

Abstract
Proper regulation of microtubule dynamics during mitosis is essential for faithful chromosome segregation. In fact, recently we discovered increased microtubule plus end assembly rates that are frequently observed in human cancer cells as an important mechanism leading to whole chromosome missegregation and chromosomal instability (CIN). However, the genetic alterations responsible for increased microtubule polymerization rates in cancer cells remain largely unknown. The identification of such lesions is hampered by the fact that determining dynamic parameters of microtubules usually involves analyses of living cells, which is technically difficult to perform in large-scale screening settings. Therefore, we sought to identify alternative options to systematically identify regulators of microtubule plus end polymerization. Here, we introduce a simple and robust phenotypic screening assay that is based on the analyses of monopolar mitotic spindle structures that are induced upon inhibition of the mitotic kinesin Eg5/KIF11. We show that increased microtubule polymerization causes highly asymmetric monoasters in the presence of Eg5/KIF11 inhibition and this phenotype can be reliably assessed in living as well as in fixed cells. Using this assay we performed a siRNA screen, in which we identify several microtubule plus end binding proteins as well as centrosomal and cortex associated proteins as important regulators of microtubule plus end assembly. Interestingly, we demonstrate that a subgroup of these regulators function in the regulation of spindle orientation through their role in dampening microtubule plus end polymerization.
Keywords: aneuploidy, chromosomal instability, chromosome segregation, microtubule assembly, microtubule dynamics, mitosis, monastrol
Introduction
Most human cancer cells show an increased rate of whole chromosome missegregation during mitosis leading to an evolvement of highly aneuploid progenitors. This cancer-associated phenotype is known as chromosomal instability (CIN) and can be found in almost all types of human cancer.1 It is believed that CIN contributes to the generation of a high genetic variability and a high adaption capability of cancer cells and thereby, supports tumorigenesis and tumor progression.2,3 However, the molecular mechanisms leading to CIN are not well understood.
Various abnormalities during mitosis have been suggested to be involved in CIN. These include the presence of supernumerary centrosomes, impaired spindle checkpoint function or abnormal microtubule-kinetochore attachments.4 Most recently, we have revealed that an increase in microtubule plus end assembly rates within mitotic spindles is not only frequently detected in chromosomally unstable cancer cells, but can also act as a trigger for CIN.5 Thus, it is of utmost interest to identify the genetic alterations that are responsible for an increase in microtubule plus end polymerization in human cancer cells. As a very first step into this direction we found that loss of the tumor suppressor CHK2 or gain of the oncogene AURKA can result in increased microtubule assembly rates during mitosis. However, the underlying mechanisms leading to increased microtubule dynamics at microtubule plus ends are still elusive.5,6
So far, regulation of microtubule plus end assembly is poorly understood, although a growing number of proteins that specifically bind to the microtubule plus ends have been identified. These microtubule plus end tracking proteins (+TIPs) include proteins such as "end binding proteins" (EBs), "adenomatous polyposis coli" (APC), "cytoplasmic linker protein of 170 kDa" (CLIP-170), "cytoplasmic linker associated proteins" (CLASPs) and members of the dynein-dynactin complex (e.g. dynein, p150Glued, Lis1), which were all shown to influence the dynamic behavior of microtubules.7 However, the underlying mechanisms of how +TIPs regulate microtubule dynamics are still poorly understood.
