examine BER efficiency, we first developed a GFP plasmid reactivation assay. We treated pEGFP-N1 vector with methylene blue and visible light (MB+VL) in vitro, which causes oxidative damage to the vector, mainly in the form of 7,8-dihydroxy-8-oxoguanine and other single base modifications.23,24 After transfection of the damaged pEGFP-N1 vector into cells, successful BER on pEGFP-N1 will restore the expression of EGFP gene, eventually turning cells fluorescent green, which can be scored by FACS (Fig. 1A). We found that the treatment with increasing concentration of MB negatively affects the restoration of EGFP in HCA2-hTERT, an immortalized foreskin fibroblast cell line, confirming that MB+VL treatment results in DNA damage (Fig. 1B). To further test if the restoration relies on BER pathway, we knocked down PARP1, a known BER factor, in HCA2-hTERT cells (Fig. 1C; Fig. S1). We then transfected the damaged pEGFP-N1 plasmid, treated with 1000 μM MB+VL, together with pDsRed2-N1, as an internal control for normalizing transfection efficiency, into control and PARP1 knocking down cells. The ratio of GFP+ cell number versus DsRed+ cell number is employed as the measure of BER efficiency. We observed a significant reduction of this ratio in PARP1 knock down cells, suggesting that this method for assaying BER efficiency is valid (Fig. 1C).
Figure 1.
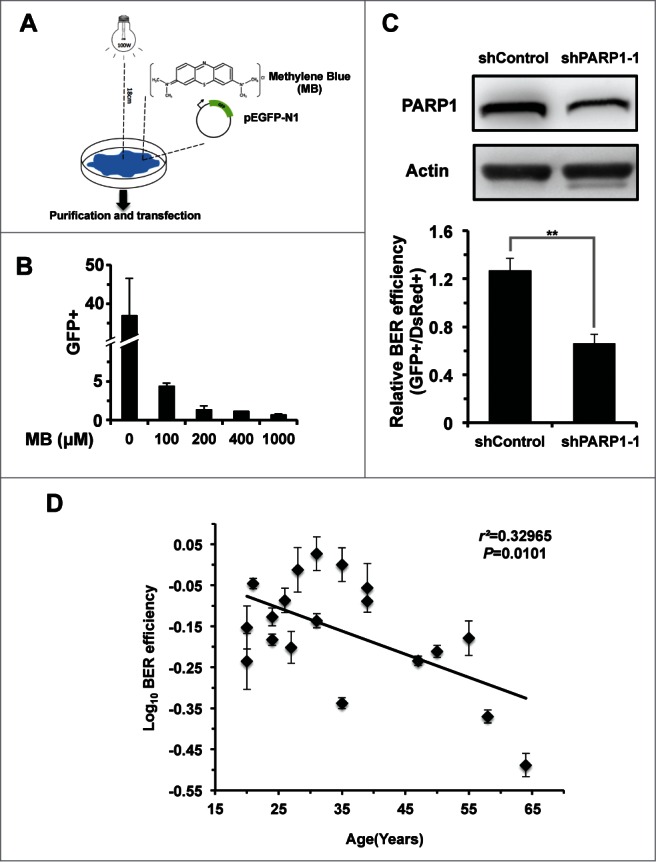
BER efficiency declines with age. (A) Schematic depiction of the plasmid reactivation assay used to analyze BER efficiency. Ten micrograms of pEGFP-N1 were mixed with methylene blue at the indicated concentrations, followed by a 60-minute irradiation treatment with visible light generated by a 100-walt bulb at a distance of 18 cm. Then the damaged pEGFP-N1 vector was purified from the mixture using the Axygen cleanup kit (Axygen, Cat. #AP-PCR-250) before 0.05 μg damaged pEGFP-N1 was transfected to fibroblasts for further FACS analysis. (B) MB+VL treatment damages the expression of EGFP gene in comparison to an untreated control. (C) Validation of the BER analysis assay. 0.05 μg of MB + VL treated pEGFP-N1 together with 0.005 μg of pDsRed2-N1, for normalizing differences in transfection efficiency, was transfected into control or PARP1 depleted HCA2-hTERT cells. Then the ratio of GFP+ cells vs. DsRed+ cells was employed as the measure of BER efficiency. (D) BER efficiency negatively correlates with age. Damaged pEGFP-N1, by MB+VL, and pDsRed2-N1 were co-transfected into foreskin fibroblast cell lines isolated from donors at different ages. When transfections were performed, all cell lines were at a population doubling (PD) number of ∼16. All experiments were repeated at least 3 times. Error bars represent standard deviation (S.D.). ** P < 0.01
To examine whether BER efficiency changes with age, we first isolated 19 foreskin fibroblast cell lines from donors of different ages, ranging from 20 to 64 y old. Using the EGFP reactivation assay, we systematically tested the BER efficiency of the 19 cell lines. This analysis revealed a significant negative correlation between BER repair efficiency and age (Fig. 1D), implying that aging may impair the BER pathway.
