

Abstract
Centrosome amplification (CA), the presence of centrosomes that are abnormally numerous or enlarged, is a well-established driver of tumor initiation and progression associated with poor prognosis across a diversity of malignancies. Pancreatic ductal adenocarcinoma (PDAC) carries one of the most dismal prognoses of all cancer types. A majority of these tumors are characterized by numerical and structural centrosomal aberrations, but it is unknown how CA contributes to the disease and patient outcomes. In this study, we sought to determine whether CA was associated with worse clinical outcomes, poor prognostic indicators, markers of epithelial-mesenchymal transition (EMT), and ethnicity in PDAC. We also evaluated whether CA could precipitate more aggressive phenotypes in a panel of cultured PDAC cell lines. Using publicly available microarray data, we found that increased expression of genes whose dysregulation promotes CA was associated with worse overall survival and increased EMT marker expression in PDAC. Quantitative analysis of centrosomal profiles in PDAC cell lines and tissue sections uncovered varying levels of CA, and the expression of CA markers was associated with the expression of EMT markers. We induced CA in PDAC cells and found that CA empowered them with enhanced invasive and migratory capabilities. In addition, we discovered that PDACs from African American (AA) patients exhibited a greater extent of both numerical and structural CA than PDACs from European American (EA) patients. Taken together, these findings suggest that CA may fuel a more aggressive disease course in PDAC patients.
Keywords: centrosome amplification, epithelial-mesenchymal transition, ethnic health disparity, pancreatic cancer, pancreatic ductal adenocarcinoma
Abbreviations
- AA
African American
- CA
Centrosome amplification
- EMT
Epithelial-mesenchymal transition
- EA
European American
- GEO
Gene Expression Omnibus
- PDAC
Pancreatic ductal adenocarcinoma
- PBS
Phosphate-buffered saline.
Introduction
Pancreatic cancer is the fourth most fatal cancer in the United States, although it constitutes only 2.7% of new cancer cases.1 Greater than 90% of all pancreatic neoplasms derive from ductal cells, and ∼85% of these are invasive PDACs.2 Over the past 30 years, improvements in survival for PDAC patients have paled in comparison with improvements for patients suffering from other cancers. This unsettling trend is expected to continue in the near future, and it is projected that by 2030 pancreatic cancer will depose colorectal cancer to become the second-leading cause of cancer-related death in the United States.3,4 A primary cause for the exceptionally high mortality of PDAC is its general lack of clinically useful prognostic markers and therapeutic targets, in contrast with many other malignancies (such as breast and lung) in which much greater strides in precision medicine have been made. Surgical resection is the only treatment to date that appreciably extends survival in PDAC, with a median survival time of ∼18 months for patients who undergo complete resection compared with only ∼6 months for patients with unresectable tumors.5 Gemcitabine is the standard of care for patients with advanced disease, although it improves survival only by about one month.6 Therefore, it is critical to identify prognostic and predictive biomarkers and clinically actionable drivers of disease aggressiveness in PDAC. Such an approach has achieved breakthroughs for other highly aggressive, treatment-refractory cancers, such as melanoma,4 by enabling risk stratification of patients and the administration of targeted therapies. A deluge of gene expression and proteomic studies has implicated ∼10% of the exome in pancreatic cancer,7 but the size and complexity of these data have so far thwarted their integration into an actionable portrait of the disease. Indeed, at present there is only one FDA-approved biomarker for pancreatic cancer, carbohydrate antigen 19-9; however, it has low prognostic value before surgery or chemotherapy, so its utility is mostly limited to post-treatment monitoring of disease progression.8 Furthermore, only one FDA-approved targeted therapy is available for PDAC, erlotinib (in combination with gemcitabine), although it improves survival by less than 2 weeks and, thus, is rarely prescribed.4 Consequently, there is a critical need for improved prognostic biomarkers and therapeutic targets for PDAC.
Biomarkers and drug targets are generally sought at “omics” levels of late, but a seemingly overlooked pool of candidates is comprised of organelles. These structures constitute the output of the cell's own integration of the staggeringly complex molecular signaling events within it, so organelles represent particularly comprehensive and clinically significant biomarkers and drug targets. Investigation of organelle-level variations among PDACs of differing aggressiveness represents entirely uncharted territory. The centrosome has emerged as a central driver of tumor aggressiveness across cancer types. CA (the presence of excessively numerous or voluminous centrosomes) can initiate tumorigenesis, engenders chromosomal instability, and precipitates invasive tumor behavior and enhanced migratory capacity.9-12 In order for cancer cells to avail themselves of the advantages of CA, however, they must prevent spindle multipolarity during mitosis, lest they succumb to fatal mitotic catastrophe. As a result, cancer cells deftly cluster supernumerary centrosomes into 2 diametrically opposed groups in order to achieve pseudo-bipolar spindle geometry and survive. The extent of CA correlates positively with aggressiveness across the entire spectrum of cancer types.13 For instance, we recently uncovered that in breast cancer CA is associated with metastatic markers, the aggressive triple-negative subtype, and worse overall and progression-free survival.12 Consequently, it is rational to suspect that CA is involved in PDAC, which is nearly unrivaled in its aggressive behavior among malignancies. Furthermore, a small study focused on centrosome abnormalities in PDAC revealed a striking difference in the profiles of centrosomes, both in terms of their number and size, as compared with normal pancreas.14 Another study observed that centrosome abnormalities (defined as supernumerary or structurally aberrant centrosomes) detected by pericentrin immunofluorescence staining were correlated with nuclear abnormalities (namely, bi- or multinucleation or the presence of giant nuclei) in a panel of pancreatic cancer cell lines.15 These studies lend credence to our rationale that CA may underlie PDAC aggressiveness, a hypothesis that we tested in the present study.
Materials and Methods
Public microarray data analysis
Robust Multi-array Average-normalized expression levels of genes whose dysregulation is known to drive CA (including AURKA, CCNA2, CCND1, CCNE2, CDK1, CEP63, CEP152, E2F1, E2F2, E2F3, LMO4, MDM2, MYCN, NDR1, NEK2, PIM1, PIN1, PLK4, RAD6, and STIL) from the primary PDACs of 42 patients were obtained from GEO series GSE28735.16 Cutoff Finder17 was used to determine optimal cut-points in individual gene expression levels to stratify patients into 2 groups based on overall survival using the log-rank test. The same data were used to determine the correlation (Pearson) between the expression levels of genes whose dysregulation drives CA (AURKA, CDK1, STIL, PIM1, PLK4, and NEK2) and EMT markers (PLAUR and MMP3), which were validated using gene expression data from 39 PDACs from GEO series GSE15471.18 SPSS software (IBM) was used for the analyses, with P<0.05 indicating statistical significance.
Clinical tissue samples
Formalin-fixed paraffin-embedded slides of PDAC and normal pancreatic tissue were procured from Emory University Hospital, whose Institutional Review Board approved all aspects of the study.