Interestingly, +TIPs have also been implicated in the regulation of mitotic spindle orientation by stabilizing the interaction of plus tips of astral microtubules with protein complexes localized at the cell cortex.8 In fact, complexes of heterotrimeric G proteins (Gα), LGN and NuMA reside at the cell cortex and interact with dynein-dynactin complexes that attach to microtubule plus tips to provide the pulling forces on astral microtubules required for proper spindle positioning.8 It is yet unknown how this interaction and the dynein-dynactin activity is regulated, but the dynamics of microtubules might play an important role. This is also supported by our recent findings showing that increased microtubule plus end polymerization rates in chromosomally instable cancer cells cause spindle orientation defects that subsequently lead to erroneous microtubule-kinetochore attachments and chromosome missegregation.5 Thus, the identification of regulators of microtubule plus end polymerization has a great potential to provide significant novel insights into both, the mechanisms of CIN in cancer cells and the mechanisms of spindle orientation. However, the systematic identification of regulators involved in microtubule plus end polymerization is difficult, because the dynamic behavior of microtubule ends is usually experimentally determined by tracking fluorescently labeled EB proteins at individual microtubule plus ends in live cells.9 It is conceivable that this approach is difficult to be performed in a large-scale screening setting, although a first attempt for this has been described.10 To systematically identify mitotic regulators of microtubule plus end assembly, we introduce here a robust and simple phenotypic screening assay that can easily be applied to fixed and living cells. Interestingly, we show that this assay can be used to systematically dissect regulators of spindle orientation that act dependently or independently of microtubule plus end dynamics.
Results
Chromosomally instable cancer cells with increased microtubule plus end assembly rates exhibit asymmetric monopolar spindles after Eg5/Kif11 inhibition
Inhibition of the mitotic kinesin Eg5 (also known as KIF11, kinesin-5 or KSP) by small molecule inhibitors is known to prevent centrosome separation at the beginning of mitosis. This leads to the formation of monoastral microtubule arrays, which are fully capable to attach to mitotic chromosomes yielding a rosette-like alignment of chromosomes around a monopolar spindle (Fig. 1A).11,12 When treating various cancer cell lines with the Eg5 inhibitors monastrol12 or dimethylenanstron (DME)13 we noticed dramatic differences in the formation of monopolar spindles. While some cell lines formed symmetric spindle microtubule arrays and rosette-like chromosome alignment, others exhibited highly asymmetric monopolar spindles associated with asymmetrically dispersed chromosomes (Fig. 1A).6 This difference was in particular apparent in a panel of colorectal cancer cell lines that differ in their chromosomal stability status14. In fact, chromosomal stable cell lines (SW48, HCT116, RKO) showed symmetric monopolar spindles associated with rosette-like chromosome alignment while chromosomally instable cell lines (SW837, SW620, SW480) displayed highly asymmetric monoasters and asymmetrically dispersed chromosomes upon DME treatment (Fig. 1B). Since we recently showed that CIN in colorectal cancer cells can be triggered by an increase in microtubule plus end assembly rates,5 we reasoned whether asymmetric monopolar spindle assembly might be a read-out for this phenotype. This hypothesis was indeed supported by our observation that transient loss of the tumor suppressor CHK2 or overexpression of AURKA, both of which represent conditions that increase microtubule plus end polymerization rates,5 was sufficient to induce asymmetric monopolar spindle assembly in the presence of DME (Figs. 1C and S1). Moreover, treatment of HCT116 and RKO cells with low doses of nocodazole increased microtubule plus end assembly rates as determined by live cell analyses tracking EB3-GFP at microtubule plus tips5 (Fig. 1D) and resulted in the formation of asymmetric monopolar spindles upon Eg5 inhibition (Fig. 1E). Hence, we found an intriguing correlation between abnormally increased microtubule assembly rates in chromosomally instable cancer cells and the formation of asymmetric monopolar spindles after treatment with Eg5 inhibitors. It is of note that our quantification of asymmetric monopolar spindles are likely to be underestimations since asymmetric spindles viewed from the top might appear as symmetric spindles in some cases (Videos S1-S3).
Figure 1.