Expression of SIRT6 but not other BER factors significantly decreases with age
To elucidate the molecular mechanisms underlying this age associated decline in BER efficiency, we analyzed the expression level of various BER factors in cells from different aged donors. We observed weak a negative correlation between each Polβ and XRCC1 expression levels and age (0.05 < P < 0 .1) while no significant correlation between any other known BER factor and age was detected (Fig. 2A and B), suggesting that changes in expression level of known BER factors is likely not responsible for the decline of BER with age.
Figure 2.
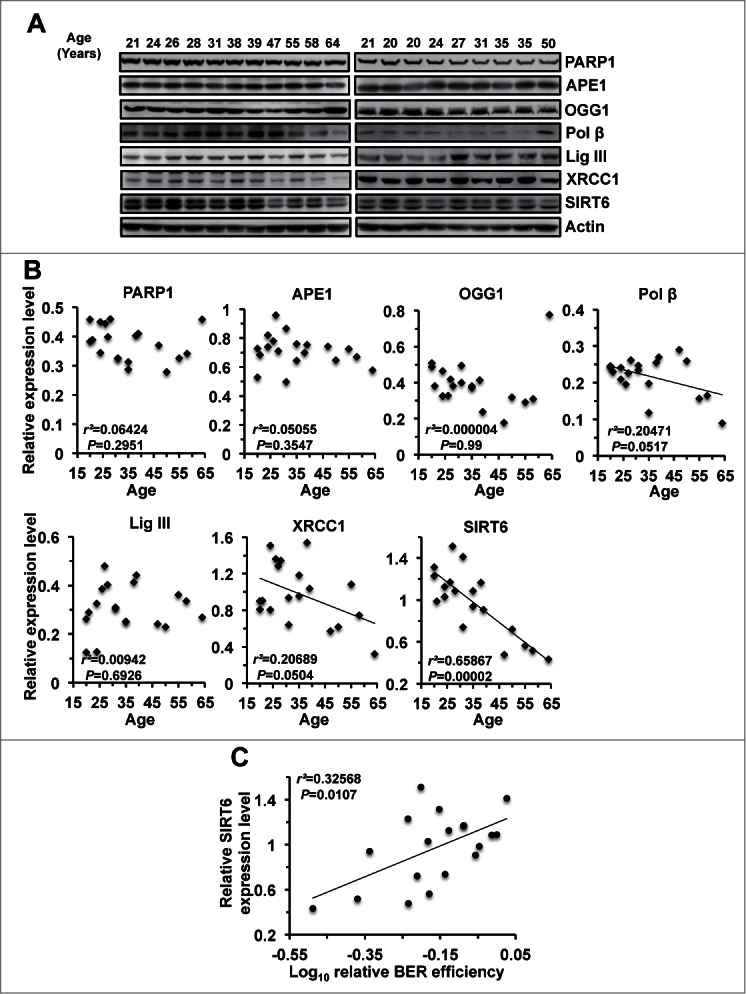
SIRT6 expression level negatively correlates with age but positively correlates with BER efficiency. (A) Western blot showing changes in BER factor expression levels with age. Nineteen cell lines, isolated from donors of different ages, were split 48 hours before being harvested for protein extract. 50 μg of protein extracts were loaded on SDS-PAGE gels for further Western blot analysis. (B) Expression levels of SIRT6, but not other major BER factors, had a significant negative correlation with age. The expression of various BER factors was quantified with ImageJ software. The quantified data, together with age, was further analyzed with Excel software. (C) Expression level of SIRT6 positively correlates with BER efficiency.
SIRT6 has been proposed to be involved in regulating BER. Therefore, we examined the expression level of SIRT6 with age. Strikingly, SIRT6 expression level exhibits an extremely strong negative correlation with age (r2 = 0.65867, P < 0.0001), suggesting that SIRT6 is possibly linked to the age associated decrease of BER. To further confirm that SIRT6 participates in BER, we examined the correlation between SIRT6 expression level and BER efficiency in skin fibroblasts extracted from patients. We observed that SIRT6 expression and BER efficiency strongly correlate (r2 = 0.32568, P < 0.05) (Fig. 2C), confirming that SIRT6 takes part in BER pathway.
SIRT6 is indispensable for BER pathway
To test if SIRT6 is required for BER, we examined BER efficiency in the presence of SIRT3 or SIRT6 overexpression (Fig. 3A). SIRT3 has been implicated in regulating the BER pathway by deacetylating OGG1,25 and we observed a significant stimulatory effect on BER by SIRT3 overexpression, suggesting that our reactivation assay is an appropriate tool for measuring BER efficiency. SIRT6 overexpression increases BER efficiency by ∼2 fold (Fig. 3B). In SIRT6 knockout MEFs (Fig. 3C), BER efficiency was reduced by ∼40% (Fig. 3D), and restoration of SIRT6 rescued the decline in SIRT6 knockout MEFs (Fig. S2). In summary, by testing BER efficiency in the presence of SIRT6 overexpression and in the absence of SIRT6, we conclude that SIRT6 is required for efficient repair by the BER pathway.
Figure 3.