Immunofluorescence staining, imaging, and scoring of clinical specimens
For immunofluorescence staining all tissue slides were deparaffinized by baking at 67°C for 2 h followed by 3 xylene washes. Slides were then rehydrated by passing them through a series of ethanol baths (100%, 95%, 70%, and 50%). Antigen retrieval was performed by incubating slides in citrate buffer (pH 6.0) in a pressure cooker at 15 psi for 3 min. Tissue samples were then incubated overnight with primary mouse antibody against γ-tubulin (1:1000 dilution) at 4°C. The samples were then washed with 3X phosphate-buffered saline (PBS) before incubating them in secondary antibody (Alexa-488 anti-mouse) at 37°C for 2 h. Samples were washed with 3X PBS and then mounted with Prolong-Gold antifade reagent that contained DAPI (Invitrogen). Tissue samples were imaged using the Zeiss LSC 700 confocal microscope, and images were processed with Zen software. The percentage of cells with CA was quantitated from 10 randomly selected fields, with ∼500 cells counted for each sample.
Immunohostochemistry, scoring, and weighted index (WI) calculation for clinical specimens
Deparaffinization and antigen retrieval were performed as described for immunofluorescence staining. Thereafter, the tissues were immunolabeled using Plk4 and MMP2 antibodies. Enzymatic antibody detection was performed with the Universal LSAB + Kit/HRP (DAKO). The staining intensity was scored as 0 = none, 1 = low, 2 = moderate, or 3 = high, and the percentage of cells from 10 randomly selected fields (∼500 cells) was determined. The product of the staining intensity and the percent of positive cells constituted the WI. Pearson's correlation coefficients between WIs were sought using SPSS software.
Cell culture
MIA PaCa-2, Capan-1, CFPAC-1, and HPAF-II cell lines were grown in Dulbecco's Modified Eagle's medium supplemented with 10% Hyclone fetal bovine serum and 1% penicillin/streptomycin. All cell lines were maintained in humidified 5% CO2 atmosphere at 37°C.
Cell lysate preparation, immunobloting, immunofluorescence staining, and confocal microscopy
Protein lysates were prepared and immunoblotting was performed as described earlier.19 Polyacrylamide gel electrophoresis was used to resolve the proteins, which were transferred onto polyvinylidene fluoride membranes (Millipore). The Pierce ECL chemiluminescence detection kit (Thermo Scientific) was used to visualize the immune-reactive bands. β-actin was used as loading control. For immunofluorescence staining, cells were grown on glass coverslips and fixed with ice-cold methanol for 10 min. Blocking was done by incubating with 2% bovine serum albumin/1XPBS/0.05% Triton X-100 at 37°C for 1 h. Coverslips were incubated in primary antibodies against γ-tubulin and α-tubulin at 1:2000 dilution for 1 h at 37°C. The cells were washed with 2% bovine serum albumin/1XPBS for 10 min at room temperature before incubating with a 1:2000 dilution of Alexa 488- or 555-conjugated secondary antibodies (Invitrogen). Cells were mounted with ProLong Gold Antifade Reagent with DAPI (Invitrogen). Antibodies against γ-tubulin and α-tubulin were from Sigma; antibodies against Aurora A and β-actin were from Cell Signaling; the antibody against centrin-2 was from Santa Cruz Biotechnology; and antibodies against Plk4 and MMP2 were from Abcam. Horseradish peroxidase-conjugated secondary antibodies were from Santa Cruz Biotechnology.
Cell migration assay
Monolayers of the aphidicolin-treated and control CFPAC-1 cells were scratched with a 200 µl pipette tip after serum starving for 8 h. Using a 20X objective, images were taken every hour using the Zeiss Axio Observer. Image J was used to define the edges of the wound and to measure wound area, and the percent change in the wound area was calculated based on the closure of the wound over time.
Boyden chamber assay
Control and aphidicolin-treated CFPAC-1 cells were collected after 48 h and resuspended in media at 5 × 104cells/ml. Transmigration assay was carried out in a Boyden chamber system. The upper wells of the chamber were loaded with 200 µl of cell suspension, and 500 µl media containing 20% FBS was added in lower chambers as a chemoattractant. Chambers were incubated for 12 h at 37°C and 5% CO2. Cells that migrated to the bottom surface of the filter were fixed with 70% methanol, stained with crystal violet, and counted using a 20X objective (10 randomly selected fields).
Statistical Methods
Unless otherwise stated in the methods and results, statistical analyses were performed using Student's t-test, and the criterion for statistical significance was P < 0.05.
Results
Increased expression of genes whose dysregulation drives CA is associated with worse overall survival in PDAC
Previous studies have reported an association between CA and chromosomal instability in PDAC.20,21 Furthermore, liver metastases exhibited a greater extent of CA than the primary tumors in an orthotopic implantation model of PDAC.21 However, the association of CA with a more aggressive disease course in PDAC patients has not been explored. As there are currently no publicly available datasets with information on CA per se, we instead analyzed expression levels of genes whose deregulation is known to drive CA (including AURKA, CCNA2, CCND1, CCNE2, CDK1, CEP63, CEP152, E2F1, E2F2, E2F3, LMO4, MDM2, MYCN, NDR1, NEK2, PIM1, PIN1, PLK4, RAD6, and STIL). Specifically, we tested the associations between Robust Multi-array Average-normalized expression levels of these genes in primary PDACs from 42 patients and overall survival using Gene Expression Omnibus (GEO) series GSE28735.16 Survival over time was estimated using the Kaplan-Meier method. Cutoff Finder, which applies the log-rank test to determine an optimal cut-point based on significance,17 was used to stratify patients into low- and high-risk groups. High expression levels of 9 genes (specifically, AURKA, CCNA2, CCNE2, CDK1, CEP152, NEK2, PIM1, PLK4, and STIL) were found to be associated with worse overall survival with P < 0.1 Associations with 5 genes, namely CDK1, NEK2, PIM1, PLK4, and STIL, were significant (P < 0.05), as depicted in Kaplan-Meier plots in Fig. 1. Consequently, CA may be associated with worse overall survival in PDAC. We found that genes whose dysregulation drives CA and clustering are upregulated in PDACs (N = 36) relative to normal pancreatic tissue (N = 12) using GEO series GSE1651522 (Fig. S1). We further found that expression levels of AURKA, CDK1, STIL, PIM1, PLK4, and NEK2 in PDACs mostly correlate with the expression of the EMT markers PLAUR and MMP3 using GEO series GSE28735 (Table 1), which we validated using GEO series GSE15471 (Table 2). These results suggest that CA may be associated with increased metastatic potential of PDACs and poor survival prospects for PDAC patients.
Figure 1.

Kaplan-Meier plots of overall survival based on low or high expression of genes whose dysregulation drives centrosome amplification in pancreatic ductal adenocarcinoma. Expression was categorized as low or high based on whether the value was below or above the following cut-points in normalized expression levels, given in parentheses here after each gene: (A) CDK1 (2.97), (B) NEK2 (3.79), (C) PIM1 (5.70), (D) PLK4 (3.47), and (E) STIL (3.06). HR = Hazard Ratio.
Table 1.