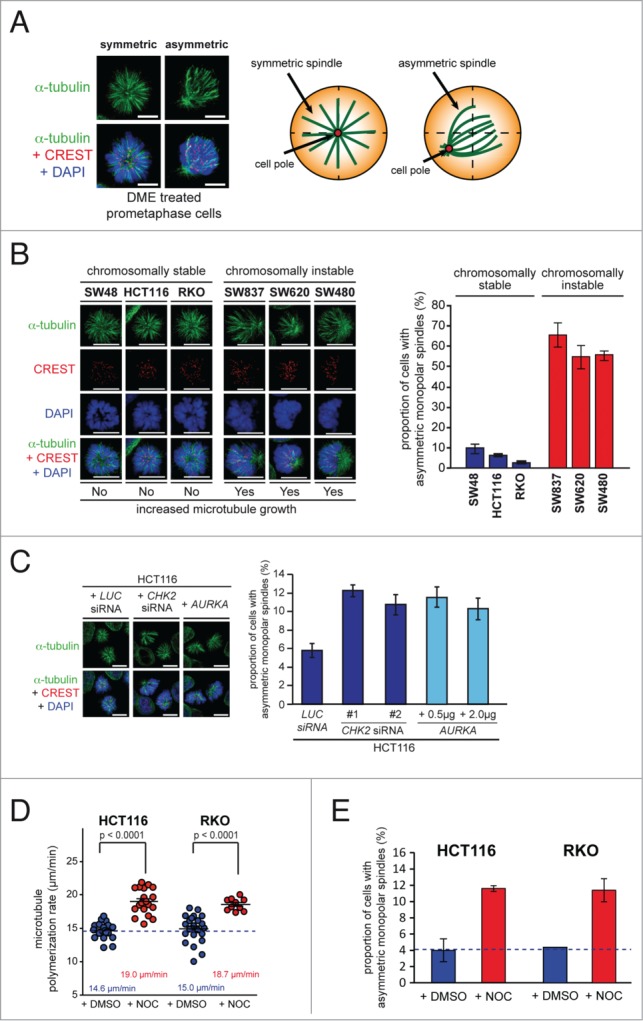
Chromosomally instable cancer cells with increased microtubule plus end assembly rates exhibit asymmetric monopolar mitotic spindles after Eg5/Kif11 inhibition. (A) Example images of symmetric and asymmetric monopolar spindles after treatment with Eg5/Kif11 inhibitors (spindles, anti-α-tubulin: green; kinetochores, CREST: red; chromosomes, DAPI: blue; scale bar, 10 μm). (B) Detection and quantification of asymmetric monopolar spindles in chromosomally stable and instable colorectal cancer cell lines after Eg5/Kif11 inhibition. Cells were treated with 2 μM of dimethylenastron (DME) for 3 hrs and monopolar spindles and mitotic chromosome alignment were detected by immunofluorescence microscopy analysis as indicated and quantified (scale bar, 10 μm; mean ± s.d.; 1500–2000 monopolar spindles from 3 independent experiments were evaluated). (C) Detection and quantification of asymmetric monopolar spindles in HCT116 cells after loss of CHK2 or upon overexpression of AURKA. Cells were transiently transfected with siRNAs or with plasmids and monopolar spindle formation was evaluated after treatment with DME as in (B). Scale bar, 10 μm. The graphs show mean values ± s.e.m. and 1500–2000 monopolar spindles from 3 independent experiments were evaluated. (D) Measurements of mitotic microtubule plus end assembly rates in chromosomally stable HCT116 and RKO cells after treatment with 0.125 nM nocodazole. Scatter dot plots show average assembly rates (20 microtubules per cell, mean ± s.e.m., t-test, n = 10–24 cells from 3 independent experiments). (E) Quantification of the proportion of HCT116 or RKO cells exhibiting asymmetrically monopolar spindles after treatment with 2 μM DME and 2.5 nM Nocodazole for 3h (mean values ± s.d.; HCT116: 3 independent experiments with a total of 1400 monopolar spindles; RKO: 2 independent experiments with a total of 500 monopolar spindles).