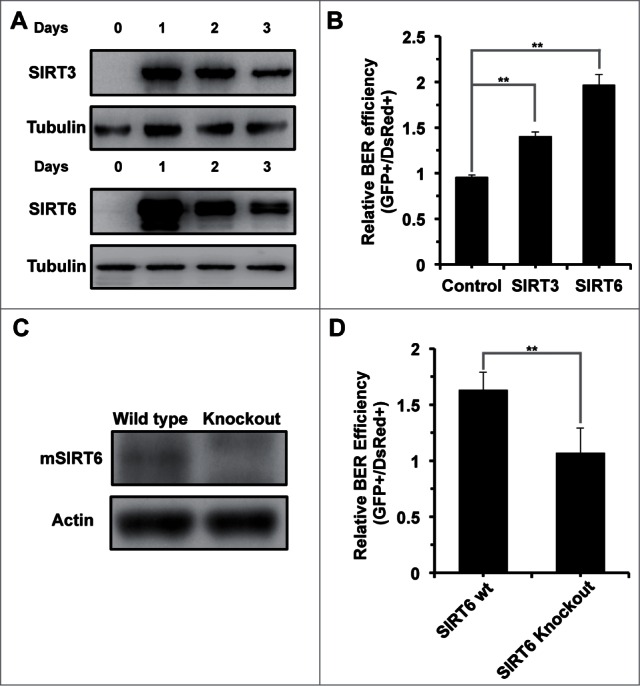
SIRT6 is required for repair via the BER pathway. (A) Transient overexpression of SIRT3 and SIRT6 in HCA2-hTERT cells. A vector for expressing SIRT3 or SIRT6 was transfected into HCA2-hTERT cells. At the indicated time points, cells were harvested for Western blot analysis of sirtuin expression. (B) Overexpression of SIRT6 promotes BER efficiency. A pcDNA3.1 control vector or a vector encoding SIRT3 or SIRT6 was co-transfected with damaged pEGFP-N1 and pDsRed2-N1. Seventy-2 hours later, cells were harvested for analysis of BER efficiency using FACSverse. (C) Western blot analysis of mSIRT6 in wild type and SIRT6 knockout MEFs. Exponentially growing MEFs were harvested for to quantify protein expression by Western blot analysis. (D) BER efficiency is reduced in SIRT6 knockout MEFs. 0.05 μg of MB+VL treated pEGFP-N1 and 0.005 μg pDsRed2-N1 were co-transfected into wild type or SIRT6 knockout MEFs 72 hours before cells were harvested for FACS analysis. All the experiments were repeated at least 3 times and error bars represent SD. ** P < 0.01
SIRT6 regulates BER in a PARP1-dependent manner
SIRT6 has 2 enzymatic activities, deacetylase and mono-ADP-ribosyl transferase, both of which are NAD+ dependent. To determine which enzymatic activity is essential to the BER pathway, we created several mutants to distinguish the 2 enzymatic activities. SIRT6 G60A has only deacetylase activity while SIRT6 R65A retains only mono-ADP-ribosyl transferase actity, and SIRT6 S56Y is catalytically inactive.16 Overexpression of 3 SIRT6 mutants failed to stimulate the BER efficiency, suggesting that both enzymatic activities are necessary for BER (Fig. 4A).
Figure 4.
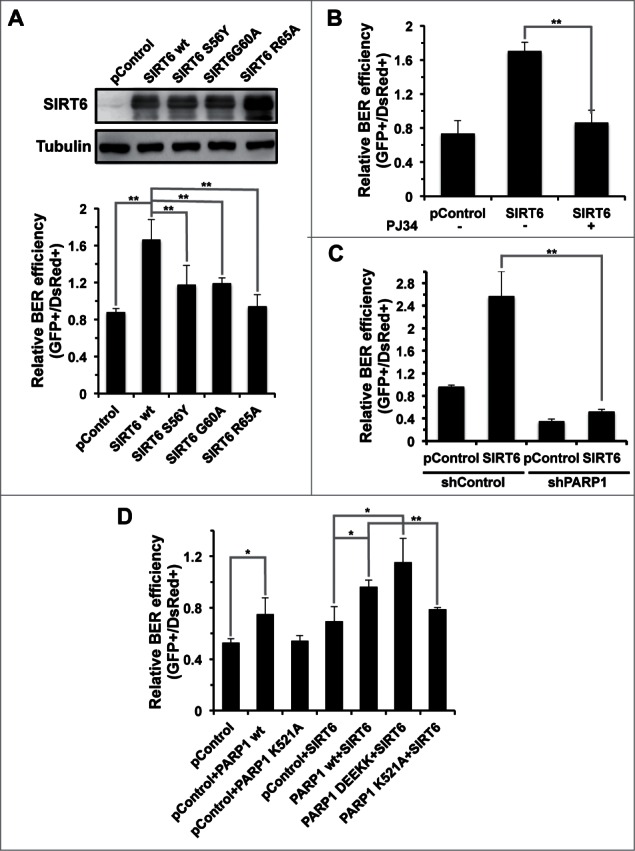
The regulation of BER by SIRT6 is mediated by PARP1. (A) The promotion of the BER pathway by SIRT6 is dependent on both its deacetylase and mono-ADP-ribosyltransferase activities. A control vector or vectors expressing SIRT6 wt or SIRT6 mutants were transfected into HCA2-hTERT cells. Cells were harvested at 24 hours post transfection for Western blot analysis of overexpression. The overexpression effect of SIRT6 wt or mutants on BER was analyzed in the same way as described above, in Figure 3. (B) PJ34 pretreatment abolishes the stimulatory effect of SIRT6 on BER. HCA2-hTERT cells were pretreated with 20 μM PJ34 for 24 hours before transfection with damaged pEGFP-N1 and pDsRed2-N1. After transfection, cells were grown in complete medium supplemented with 20 μM PJ34 for 72 hours before analysis of BER efficiency. (C) SIRT6 fails to stimulate BER in PARP1 depleted HCA2-hTERT cells. A control vector or a SIRT6 expression vector was co-transfected with damaged pEGFP-N1 and pDsRed2-N1 into control HCA2-hTERT or PARP1 depleted HCA2-hTERT. FACS analysis was performed 72 hours later. (D) The K521 site in PARP1 is essential for activation of BER by SIRT6. A control plasmid, PARP1 wt or PARP1 mutant plasmid together with a SIRT6 expression vector were cotransfected into PARP1 depleted HCA2-hTERT cells for analysis of BER efficiency. PARP1 DEEKK represents PARP1 with D387, E488, E491, K498, and K524 mutated to Alanine. All experiments were repeated at least three times. Error bars represent S.D.. ** P < 0.01; * P < 0.05