Correlation between the expression levels of genes whose dysregulation drives centrosome amplification (AURKA, CDK1, STIL, PIM1, PLK4, and NEK2), that contribute to centrosome structure (CETN2), or that indicate epithelial-mesenchymal transition (PLAUR and MMP3). Normalized gene expression levels from breast tumors in GEO series GSE28735 were used for the statistical analysis
| AURKA | CDK1 | STIL | PIM1 | PLK4 | NEK2 | CETN2 | ||
|---|---|---|---|---|---|---|---|---|
| PLAUR | Pearson Correlation | 0.311 | 0.395 | 0.305 | 0.539 | 0.237 | 0.359 | 0.403 |
| Sig. (2-tailed) | 0.037 | 0.007 | 0.042 | 0.000 | 0.117 | 0.015 | 0.006 | |
| N | 45 | 45 | 45 | 45 | 45 | 45 | 45 | |
| MMP3 | Pearson Correlation | 0.142 | 0.271 | 0.506 | −0.058 | 0.248 | 0.376 | 0.514 |
| Sig. (2-tailed) | 0.353 | 0.072 | 0.000 | 0.705 | 0.100 | 0.011 | 0.000 | |
| N | 45 | 45 | 45 | 45 | 45 | 45 | 45 | |
Table 2.
Correlation between the expression levels of genes whose dysregulation drives centrosome amplification (AURKA, CDK1, STIL, PIM1, PLK4, and NEK2), that contribute to centrosome structure (CETN2), or that indicate epithelial-mesenchymal transition (PLAUR and MMP3). Normalized gene expression levels from breast tumors in GEO series GSE15471 were used for the statistical analysis
| AURKA | CDK1 | STIL | PIM1 | PLK4 | NEK2 | CETN2 | ||
|---|---|---|---|---|---|---|---|---|
| PLAUR | Pearson Correlation | 0.567 | 0.590 | 0.578 | 0.436 | 0.578 | 0.454 | 0.414 |
| Sig. (2-tailed) | 0.000 | 0.000 | 0.000 | 0.005 | 0.000 | 0.004 | 0.009 | |
| N | 39 | 39 | 39 | 39 | 39 | 39 | 39 | |
| MMP3 | Pearson Correlation | 0.388 | 0.429 | 0.256 | 0.003 | 0.450 | 0.472 | 0.019 |
| Sig. (2-tailed) | 0.015 | 0.006 | 0.116 | 0.985 | 0.004 | 0.002 | 0.909 | |
| N | 39 | 39 | 39 | 39 | 39 | 39 | 39 | |
We next sought correlations between protein levels of an EMT marker (MMP2) and CA driver (Plk4) in PDAC samples. To this end, we first immunohistochemically stained 54 PDACs and uninvolved adjacent normal tissue for Plk4 and MMP2 and calculated WIs. The staining intensity was scored as 0 = none, 1 = low, 2 = moderate, or 3 = high, and the percentage of positive cells (i.e., with 1+ staining intensity) from 10 randomly selected fields (∼500 cells) was determined. The product of the staining intensity and the percent of positive cells constituted the WI. Descriptive statistics regarding patient and clinicopathological characteristics and biomarker WIs are given in Table 3. Both Plk4 and MMP2 proteins were overexpressed in PDACs (Fig. 2). In addition, a positive Pearson's correlation was found between these markers in PDACs (r = 0.460, P < 0.001).
Table 3.
Descriptive statistics for patient and clinicopathologic characteristics in the analysis of MMP2 and Plk4 levels in tumors and matched normal tissue. SD = standard deviation; WI = weighted index
| Variable | Level | Number | Percentage |
|---|---|---|---|
| Race | African American | 23 | 42.6 |
| European American | 31 | 57.4 | |
| Tumor Size (cm) | ≤2 | 8 | 14.8 |
| >2 | 46 | 85.2 | |
| Tumor Size (cm) | Median | 3.0 | |
| Mean | 3.6 | ||
| Maximum | 12.0 | ||
| Minimum | 0.3 | ||
| SD | 2.0 | ||
| Well | 10 | 18.5 | |
| Differentiation | Moderate | 31 | 57.4 |
| Poor | 13 | 24.07 | |
| MMP2 WI | Median | 6.0 | |
| Mean | 4.2 | ||
| Maximum | 9 | ||
| Minimum | 0 | ||
| SD | 3.1 | ||
| Plk4 WI | Median | 2.0 | |
| Mean | 2.1 | ||
| Maximum | 9.0 | ||
| Minimum | 0 | ||
| SD | 2.2 | ||
| MMP2 WI | Low (<2) | 13 | 24.1 |
| Moderate (2–6) | 34 | 63.0 | |
| High (>6) | 7 | 13.0 | |
| Plk4 WI | Low (<2) | 25 | 46.3 |
| Moderate (2–6) | 27 | 50.0 | |
| High (>6) | 2 | 3.7 | |
| Perinuerual invasion | Yes | 43 | 79.6 |
| No | 10 | 18.5 | |
| Lymphovascular invasion | Yes | 26 | 48.1 |
| No | 28 | 51.9 | |
| Age at diagnosis | Median | 64.0 | |
| Mean | 62.1 | ||
| Maximum | 84.0 | ||
| Minimum | 35.0 | ||
| SD | 9.9 | ||
| Duodenal invasion | Yes | 20 | 37.0 |
| No | 34 | 63.0 | |
| Soft tissue involvement | Yes | 37 | 68.5 |
| No | 17 | 31.5 | |
| 1 | 2 | 3.07 | |
| 2 | 8 | 14.8 | |
| Stage T | 3 | 38 | 70.7 |
| 4 | 3 | 5.05 | |
| Missing | 3 | 5.05 | |
| 1 | 30 | 55.5 | |
| Stage N | 0 | 21 | 38.8 |
| Missing | 3 | 5.05 | |
| 1 | 48 | 88.8 | |
| Stage M | 0 | 6 | 11.1 |
Figure 2.
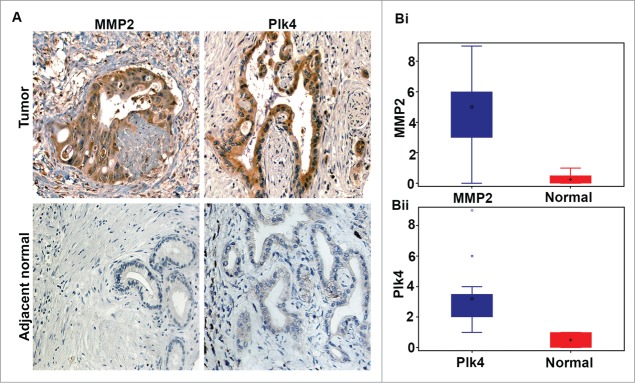
Pancreatic ductal adenocarcinomas exhibit higher expression of Plk4 and MMP2 than normal pancreas. (A) Representative micrographs showing immunohistochemical staining for Plk4 (a protein whose overexpression drives centrosome amplification) and MMP2 (an epithelial-mesenchymal transition marker) in uninvolved adjacent normal and tumor tissue from grade-matched pancreatic ductal adenocarcinoma (PDAC) patients. (Bi) Box-and-whisker plot depicting the MMP2 weighted index in PDACs and normal pancreas. (Bii) Box-and-whisker plot depicting the Plk4 weighted index in PDACs and normal pancreas.