Asymmetric monopolar spindle formation after Eg5 inhibition is a robust indicator for increased microtubule assembly rates during mitosis
To test whether the asymmetric monopolar spindle formation upon Eg5 inhibition is indeed a consequence of increased microtubule plus end polymerization, we restored proper microtubule growth rates in chromosomally instable cancer cells. As shown in our previous work,5 this can be achieved by various means including treatment with low doses of Taxol®, which stabilizes microtubule plus ends,15,or by partial repression of the microtubule polymerase ch-TOG/CKAP5 that catalyzes the incorporation of α/β-tubulin subunits at the microtubule plus ends.16 Indeed, restoring proper microtubule plus end assembly rates by low doses of Taxol® in chromosomally instable colorectal cancer cells as well as in CHK2 deficient or AURKA overexpressing HCT116 cells efficiently suppressed the formation of asymmetric monoasters (Figs. 2A and B). Similarly, partial depletion of ch-TOG/CKAP5 also suppressed the formation of asymmetric monopolar spindles in CHK2 deficient or AURKA overexpressing cells that exhibit otherwise increased microtubule polymerization rates (Figs. 2C and S2). Moreover, since an elevated level of Aurora-A kinase activity was shown to be involved in mediating an increase in microtubule plus end assembly rates in colorectal cancer cells5 we analyzed monopolar spindle formation after inhibition of Aurora-A by the small molecule inhibitor MLN8054.17 Similar to the Taxol® treatments, selective inhibition of Aurora-A fully suppressed the formation of asymmetric monopolar spindles in the presence of DME in cancer cells with increased microtubule assembly rates (Fig. 2D). This result was also confirmed in living cells where asymmetric monopolar spindles were rapidly reverted into symmetric monopolar microtubule arrays upon MLN8054 or Taxol® treatment within a few minutes (Fig. 2E, F and Videos S4 and S5). These findings demonstrate that increased microtubule plus end assembly rates result in asymmetric monopolar spindle arrays in the presence of Eg5 inhibition. Moreover, our results indicate that detection of asymmetric monopolar spindles might be used as a robust readout for abnormally increased microtubule assembly rates in fixed and living cells and opens up the opportunity to systematically screen for regulators of microtubule plus end assembly.
Figure 2 (See previous page).
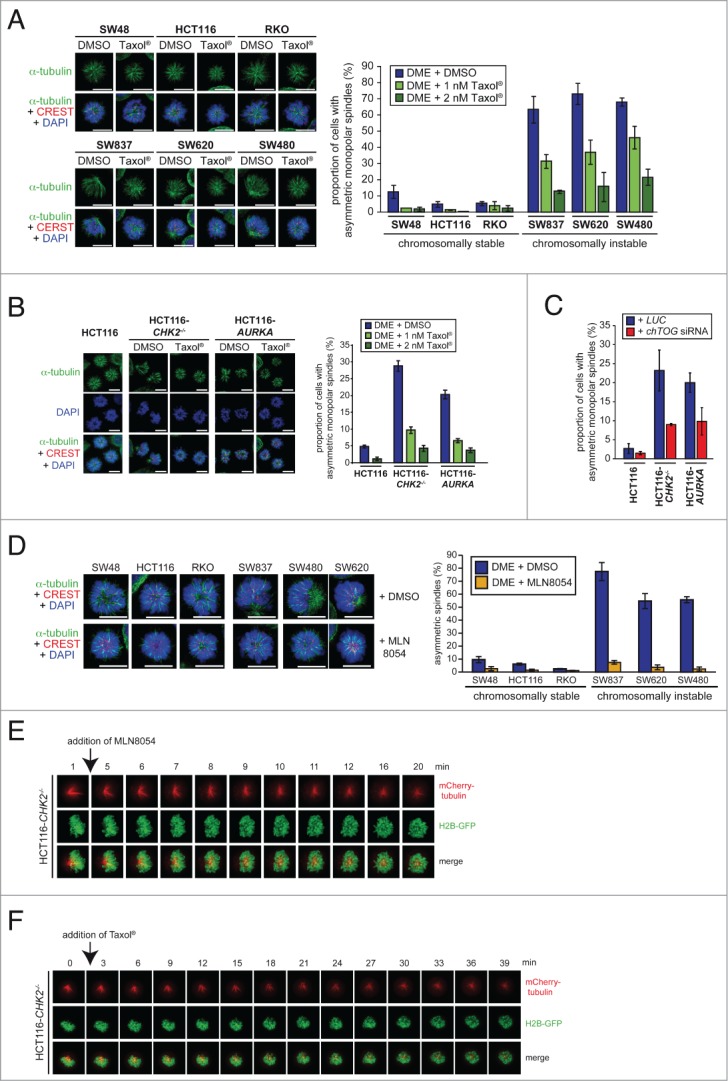
Asymmetric monopolar spindle formation after Eg5 inhibition is a robust indicator for increased microtubule assembly rates during mitosis. (A) Suppression of asymmetric monopolar spindle formation after restoration of normal microtubule plus end assembly rates by low dose Taxol® treatment. The indicated cell lines were treated with 2 μM DME in the absence or presence of Taxol® and cells with monopolar spindles were quantified by immunofluorescence analysis. Representative images are shown (spindles, anti-α-tubulin: green; kinetochores, CREST:red; chromosomes, DAPI: blue; scale bar, 10 μm). The graphs show mean values ± s.d. and 650–1300 monopolar spindles from 2–3 independent experiments were evaluated. (B) Suppression of monopolar spindle formation in CHK2 deficient or AURKA