Our previous report indicates that SIRT6 relies on its mono-ADP-ribosyl transferase activity to activate PARP1, which in turn stimulates repair of DNA double strand breaks.16 The participation of PARP1 in the BER process has been well characterized. It localizes to the damage site when single stranded DNA breaks are generated during the BER process, and helps to recruit downstream repair factors in order to complete the repair. We therefore examined if SIRT6 stimulates BER in a PARP1 dependent manner. Pretreating HCA2-hTERT cells with PJ34, a potent PARP1 inhibitor, suppressed the BER stimulation mediated by SIRT6 (Fig. 4B). In addition, in PARP1 knocking down cells, overexpression of SIRT6 failed to activate the BER pathway (Fig. 4C). From these results, we conclude that, similar to its regulatory role in DNA DSB repair, SIRT6 promotes BER in a PARP1 dependent manner.
We previously demonstrated that SIRT6 mono-ADP-ribosylates PARP1 at lysine 521, thereby stimulating the poly-ADP-ribosyl polymerase activity of PARP1 and eventually promoting DNA DSB repair. To test if SIRT6 activates BER via the same mechanism, we overexpressed SIRT6 and wild type PARP1 or its mutant versions in PARP1 depleted cells. We observed that BER was rescued by overexpression of PARP1 wt but not PARP1 K521A mutant in PARP1 depleting cells, suggesting that the site K521 is essential for the function of PARP1 in BER. We next found that restoration of PARP1 rescued the stimulatory effect on BER by SIRT6, further confirming that PARP1 mediates the BER activation by SIRT6. In addition, PARP1 K521A mutant overexpression impairs the stimulatory effect on BER while PAPR1 DEEKK, a mutant with other 5 ribosylation sites mutated (D387A, E488A, E491A, K498A, and K524A), retained the stimulatory effect, suggesting that SIRT6 modifies the site of K521 to activate PARP1 (Fig. 4D).
SIRT6 overexpression rescues the decline of BER in aging cells
Since the expression of XRCC1, Pol β and SIRT6 decline with aging, we tested if BER efficiency could be restored by supplementing these 3 factors. The ORFs of XRCC1 and Pol β were cloned into an expression vector with a CMV promoter. The vectors encoding the 3 factors, together with damaged pEGFP-N1 and the internal control pDsRed2-N1, were cotransfected into 2 cell lines derived from young (FF26, 26 y old) and old (FF64, 64 y old) donors (Fig. 5A), we examined the repair efficiency by FACS (Fig. 5B). In young cells, we did not observe any significant change resulting from XRCC1 or Pol β overexpression. Similar to the data acquired from immortalized HCA2 cells, SIRT6 overexpression stimulated BER efficiency by 63%. By contrast, in old cells, we detected a weak but significant increase of BER efficiency of 30% when XRCC1 was overexpressed. More remarkably, overexpression of SIRT6 in old cells greatly stimulated BER efficiency by 2.fold3-. We further examined if the stimulation of BER efficiency by SIRT6 in old cells was dependent on its enzymatic activities. Similar to the previous results, the 3 SIRT6 mutants failed to stimulate BER efficiency (Fig. S3), confirming both activities are required for the rescue in old cells.
Figure 5.