Amplified centrosomes enhance the motility and invasiveness of PDAC cells
Having confirmed an association between a protein whose overexpression drives CA and a marker of EMT, we were interested in exploring how CA may transform non-invasive pancreatic tumors into aggressive ones that metastasize. Thus, we examined whether CA can enhance the motility and invasiveness of pancreatic cancer cells. To this end, we first screened 3 well-established pancreatic cancer cell lines (namely, MIA PaCa-2, CFPAC-1, and HPAFII) by immunostaining centrosomes (γ-tubulin, green) and microtubules (α-tubulin, red) and counterstaining nuclei with DAPI (blue) (Fig. 3A). We found that HPAFII cells exhibited the greatest extent of numerical CA (∼20% of cells), followed by MIA Paca-2 (∼15% of cells) and CFPAC-1 (∼10% of cells) (Fig. 3B). We also evaluated the expression of centrosome-related proteins in these cell lines using immunoblotting methods. We found that the cell lines with high CA expressed elevated levels of centrosome structural proteins (centrin-2 and γ-tubulin) and proteins whose dysregulation is known to drive CA (Aurora A and Plk4) (Fig. 3C). We next asked if aberrations in centrosome number translate into aberrations in mitotic spindle geometry. We found that all 3 cell lines exhibited a significantly lower proportion of cells with multipolar spindles in comparison with the proportion of cells with supernumerary centrosomes (Fig. 3B). This discordance corroborates the hypothesis that cancer cells deal with supernumerary centrosomes by clustering them to form pseudobipolar spindles. Taken together, our data suggest that cultured pancreatic cancer cells are characterized by CA, which they are generally successful in managing by executing centrosome clustering.
Figure 3.
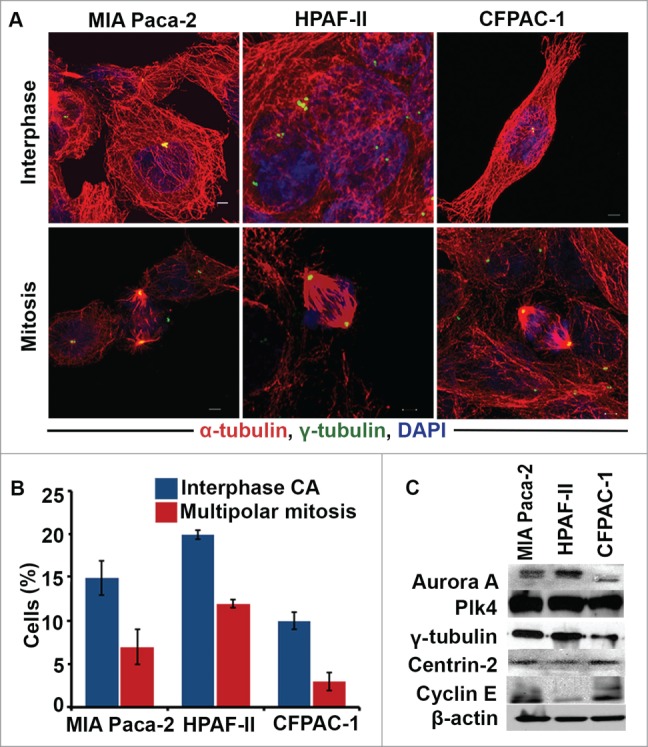
Pancreatic cancer cells with high levels of centrosome amplification (CA) express high levels of centrosome structural proteins and proteins whose dysregulation drives CA. (A) Immunofluorescence micrographs showing MIA PaCa-2, HIFA-II, and CFPAC-1 cells in interphase and mitosis stained for γ-tubulin (green), α-tubulin (red), and nuclei (blue). (B) Bar graphs representing the percentage of cells with CA in MIA PaCa-2, CFPAC-1, and HPFA-II cells. (C) Immunoblots depicting the levels of centrosome structural proteins (γ-tubulin and centrin-2) and proteins whose dysregulation drives CA (Aurora A, Plk4, and Cyclin E) in MIA PaCa-2, HPFA-II, and CFPAC-1 cells.
Next, we were curious to learn whether inducing CA via pharmacological means could enhance the motility of pancreatic cancer cells with relatively low levels of CA. To this end, we induced CA in CFPAC-1 pancreatic cancer cells (∼10% of which have CA, the lowest level in the lines we surveyed) by treating them with 25 µM aphidicolin for 48 h. Aphidicolin arrests cells in G1/S phase by inhibiting DNA polymerase.23,24 After treatment ∼22% of cells exhibited amplified centrosomes. We then performed a wound-healing assay, which revealed that pharmacological induction of CA stimulated migration, as the wound was filled in about half the time taken by control cells (Fig. 4A and Bi). Thereafter, we examined the invasive capabilities of cells with supernumerary centrosomes by performing a classical Boyden chamber assay. We observed that 80% of the CFPAC-1 cells in which CA was induced invaded the Matrigel in 12 h in contrast with only 53% of control cells (that is, CFPAC-1 cells not treated with aphidicolin) (Figs. 4Bii and C). We confirmed that CA was induced in aphidicolin-treated cells by immunoblotting for centrin-2 and Plk4 levels (Fig. 4D). In addition, we noted that cells treated with aphidicolin expressed higher levels of N-cadherin (Fig. 4D), suggesting that these cells may have attained a more mesenchymal phenotype. In summary, our findings that CA upregulates N-cadherin levels, invasive capacity, and wound-healing imply that CA may contribute to EMT in pancreatic cancer cells.
Figure 4.

Cells with centrosome amplification (CA) migrate more rapidly in a wound-healing assay. (A) Bright-field micrographs showing the wound-healing capacity of CFPAC-1 control (untreated) and aphidicolin-treated cells at 0, 12, and 36 h. (Bi) Bar graphs representing the percent of aphidicolin-treated and control (untreated) CFPAC-1 cells in the wound. (Bii) Bar graphs representing the percent of aphidicolin-treated and control CFPAC-1 cells that invaded the Boyden chamber after 24 h. (C) Brightfield micrographs showing CFPAC-1 control and aphidicolin-treated cells that invaded the Boyden chamber after 24 h. (D) Immunoblots of a centrosome structural protein (centrin-2), a protein whose dysregulation drives CA (Plk4), and epithelial-mesenchymal transition markers (N-cadherin and MMP2) in CFPAC-1 control and aphidicolin-treated cells.