overexpressing cells after treatment with low doses of Taxol®. Cells were treated as in (A) and the proportion of cells with asymmetric monopolar spindles were assessed by immunofluorescence microscopy and quantified. Representative images are shown (spindles, anti-α-tubulin: green; kinetochores, CREST:red; chromosomes, DAPI: blue; scale bar, 10 μm). The graphs show mean values ± s.d.; 3 independent experiments with a total of 1500 monopolar spindles evaluated. (C) Suppression of monopolar spindle formation in CHK2 deficient or AURKA overexpressing cells after partial loss of ch-TOG. Cells were transfected with siRNAs targeting CH-TOG treated with DME and the proportion of cells with asymmetric monopolar spindles were quantified (mean ± s.d.; 2 independent experiments with a total of 1000 monopolar spindles evaluated). (D) Suppression of asymmetric monopolar spindle formation after restoration of normal microtubule plus end assembly rates by inhibition of Aurora-A. The indicated cell lines were treated with 2 μM DME and 0.5 μM MLN8054 for 2.5 h and asymmetric monopolar spindle formation was assessed. Representative immunofluorescence microscopy images are shown (spindles, anti-α-tubulin: green; kinetochores, CREST: red; chromosomes, DAPI: blue; scale bar, 10 μm). The graphs show mean values ± s.d. (2–4 independent experiments with a total of 430–1680 monopolar spindles evaluated). (E and F) Live cell video image sequences showing HCT116-CHK2-/- cells expressing GFP-tagged histone H2B and mCherry-tagged α-tubulin and treated with 2 μM DME for 2 h and subsequently with 0.5 μM MLN8054 (E) or 2 nM Taxol® (F). Live cell videos according to these images are provided as supplementary videos 1 and 2.
Identification of Cep192 as a positive regulator of microtubule plus end assembly
Our previous work demonstrated that an increase in Aurora-A kinase activity localized at mitotic centrosomes can trigger an increase in mitotic microtubule plus end assembly rates.5 During mitosis, Aurora-A can associate with different co-factors including TPX2 and Cep192, which direct the kinase to mitotic spindles and centrosomes, respectively.18,19 Hence, we reasoned whether these co-factors are also involved in the regulation of microtubule plus end assembly. To test this, we expressed YFP-tagged TPX2 or GFP-tagged CEP192 in chromosomally stable HCT116 cells (Fig. S3) and detected, in agreement with previous findings,18,19 spindle localization for TPX2 and centrosomal localization for Cep192 (Fig. 3A). By performing the monopolar spindle assay, we found an induction of asymmetric monopolar spindles upon expression of CEP192, but not after expression of TPX2 (Fig. 3B). Importantly, the formation of asymmetric monopolar spindles after expression of CEP192 could be fully suppressed by treatment with Taxol® or MLN8054 indicating that the phenotype is induced by an Aurora-A dependent increase in microtubule polymerization (Fig. 3B). To verify this, we tracked mCherry-EB3 in living cells and confirmed indeed an increase in the microtubule polymerization rates upon expression of CEP192 (Fig. 3C). Thus, by increasing the expression of CEP192 the monopolar spindle assay reliably predicted a role of Cep192 as a positive regulator of microtubule plus end assembly.