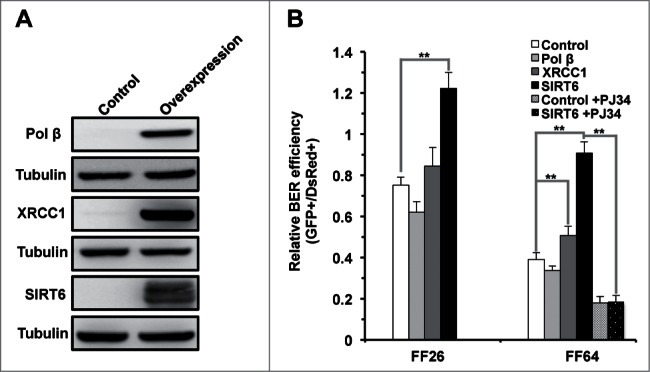
SIRT6 but not XRCC1 or Pol β rescues the decline of BER in old foreskin fibroblast cells. (A) Western blot analysis of XRCC1, Pol β and SIRT6 in the foreskin fibroblasts isolated from a 64 y old donor (FF 64). Vectors expressing the 3 genes were respectively transfected into the cells 24 hours before being harvested for protein extraction. Western blotting was in turn performed with the indicated antibodies. (B) SIRT6 restores the decrease of BER efficiency in the old fibroblasts in PARP1 dependent manner. Vectors expressing XRCC1, Pol β or SIRT6 with damaged pEGFP-N1 and pDsRed2-N1 were cotransfected into fibroblasts isolated from a 26 y and a 64 y old donors (FF26 and F64, respectively). Three days later, cells were harvested for FACS analysis. For PJ34 treatment experiments, PJ34 at 20 μM was supplemented to the complete growth medium for 24 hours before and 72 hours post transfection. The same transfection as Figure 4B was performed in FF64 cells. On day 3 post transfection, cells were harvested for FACS analysis. All the experiments were repeated at least 3 times. Error bars represent S.D.. ** P < 0.01
We demonstrated that SIRT6 mediates repair of DNA by the BER pathway, in a PARP1 dependent manner, in immortalized fibroblasts. We next tested if the restoration of BER efficiency by SIRT6 in aged cells was similarly dependent on PARP1. Accordingly, we pretreated the old cell line with PJ34 and analyzed the repair efficiency in the presence of SIRT6 overexpression. Consistent with our previous findings, inhibition of PARP1 by PJ34 suppressed the restoration of BER by SIRT6 (Fig. 5B), suggesting that SIRT6 rescues the decline of BER in old cells in a PARP1 dependent fashion.
Discussion
Despite a number of studies conducted in rodent models which have indicated that aging is associated with a dysfunctional BER pathway, the lack of an appropriate tool for analyzing BER efficiency and the difficulty associated with acquiring a sufficient number of human cell lines at different ages, very limited evidence was provided to link the aging and decline of BER efficiency, particularly in humans. Here, for the first time, with 19 foreskin fibroblast cell lines isolated from donors at different ages, we systematically analyzed the change of BER efficiency during aging. We successfully developed a novel fluorescent gene reactivation assay for measuring the BER efficiency, which is more quantitative and rapid than previously reported BER detection assays, such as quantifying the AP sites using biotin-containing aldehyde-reactive probes10 or estimating the BER glycosylase activity with biochemical approaches.5,26,27 Using this novel assay, we observed a trend of declining BER efficiency with age. The decrease in BER efficiency may result in a high rate of DNA mutations, which may lead to mutations in DNA repair genes, initiating a vicious cycle of inefficient BER, followed by accumulation of DNA damage, which in turn leads to more inefficient BER and ultimately culminates in cellular senescence and organismal aging.
Most of mechanistic studies on age related change in BER have been focused on the glycosylase activities and the the inducibility and the translocation of BER factors in response to DNA damages. 28 Whether the expression level change of BER factors contributes to aging has not been defined. Our analysis indicates that the compromised BER efficiency during aging is unlikely to be caused by expression changes of well-known BER factors, at least in human foreskin fibroblasts. First, we only observed a weak correlation between XRCC1, Pol β and age, and no correlation between PARP1, APE1, OGG1 or Lig III and age. Additionally, restoring XRCC1 or Pol β in old cells failed to restore the BER efficiency to a similar repair capability, such as that which is seen in young cells. However, strikingly, expression levels of SIRT6, which was first characterized as a BER factor, negatively correlate with age and positively correlate with BER efficiency, suggesting that SIRT6 regulates the decline of BER with age. Overexpression of SIRT6 in old cells rescues the decline of BER efficiency to the level of young cells. Our further investigation on the molecular mechanism by which SIRT6 mediates BER finally solved the mystery of how SIRT6 regulates this critical repair pathway. Regulation of both DNA DSBs and BER by SIRT6 is mediated by PARP1. The differences lay on the steps post PARP1 recruitment to DNA damage sites. For DNA DSBs, the activation of PARP1 may facilitate the recruitment of Rad51 and NBS1. For BER, it may accelerate the recruitment of XRCC1.