Amplified centrosomes are associated with duodenal invasion and distinguish AA PDACs from EA PDACs
A previous study of 13 PDACs uncovered that a greater proportion of PDAC cells have CA, both numerical and structural, than normal pancreas.25 However, the association of CA with poor prognostic indicators in pancreatic cancer is unknown. To address this, we examined the centrosomal profiles of 64 PDACs and sought associations with clinicopathologic parameters including age, sex, ethnicity, tumor size, grade/extent of differentiation, stage, lymphovascular and perineural invasion, and lymph node metastasis. Descriptive statistics regarding patient and clinicopathologic characteristics are given in Table 4. Formalin-fixed tissue sections from the PDACs were immunostained for centrosomes (γ-tubulin) and nuclei were counterstained with DAPI (Fig. 5A). Centrosome number and volume were quantitated in tumor areas pre-marked by a gastrointestinal pathologist using confocal microscopy. Tumor cells with more than 2 centrosomes or centrosomes greater than 0.56 μm3 in volume were regarded as having numerical or structural CA, respectively. The volume of 0.56 μm3 was used as a cut-point because that was the maximum centrosome volume found in normal pancreatic cells using the 3D volume measurement module from the Zeiss imaging software. The percentage of cells exhibiting centrosomal aberrations was quantitated from 10 randomly selected fields (∼500 cells) for each sample. The mean volume of the γ-tubulin spots observed in pancreatic cancer tissues was 1.75 μm3, which was ∼9 times greater than the mean volume in normal pancreatic cells. Pancreatic tumors also exhibited extensive numerical CA, with ∼25–40% of cells bearing extra centrosomes, unlike normal pancreatic tissue, in which only ∼5% of cells had extra centrosomes. When we compared the extent of CA between tumors of different levels of differentiation, we found that moderately differentiated tumors exhibited the highest CA when compared with well- and poorly differentiated tumors (Fig. 5B); however, the results were not statistically significant, perhaps due to the paucity of well-differentiated tumors in our data set (N = 6), as PDACs are most often moderately to poorly differentiated. While CA was strongly associated with duodenal invasion in well-differentiated PDACs (r = 0.772, p = 0.042), it was not associated with tumor size, stage, perineural or lymphovascular invasion, or number of positive lymph nodes in PDACs of any degree of differentiation. In summary, CA clearly differentiates PDACs from adjacent normal tissue and is associated with duodenal invasion in well-differentiated PDACs.
Table 4.
Descriptive statistics for patient and clinicopathologic characteristics in the analysis of centrosome amplification in tumors and matched normal tissue
| Variable | Level | Number | Percentage |
|---|---|---|---|
| Race | African American | 30 | 46.9 |
| European American | 34 | 53.1 | |
| Centrosome amplification (percent) | Low (<10) | 14 | 21.9 |
| Moderate (10–40) | 38 | 59.4 | |
| High (>40) | 12 | 15.6 | |
| Tumor Differentiation | Well | 6 | 9.4 |
| Moderate | 40 | 62.5 | |
| Poor | 14 | 21.9 | |
| Tumor size (cm) | ≤2 | 10 | 14.5 |
| >2 | 54 | 78.3 | |
| Tumor size (cm) | Median | 3.1 | |
| Mean | 3.6 | ||
| Maximum | 12.0 | ||
| Minimum | 1.0 | ||
| SD | 1.9 | ||
| Age at diagnosis | Median | 66.0 | |
| Mean | 64.4 | ||
| Maximum | 87.0 | ||
| Minimum | 35.0 | ||
| SD | 10.8 | ||
| Centrosome amplification (percent) | Median | 36.3 | |
| Mean | 36.2 | ||
| Maximum | 72.2 | ||
| Minimum | 1.4 | ||
| SD | 18.0 | ||
| Duodenal invasion | Yes | 32 | 50 |
| No | 32 | 50 | |
| Soft tissue involvement | Yes | 50 | 50 |
| No | 14 | 21.9 | |
| Perineural invasion | Yes | 55 | 85.9 |
| No | 10 | 15.6 | |
| Lymphovascular invasion | Yes | 26 | 40.6 |
| No | 38 | 59.4 | |
| 0 | 1 | 1.5 | |
| 1 | 7 | 3.1 | |
| Stage T | 2 | 5 | 9.2 |
| 3 | 51 | 79.6 | |
| 1 | 42 | 65.2 | |
| Stage N | 0 | 22 | 34.5 |
| 1 | 56 | 87.5 | |
| Stage M | 0 | 8 | 12.5 |
Figure 5.
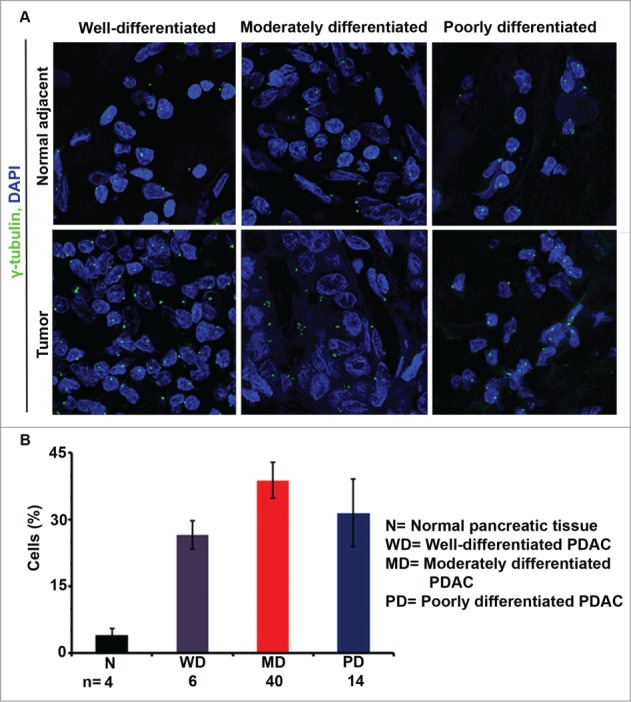
Centrosome amplification (CA) in pancreatic ductal adenocarcinomas (PDACs) and normal pancreatic tissue. Representative confocal micrographs depicting centrosomal profiles in well-, moderately, and poorly differentiated PDACs and adjacent normal pancreatic tissue. Centrosomes were immunostained (γ-tubulin, green) and nuclei were counterstained with DAPI (blue). (B) Bar graph representation of the percentage of cells showing CA in well-, moderately, and poorly differentiated PDACs and normal pancreatic tissue samples. ∼500 cells were counted in each case.
Finally, because AA ethnicity is a risk factor for the development of PDAC,26 we were interested in determining whether centrosomal profiles from AA patients differed from those of EA patients. Interestingly, when we immunostained moderately differentiated pancreatic tumor samples from AA and EA PDAC patients (N = 20 for each group) (Fig. 6A) and compared their centrosomal profiles, we found that numerical and structural CA in AA tumors were significantly higher than in EA tumors (Fig. 6B and C).
Figure 6.
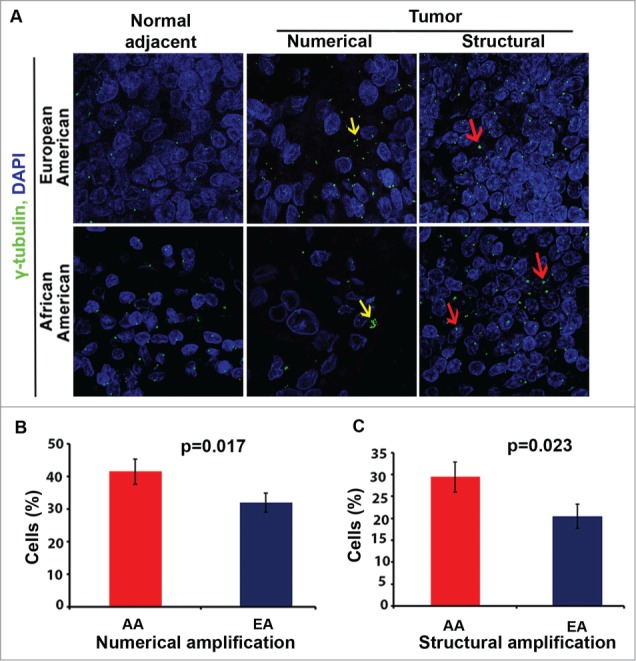
Centrosome amplification (CA) in pancreatic ductal adenocarcinomas (PDACs) and normal pancreas specimens from African American (AA) and European American (EA) patients. (A) Representative confocal micrographs depicting centrosomal profiles in grade-matched AA and EA PDACs and adjacent normal tissues. Centrosomes were immunostained for γ-tubulin (green) and nuclei were counterstained with DAPI (blue). (B) Bar graphs representing the percent of cells showing numerical CA in AA and EA tissue samples. (C) Bar graphs representing the percent of cells showing structural CA in AA and EA PDAC tissue samples. ∼500 cells were counted in each case.
Discussion
Five-year survival rates in PDAC hover around 5% notwithstanding about a half century of research into the etiology of its aggressive nature and potential therapeutic interventions.27 Out of this burgeoning body of research has emerged an appreciation of the remarkably complex mutational landscape of PDACs, their extensive intratumor heterogeneity due to chromosomal instability, and their extraordinary propensity to metastasize,27,28 although this knowledge has not translated into considerable improvements in patient outcomes. CA is a well-established mediator of chromosomal instability,29 and the extent of CA correlates with chromosomal instability in pancreatic cancer cells.30 CA has also been demonstrated to trigger cellular invasiveness and augmented migratory capabilities in an oncogene-like manner11,12 and is associated with markers of aggressiveness and worse prognosis.12,13 Previous work has identified that the vast majority of PDACs exhibit CA,20 suggesting that CA is a hallmark of these tumors. Therefore, it seems likely that in PDAC CA is at least partly responsible for intratumor heterogeneity and metastasis. Our study is the first to demonstrate that high expression levels of genes whose dysregulation drives CA are associated with worse overall survival and EMT marker expression in PDAC, CA is significantly associated with duodenal invasion in well-differentiated PDACs, and induction of CA in PDAC cell lines promotes motility, invasiveness, and EMT marker expression.
Previous studies have shown that cancer cells manage the excessive centrosomal load by forming juxtanuclear supercentrosomal clusters, which they maintain all through interphase and then disperse transiently in prophase, followed soon by tight reclustering.31 Centrosome declustering drugs, such as the non-toxic antifungal griseofulvin and antitussive noscapine, disaggregate the centrosomal clusters, forcing the mitotic spindle to assume a persistently high-grade multipolar configuration that is incompatible with cell survival.32 A previous study discovered that the CFPAC-1 cell line exhibits considerable centrosome clustering, with more than half of mitoses in cells with CA assuming a pseudobipolar configuration,33 similar to our findings. Together, these findings suggest that pancreatic cancer cells tend to cluster their supernumerary centrosomes. As a corollary, it seems that centrosome declustering drugs could prove advantageous in PDAC, an intriguing hypothesis that merits testing.
We discovered that AA PDACs exhibited more extensive numerical and structural CA than EA PDACs. The age-adjusted incidence of PDAC is ∼30% higher among AAs as compared with EAs,34 and AA race is an established risk factor for PDAC.26 Various socioeconomic and lifestyle factors that may contribute to the development of PDAC are more common in the AA population,26 as are certain K-Ras mutations and possibly strong HER2 expression.35 Intriguingly, it has been demonstrated that K-RasG12D induces CA in mammary epithelial cells36 and head and neck papilloma cells.37 One study found that this mutation is more prevalent among AAs than EAs (47% vs. 34%), although the difference did not reach statistical significance,35 perhaps due to the relatively small sample size. K-RasG12V, on the other hand, was significantly more prevalent among AAs in this study, but we are unaware of any data regarding the impact of this mutation on CA. What might be considered indirect evidence that K-RasG12V promotes CA is the recent finding by Hu and colleagues that this mutation increases the frequency of multipolar anaphases and apoptosis following treatment of ED-1 murine lung cancer cells with seliciclib, a cyclin-dependent kinase inhibitor.38 CA renders cancer cells vulnerable to multipolar mitosis,32 so cells undergoing multipolar anaphase might have supernumerary centrosomes, although this was not directly tested in that study. The relationship between ethnicity and mutations in genes whose dysregulation is known to drive CA that are common in PDAC (e.g., TP53,39,40 SMAD4,37 CDKN2A,41 and CHEK2,42 depicted in Fig. S2) merits investigation. One study found that a greater proportion of AA PDACs displayed strong HER2 expression than EA PDACs,35 and HER2 overexpression is associated with CA in breast cancer;43,44 thus, HER2 overexpression may contribute to the greater extent of CA we uncovered in AA PDACs. More research is needed to confirm the CA-promoting role of ethnicity-associated gene amplifications and mutations in PDAC, although it is tempting to speculate that they underlie the differences in centrosome profiles we observed between AAs and EAs. The diversity of factors that may confer increased PDAC risk that are associated with AA ethnicity are depicted in Fig. S3. Although CA is a well-defined risk factor for cancer aggressiveness,32 the literature reports that AAs with PDAC do not experience worse overall survival in multivariate analyses accounting for a variety of factors, such as treatment received and socioeconomic status, as detailed by a recent review of 9 relevant studies.45 This discrepancy cannot be resolved based on existing data and deserves further exploration. Regardless, it stands to reason that AA ethnicity might predict therapeutic response to declustering drugs, and clinical trials testing these drugs clearly should consider ethnicity in their assessment of drug efficacy.
In summary, our microarray analysis suggests that higher levels of certain genes whose dysregulation promotes CA are associated with worse overall survival, although further study is needed to confirm that CA itself is indeed associated with worse clinical outcomes. In line with these in silico results, we found that induction of CA in PDAC cell lines resulted in more aggressive cellular behavior, such as increased motility and invasiveness. For the most part, however, we did not find that CA was associated with worse clinicopatholgic features in PDACs aside from duodenal invasion in well-differentiated tumors. A larger sample size is needed to confirm these immunohistochemical findings, which seem generally to conflict with our in silico and in vitro findings. It is possible that the relationship between CA and clinicopathologic features is complex, and some weighted combination of the numerical and structural CA values would be more strongly associated with clinicopathologic features like the extent of differentiation, tumor size, and lymph node positivity, which should be a focus of future work. Ultimately, our study establishes CA, a long-standing cancer cell-selective trait, as a quantifiable cell biological property in PDAC that undoubtedly merits further investigation.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors gratefully acknowledge Sergey Klimov for assisting with statistical analysis and Shristi Bhattarai and Ansa Riaz for helping to procure patient information.