SIRT6 has both deacetylase and mono-ADP-ribosyltransferase activities. Our data indicates that in addition to mono-ADP-ribosyltransferase activity, SIRT6 as a deacetylase is also essential for BER pathway. Unfortunately, we did not identify any BER protein which could be deacetylated by SIRT6 (data not shown). We propose that SIRT6 might deacetylate a non-BER factor to indirectly regulate BER. For instance, by deacetylation, it might activate a kinase in response to DNA damage, which mediates the further activation of BER pathway. Recent advances in our understanding of epigenetic changes with aging have indicated that aged cells have an impaired capacity to induce the re-localization of chromatin remodelers to DNA damage sites.29 SIRT6 deacetylation activity might also be essential for the recruitment of chromatin remodelers to DNA damage sites in response to stresses. Alternatively, the deacetylation of chromatin remodelers by SIRT6 might be a critical step in remodeling nucleosomes on the DNA so that the repair machinery may function efficiently.
Recent advances of studies on SIRT6 have revealed that it is a crucial factor in the context of aging and aging associated diseases. SIRT6 overexpression extends mice lifespan and healthspan.14,30 SIRT6 is a tumor suppressor31 and its overexpression selectively kills different types of cancer cells.32 Here, in summary, we uncovered the regulatory mechanisms of BER by SIRT6 and provide another piece of evidence demonstrating that SIRT6 contributes to the maintenance of genomic stability, thereby possibly delaying aging and suppressing tumorigenesis. Therefore, developing a way of activating SIRT6 might open new avenue for treating aging associated diseases or improving the healthspan of human beings.
Materials and Methods
Cell culture
All primary cells were grown in MEM medium (Hyclone, Cat. #SH30234) supplemented with 15% fetal bovine serum (Life Technologies, Cat. # 16000), 1 × nonessential amino acid (Hyclone, Cat. # SH3023801) and 1% penicillin/streptomycin (Hyclone, cat. #SV30010). The cultures were maintained in a 5% CO2 and 3% O2 humidified incubator (Thermo Fisher Heracell 240i) at 37°C.
Generation of cell lines from donors at different ages
The use of human subjects was with the consent of donors, and was approved by the School of Life Sciences and Technology at Tongji University. All volunteer donors were in good health conditions when circumcision surgeries were performed. Fibroblasts were isolated from acquired tissues within 24 h post surgery. The isolation procedure of the human skin fibroblasts was as described.33
Plasmids and antibodies
The ORFs of XRCC1 and Pol β were amplified from cDNA of 293 cells and subsequently cloned into the pEGFP-N1 backbone by replacing EGFP ORF with SalI and NotI restriction enzymes. The information of other vectors encoding SIRT6 wt, SIRT6 mutants, PARP1 wt and PARP1 mutants is as described.16 The vector expressing SIRT3 was acquired from Addgene (Plasmid #13814). The pLKO1 vector was utilized to clone viral shRNA vector for knocking down endogenous PARP1. The sequences of PARP1 shRNAs are CGACCTGATCTGGAACATCAA and CCGAGAAATCTCTTACCTCAA. Antibodies recognizing hSIRT6 (Abcam, ab62738), mSIRT6 (Abcam, ab62738), SIRT3 (Abclonal, A5718), PARP1 (Abcam, ab6079), OGG1 (Abclonal, A2268), APE1 (Abclonal, A1117), XRCC1 (Abcam, ab1838), LigIII (Abclonal, A1887), Pol β (Abclonal, A1681), Actin-HRP (Santa Cruz, sc-47778), Tubulin (CMCTAG, AT0050) were used for Western blotting.
Transfections
All primary cells were seeded at a density of 1 × 105 cells/well on a 6-well plate, 48 hours before electroporation. Transfections were performed using NHDF solutions with the program of DT-130 on a Lonza 4D machine.
Western blot
Protein extracts were prepared at indicated time points, as described in figure legends. Thirty to 60 μg of protein extracts were loaded to SDS-PAGE gels before the proteins were transferred with Trans-blot machine (Bio-Rad, US). Western blots were subsequently performed with indicated antibodies.
FACS analysis
Cells were harvested 72 hours post transfection, resuspended in 1 × PBS and stored on ice before FACS analysis on FACS verse (BD Biosciences, USA). At least 20,000 cells were analyzed for each repeat of the experiments.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors would like to express their gratitude to the FACS core facility at Tongji University for the technical discussions and help.
Funding
The work was supported by grants from National Basic Research Program of China (973 Program, Grant No. 2013CB967600, 2015CB964800), the National Science Foundation of China (Grant No. 31371396), Shanghai Pujiang Program (Grant No. 13PJ1408300), the 1000 Youth Talents Program, the Open Project Program of State Key Laboratory of Natural Medicines, China Pharmaceutical University (Grant No. SKLNMKF201404) to Z.M. and the Cultural Program for Outstanding Young Talents in Tongji University, the National Science Foundation for Post-doctoral Scientists of China (Grant No. 2014M551446) to L.Z.
Supplemental Material
Supplemental data for this article can be accessed on thepublisher's website.