Funding
This study was supported by grants to RA from the National Cancer Institutes of Health (U01 CA179671 and R01 CA169127), a graduate fellowship to KM from the Second Century Initiative Program at Georgia State University, and a graduate fellowship to AO from the Molecular Basis of Disease Program at Georgia State University.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin 2013; 63:11-30; PMID:23335087 [DOI] [PubMed] [Google Scholar]
- 2.Klimstra DS, Adsay NV. CHAPTER 35 - Tumors of the Pancreas and Ampulla of Vater : Goldblum RDOR, ed. Surgical Pathology of the GI Tract, Liver, Biliary Tract, and Pancreas (Second Edition). Philadelphia: W.B. Saunders, 2009:909-60. [Google Scholar]
- 3.Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 2014; 74:2913-21; PMID:24840647; http://dx.doi.org/ 10.1158/0008-5472.CAN-14-0155 [DOI] [PubMed] [Google Scholar]
- 4.Garrido-Laguna I, Hidalgo M. Pancreatic cancer: from state-of-the-art treatments to promising novel therapies. Nat Rev Clin Oncol 2015; 12(6):319-34 advance online publication; PMID:25824606 [DOI] [PubMed] [Google Scholar]
- 5.Bachmann J, Michalski CW, Martignoni ME, Büchler MW, Friess H. Pancreatic resection for pancreatic cancer. HPB 2006; 8:346-51; PMID:18333087; http://dx.doi.org/ 10.1080/13651820600803981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thota R, Pauff JM, Berlin JD. Treatment of metastatic pancreatic adenocarcinoma: a review. Oncology (Williston Park) 2014; 28:70-4; PMID:24683721 [PubMed] [Google Scholar]
- 7.Harsha HC, Kandasamy K, Ranganathan P, Rani S, Ramabadran S, Gollapudi S, Balakrishnan L, Dwivedi SB, Telikicherla D, Selvan LD, et al.. A compendium of potential biomarkers of pancreatic cancer. PLoS Med 2009; 6:e1000046; PMID:19360088; http://dx.doi.org/ 10.1371/journal.pmed.1000046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fong ZV, Winter JM. Biomarkers in pancreatic cancer: diagnostic, prognostic, and predictive. Cancer J 2012; 18:530-8; PMID:23187839; http://dx.doi.org/ 10.1097/PPO.0b013e31827654ea [DOI] [PubMed] [Google Scholar]
- 9.Basto R, Brunk K, Vinadogrova T, Peel N, Franz A, Khodjakov A, Raff JW. Centrosome amplification can initiate tumorigenesis in flies. Cell 2008; 133:1032-42; PMID:18555779; http://dx.doi.org/ 10.1016/j.cell.2008.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ganem NJ, Godinho SA, Pellman D. A mechanism linking extra centrosomes to chromosomal instability. Nature 2009; 460:278-82; PMID:19506557; http://dx.doi.org/ 10.1038/nature08136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Godinho SA, Picone R, Burute M, Dagher R, Su Y, Leung CT, Polyak K, Brugge JS, Thery M, Pellman D. Oncogene-like induction of cellular invasion from centrosome amplification. Nature 2014; 510:167-71; PMID:24739973; http://dx.doi.org/ 10.1038/nature13277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pannu V, Mittal K, Cantuaria G, Reid MD, Li X, Donthamsetty S, McBride M, Klimov S, Osan R, Gupta MV, et al.. Rampant centrosome amplification underlies more aggressive disease course of triple negative breast cancers. Oncotarget 2015; 6:10487-97; PMID:25868856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan JY. A clinical overview of centrosome amplification in human cancers. Int J Biol Sci 2011; 7:1122-44; PMID:22043171; http://dx.doi.org/ 10.7150/ijbs.7.1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sato N, Mizumoto K, Nakamura M, Nakamura K, Kusumoto M, Niiyama H, Ogawa T, Tanaka M. Centrosome abnormalities in pancreatic ductal carcinoma. Clin Cancer Res 1999; 5:963-70; PMID:10353727 [PubMed] [Google Scholar]
- 15.Zhu J, Abbruzzese JL, Izzo J, Hittelman WN, Li D. AURKA amplification, chromosome instability, and centrosome abnormality in human pancreatic carcinoma cells. Cancer Genet Cytogenet 2005; 159:10-7; PMID:15860351; http://dx.doi.org/ 10.1016/j.cancergencyto.2004.09.008 [DOI] [PubMed] [Google Scholar]
- 16.Zhang G, He P, Tan H, Budhu A, Gaedcke J, Ghadimi BM, Ried T, Yfantis HG, Lee DH, Maitra A, et al.. Integration of metabolomics and transcriptomics revealed a fatty acid network exerting growth inhibitory effects in human pancreatic cancer. Clin Cancer Res 2013; 19:4983-93; PMID:23918603; http://dx.doi.org/ 10.1158/1078-0432.CCR-13-0209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Budczies J, Klauschen F, Sinn BV, Gyorffy B, Schmitt WD, Darb-Esfahani S, Denkert C. Cutoff Finder: a comprehensive and straightforward Web application enabling rapid biomarker cutoff optimization. PLoS One 2012; 7:e51862; PMID:23251644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Badea L, Herlea V, Dima SO, Dumitrascu T, Popescu I. Combined gene expression analysis of whole-tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tumor epithelia. Hepatogastroenterology 2008; 55:2016-27; PMID:19260470 [PubMed] [Google Scholar]
- 19.Pannu V, Rida PC, Ogden A, Turaga RC, Donthamsetty S, Bowen NJ, Rudd K, Gupta MV, Reid MD, Cantuaria G, et al.. HSET overexpression fuels tumor progression via centrosome clustering-independent mechanisms in breast cancer patients. Oncotarget 2015; 6:6076-91; PMID:25788277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sato N, Mizumoto K, Nakamura M, Nakamura K, Kusumoto M, Niiyama H, Ogawa T, Tanaka M. Centrosome abnormalities in pancreatic ductal carcinoma. Clin Cancer Res 1999; 5:963-70; PMID:10353727 [PubMed] [Google Scholar]
- 21.Shono M, Sato N, Mizumoto K, Maehara N, Nakamura M, Nagai E, Tanaka M. Stepwise Progression of Centrosome Defects Associated with Local Tumor Growth and Metastatic Process of Human Pancreatic Carcinoma Cells Transplanted Orthotopically into Nude Mice. Lab Invest 0000; 81:945-52 [DOI] [PubMed] [Google Scholar]