References
- 1. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell 2013; 153:1194-217; PMID:23746838; http://dx.doi.org/ 10.1016/j.cell.2013.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tigges J, Krutmann J, Fritsche E, Haendeler J, Schaal H, Fischer JW, Kalfalah F, Reinke H, Reifenberger G, Stuhler K, et al. The hallmarks of fibroblast ageing. Mech Ageing Dev 2014; 138:26-44; PMID:24686308; http://dx.doi.org/ 10.1016/j.mad.2014.03.004 [DOI] [PubMed] [Google Scholar]
- 3. Lindahl T. Instability and decay of the primary structure of DNA. Nature 1993; 362:709-15; PMID:8469282; http://dx.doi.org/ 10.1038/362709a0 [DOI] [PubMed] [Google Scholar]
- 4. Maynard S, Schurman SH, Harboe C, de Souza-Pinto NC, Bohr VA. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis 2009; 30:2-10; PMID:18978338; http://dx.doi.org/ 10.1093/carcin/bgn250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wilson DM, 3rd, Bohr VA. The mechanics of base excision repair, and its relationship to aging and disease. DNA Rep 2007; 6:544-59; PMID:17112792; http://dx.doi.org/ 10.1016/j.dnarep.2006.10.017 [DOI] [PubMed] [Google Scholar]
- 6. Caldecott KW. Mammalian single-strand break repair: mechanisms and links with chromatin. DNA Rep 2007; 6:443-53; PMID:17118715; http://dx.doi.org/ 10.1016/j.dnarep.2006.10.006 [DOI] [PubMed] [Google Scholar]
- 7. Cabelof DC, Raffoul JJ, Yanamadala S, Ganir C, Guo Z, Heydari AR. Attenuation of DNA polymerase beta-dependent base excision repair and increased DMS-induced mutagenicity in aged mice. Mutat Res 2002; 500:135-45; PMID:11890943; http://dx.doi.org/ 10.1016/S0027-5107(02)00003-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Intano GW, Cho EJ, McMahan CA, Walter CA. Age-related base excision repair activity in mouse brain and liver nuclear extracts. J Gerontol A Biol Sci Med Sci 2003; 58:205-11; PMID:12634285; http://dx.doi.org/ 10.1093/gerona/58.3.B205 [DOI] [PubMed] [Google Scholar]
- 9. Krishna TH, Mahipal S, Sudhakar A, Sugimoto H, Kalluri R, Rao KS. Reduced DNA gap repair in aging rat neuronal extracts and its restoration by DNA polymerase beta and DNA-ligase. J Neurochem 2005; 92:818-23; PMID:15686483; http://dx.doi.org/ 10.1111/j.1471-4159.2004.02923.x [DOI] [PubMed] [Google Scholar]
- 10. Atamna H, Cheung I, Ames BN. A method for detecting abasic sites in living cells: age-dependent changes in base excision repair. Proc Natl Acad Sci U S A 2000; 97:686-91; PMID:10639140; http://dx.doi.org/ 10.1073/pnas.97.2.686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Szczesny B, Bhakat KK, Mitra S, Boldogh I. Age-dependent modulation of DNA repair enzymes by covalent modification and subcellular distribution. Mech Ageing Dev 2004; 125:755-65; PMID:15541770; http://dx.doi.org/ 10.1016/j.mad.2004.07.005 [DOI] [PubMed] [Google Scholar]
- 12. Szczesny B, Hazra TK, Papaconstantinou J, Mitra S, Boldogh I. Age-dependent deficiency in import of mitochondrial DNA glycosylases required for repair of oxidatively damaged bases. Proc Natl Acad Sci U S A 2003; 100:10670-5; PMID:12960370; http://dx.doi.org/ 10.1073/pnas.1932854100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lin SJ, Defossez PA, Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science 2000; 289:2126-8; PMID:11000115; http://dx.doi.org/ 10.1126/science.289.5487.2126 [DOI] [PubMed] [Google Scholar]
- 14. Kanfi Y, Naiman S, Amir G, Peshti V, Zinman G, Nahum L, Bar-Joseph Z, Cohen HY. The sirtuin SIRT6 regulates lifespan in male mice. Nature 2012; 483:218-21; PMID:22367546; http://dx.doi.org/ 10.1038/nature10815 [DOI] [PubMed] [Google Scholar]
- 15. Kaidi A, Weinert BT, Choudhary C, Jackson SP. Human SIRT6 promotes DNA end resection through CtIP deacetylation. Science 2010; 329:1348-53; PMID:20829486; http://dx.doi.org/ 10.1126/science.1192049 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16. Mao Z, Hine C, Tian X, Van Meter M, Au M, Vaidya A, Seluanov A, Gorbunova V. SIRT6 promotes DNA repair under stress by activating PARP1. Science 2011; 332:1443-6; PMID:21680843; http://dx.doi.org/ 10.1126/science.1202723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mao Z, Tian X, Van Meter M, Ke Z, Gorbunova V, Seluanov A. Sirtuin 6 (SIRT6) rescues the decline of homologous recombination repair during replicative senescence. Proc Natl Acad Sci U S A 2012; 109:11800-5; PMID:22753495; http://dx.doi.org/ 10.1073/pnas.1200583109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McCord RA, Michishita E, Hong T, Berber E, Boxer LD, Kusumoto R, Guan S, Shi X, Gozani O, Burlingame AL, et al. SIRT6 stabilizes DNA-dependent protein kinase at chromatin for DNA double-strand break repair. Aging 2009; 1:109-21; PMID:20157594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Toiber D, Erdel F, Bouazoune K, Silberman DM, Zhong L, Mulligan P, Sebastian C, Cosentino C, Martinez-Pastor B, Giacosa S, et al. SIRT6 recruits SNF2H to DNA break sites, preventing genomic instability through chromatin remodeling. Mol Cell 2013; 51:454-68; PMID:23911928; http://dx.doi.org/ 10.1016/j.molcel.2013.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lombard DB. Sirtuins at the breaking point: SIRT6 in DNA repair. Aging 2009; 1:12-6; PMID:20157593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Van Meter M, Mao Z, Gorbunova V, Seluanov A. Repairing split ends: SIRT6, mono-ADP ribosylation and DNA repair. Aging 2011; 3:829-35; PMID:21946623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mostoslavsky R, Chua KF, Lombard DB, Pang WW, Fischer MR, Gellon L, Liu P, Mostoslavsky G, Franco S, Murphy MM, et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 2006; 124:315-29; PMID:16439206; http://dx.doi.org/ 10.1016/j.cell.2005.11.044 [DOI] [PubMed] [Google Scholar]
- 23. Floyd RA, West MS, Eneff KL, Schneider JE. Methylene blue plus light mediates 8-hydroxyguanine formation in DNA. Arch Biochem Biophys 1989; 273:106-11; PMID:2502945; http://dx.doi.org/ 10.1016/0003-9861(89)90167-7 [DOI] [PubMed] [Google Scholar]
- 24. Epe B, Pflaum M, Boiteux S. DNA damage induced by photosensitizers in cellular and cell-free systems. Mutati Res 1993; 299:135-45; PMID:7683082; http://dx.doi.org/ 10.1016/0165-1218(93)90091-Q [DOI] [PubMed] [Google Scholar]
- 25. Cheng Y, Ren X, Gowda AS, Shan Y, Zhang L, Yuan YS, Patel R, Wu H, Huber-Keener K, Yang JW, et al. Interaction of Sirt3 with OGG1 contributes to repair of mitochondrial DNA and protects from apoptotic cell death under oxidative stress. Cell Death Dis 2013; 4:e731; PMID:23868064; http://dx.doi.org/ 10.1038/cddis.2013.254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen D, Cao G, Hastings T, Feng Y, Pei W, O'Horo C, Chen J. Age-dependent decline of DNA repair activity for oxidative lesions in rat brain mitochondria. J Neurochem 2002; 81:1273-84; PMID:12068075; http://dx.doi.org/ 10.1046/j.1471-4159.2002.00916.x [DOI] [PubMed] [Google Scholar]
- 27. Imam SZ, Karahalil B, Hogue BA, Souza-Pinto NC, Bohr VA. Mitochondrial and nuclear DNA-repair capacity of various brain regions in mouse is altered in an age-dependent manner. Neurobiol Aging 2006; 27:1129-36; PMID:16005114; http://dx.doi.org/ 10.1016/j.neurobiolaging.2005.06.002 [DOI] [PubMed] [Google Scholar]
- 28. Gorbunova V, Seluanov A, Mao Z, Hine C. Changes in DNA repair during aging. Nucleic Acids Res 2007; 35:7466-74; PMID:17913742; http://dx.doi.org/ 10.1093/nar/gkm756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pegoraro G, Misteli T. The central role of chromatin maintenance in aging. Aging 2009; 1:1017-22; PMID:20157584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kanfi Y, Peshti V, Gil R, Naiman S, Nahum L, Levin E, Kronfeld-Schor N, Cohen HY. SIRT6 protects against pathological damage caused by diet-induced obesity. Aging Cell 2010; 9:162-73; PMID:20047575; http://dx.doi.org/ 10.1111/j.1474-9726.2009.00544.x [DOI] [PubMed] [Google Scholar]
- 31. Sebastian C, Zwaans BM, Silberman DM, Gymrek M, Goren A, Zhong L, Ram O, Truelove J, Guimaraes AR, Toiber D, et al. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell 2012; 151:1185-99; PMID:23217706; http://dx.doi.org/ 10.1016/j.cell.2012.10.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Van Meter M, Mao Z, Gorbunova V, Seluanov A. SIRT6 overexpression induces massive apoptosis in cancer cells but not in normal cells. Cell Cycle 2011; 10:3153-8; PMID:21900744; http://dx.doi.org/ 10.4161/cc.10.18.17435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Seluanov A, Hine C, Bozzella M, Hall A, Sasahara TH, Ribeiro AA, Catania KC, Presgraves DC, Gorbunova V. Distinct tumor suppressor mechanisms evolve in rodent species that differ in size and lifespan. Aging Cell 2008; 7:813-23; PMID:18778411; http://dx.doi.org/ 10.1111/j.1474-9726.2008.00431.x [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.