- 22.Pei H, Li L, Fridley BL, Jenkins GD, Kalari KR, Lingle W, Petersen G, Lou Z, Wang L. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell 2009; 16:259-66; PMID:19732725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pedrali-Noy G, Spadari S, Miller-Faures A, Miller AO, Kruppa J, Koch G. Synchronization of HeLa cell cultures by inhibition of DNA polymerase alpha with aphidicolin. Nucleic Acids Res 1980; 8:377-87; PMID:6775308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ichikawa A, Negishi M, Tomita K, Ikegami S. Aphidicolin: a specific inhibitor of DNA synthesis in synchronous mastocytoma P-815 cells. Jpn J Pharmacol 1980; 30:301-8; PMID:6779040 [DOI] [PubMed] [Google Scholar]
- 25.Sato N, Mizumoto K, Nakamura M, Nakamura K, Kusumoto M, Niiyama H, Ogawa T, Tanaka M. Centrosome Abnormalities in Pancreatic Ductal Carcinoma. Clin Cancer Res 1999; 5:963-70; PMID:10353727 [PubMed] [Google Scholar]
- 26.Yeo TP. Demographics, epidemiology, and inheritance of pancreatic ductal adenocarcinoma. Semin Oncol 2015; 42:8-18; PMID:25726048 [DOI] [PubMed] [Google Scholar]
- 27.Waddell N, Pajic M, Patch A-M, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, et al.. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015; 518:495-501; PMID:25719666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Samuel N, Hudson TJ. The molecular and cellular heterogeneity of pancreatic ductal adenocarcinoma. Nat Rev Gastroenterol Hepatol 2012; 9:77-87 [DOI] [PubMed] [Google Scholar]
- 29.Ganem NJ, Godinho SA, Pellman D. A Mechanism Linking Extra Centrosomes to Chromosomal Instability. Nature 2009; 460:278-82; PMID:19506557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sato N, Mizumoto K, Nakamura M, Maehara N, Minamishima YA, Nishio S, Nagai E, Tanaka M. Correlation between centrosome abnormalities and chromosomal instability in human pancreatic cancer cells. Cancer Genet Cytogenet 2001; 126:13-9; PMID:11343773 [DOI] [PubMed] [Google Scholar]
- 31.Pannu V, Rida PCG, Celik B, Turaga RC, Ogden A, Cantuaria G, Gopalakrishnan J, Aneja R. Centrosome-declustering drugs mediate a two-pronged attack on interphase and mitosis in supercentrosomal cancer cells. Cell Death Dis 2014; 5:e1538; PMID:25412316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ogden A, Rida PC, Aneja R. Let's huddle to prevent a muddle: centrosome declustering as an attractive anticancer strategy. Cell Death Differ 2012; 19:1255-67; PMID:22653338; http://dx.doi.org/ 10.1038/cdd.2012.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kwon M, Godinho SA, Chandhok NS, Ganem NJ, Azioune A, Thery M, Pellman D. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev 2008; 22:2189-203; PMID:18662975; http://dx.doi.org/ 10.1101/gad.1700908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Howlader N NA, Krapcho M, Garshell J, Miller D, Altekruse SF, Kosary CL, Yu M, Ruhl J, Tatalovich Z, Mariotto A, et al (eds). SEER Cancer Statistics Review, 1975-2012, National Cancer Institute; Bethesda, MD, http://seer.cancer.gov/csr/1975_2012/, based on November 2014 SEER data submission, posted to the SEER web site, April 2015. [Google Scholar]
- 35.Pernick NL, Sarkar FH, Philip PA, Arlauskas P, Shields AF, Vaitkevicius VK, Dugan MC, Adsay NV. Clinicopathologic analysis of pancreatic adenocarcinoma in African Americans and Caucasians. Pancreas 2003; 26:28-32; PMID:12499914; http://dx.doi.org/ 10.1097/00006676-200301000-00006 [DOI] [PubMed] [Google Scholar]
- 36.Zeng X, Shaikh FY, Harrison MK, Adon AM, Trimboli AJ, Carroll KA, Sharma N, Timmers C, Chodosh LA, Leone G, et al.. The Ras oncogene signals centrosome amplification in mammary epithelial cells through cyclin D1/Cdk4 and Nek2. Oncogene 2010; 29:5103-12; PMID:20581865; http://dx.doi.org/ 10.1038/onc.2010.253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bornstein S, White R, Malkoski S, Oka M, Han G, Cleaver T, Reh D, Andersen P, Gross N, Olson S, et al.. Smad4 loss in mice causes spontaneous head and neck cancer with increased genomic instability and inflammation. J Clin Invest 2009; 119:3408-19; PMID:19841536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu S, Danilov AV, Godek K, Orr B, Tafe LJ, Rodriguez-Canales J, Behrens C, Mino B, Moran CA, Memoli VA, et al.. CDK2 Inhibition Causes Anaphase Catastrophe in Lung Cancer through the Centrosomal Protein CP110. Cancer Res 2015; 75(10):2029-38; PMID:25808870; http://dx.doi.org/ 10.1158/0008-5472.CAN-14-1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tarapore P, Fukasawa K. Loss of p53 and centrosome hyperamplification. Oncogene 2002; 21:6234-40; PMID:12214254; http://dx.doi.org/ 10.1038/sj.onc.1205707 [DOI] [PubMed] [Google Scholar]
- 40.Fukasawa K. P53, cyclin-dependent kinase and abnormal amplification of centrosomes. Biochim Biophys Acta 2008; 1786:15-23; PMID:18472015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McDermott KM, Zhang J, Holst CR, Kozakiewicz BK, Singla V, Tlsty TD. p16(INK4a) Prevents Centrosome Dysfunction and Genomic Instability in Primary Cells. PLoS Biology 2006; 4:e51; PMID:16464125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang HW, Kim T-M, Song SS, Shrinath N, Park R, Kalamarides M, Park PJ, Black PM, Carroll RS, Johnson MD. Alternative Splicing of CHEK2 and Codeletion with NF2 Promote Chromosomal Instability in Meningioma. Neoplasia (New York, NY) 2012; 14:20-8; http://dx.doi.org/ 10.1593/neo.111574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guo H-Q, Gao M, Ma J, Xiao T, Zhao L-l, Gao Y, Pan Q-J. Analysis of the cellular centrosome in fine-needle aspirations of the breast. Breast Cancer Res 2007; 9:R48-R; PMID:17662154; http://dx.doi.org/ 10.1186/bcr1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Montagna C, Andrechek ER, Padilla-Nash H, Muller WJ, Ried T. Centrosome abnormalities, recurring deletions of chromosome 4, and genomic amplification of HER2/neu define mouse mammary gland adenocarcinomas induced by mutant HER2/neu. Oncogene 2002; 21:890-8; PMID:11840334; http://dx.doi.org/ 10.1038/sj.onc.1205146 [DOI] [PubMed] [Google Scholar]
- 45.Khawja SN, Mohammed S, Silberfein EJ, Musher BL, Fisher WE, Van Buren G 2nd. Pancreatic cancer disparities in african americans. Pancreas 2015; 44:522-7; PMID:25872128; http://dx.doi.org/ 10.1097/MPA.0000000000000323 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.