Figure 3.
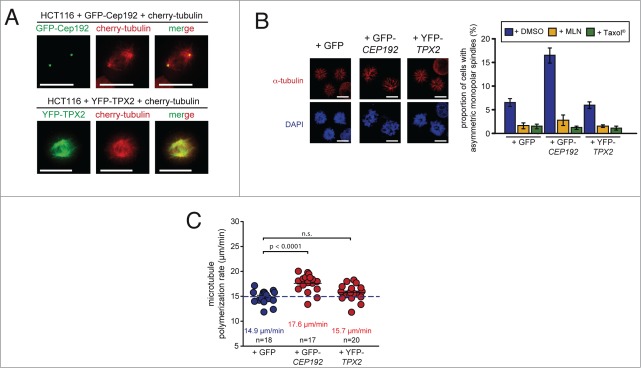
Identification of Cep192 as a regulator of microtubule plus end assembly. (A) Representative examples of HCT116 cells expressing mCherry-tagged α-Tubulin and GFP-tagged Cep192 (upper panel) or YFP-tagged TPX2 (lower panel). Scale bar, 10 μm. (B) Detection and quantification of HCT116 cells overexpressing CEP192 or TPX2 and showing asymmetric monopolar spindles after Eg5/Kif11 inhibition. Cells were transfected with plasmids expressing GFP-Cep192 or YFP-TPX2 and asymmetric monopolar spindles were evaluated by immunofluorescence analysis in the presence of 2 μM DME. Representative examples are given (spindles, anti-α-tubulin: green; kinetochores, CREST:red; chromosomes, DAPI: blue; scale bar, 10 μm). The graphs show the quantification of the proportion of cells showing asymmetric monopolar spindles in the absence or presence of 2 nM Taxol® or 0.5 μM MLN8054 (mean values ± s.e.m.; 3–8 independent experiments with a total of 1500–4000 monopolar spindles evaluated). (C) Determination of mitotic microtubule plus-end assembly rates in response to overexpression of Cep192 or TPX2. HCT116 cells expressing GFP-Cep192 or YPF-TPX2 were transfected with mCherry-EB3 and microtubule growth rates were determined in living cells. The scatter dot plots show average assembly rates (20 microtubules per cell, mean ± s.e.m., t-test, n = 17–20 cells from 3 independent experiments).
Systematic screening for regulators of microtubule plus end assembly
In addition to identifying positive regulators of microtubule plus end assembly such as Aurora-A or Cep192 we reasoned that the monopolar spindle assay should also be useful to identify negative regulators of microtubule assembly. To identify such regulators, we used siRNAs targeting 24 pre-selected candidate genes previously implicated in centrosomal regulation, microtubule plus end binding or spindle orientation. After siRNA transfection, we verified the efficiency of the siRNAs (Fig. S4) and performed the monopolar spindle assay (as outlined in Fig. 4A). Upon microscopic evaluation of asymmetric monopolar spindle formation in fixed cells, we identified 11 siRNAs that induced asymmetric monopolar spindles (Fig. 4B) indicating that those genes might be negative regulators of microtubule plus end assembly. Among these candidates are +TIPs such as the CLASPs, CLIP-170, dynein, Lis1, p150glued, but also the centrosomal or cortex associated proteins Cep170, PCM-1, NuMA and Gα1–3 suggesting that microtubule plus end assembly is not only regulated at the plus tip, but also by centrosomes and the cell cortex.
Figure 4.

Screening for regulators of microtubule plus end assembly. (A) Experimental outline for the identification of regulators of microtubule plus end assembly. (B) Identification of regulators of microtubule plus end assembly using the monopolar spindle assay. HCT116 cells were transfected with the indicated siRNAs and the proportion of cells with asymmetric monopolar spindles was determined after treatment with DME for 3 h. The graphs show mean values ± s.d. (3 independent experiments with a total of 1500 monopolar spindles evaluated). (C) Outline for the calculation of a spindle pole displacement factor (SPDF). (D) Determination of the spindle pole displacement factor (SPDF) in HCT116 cells transfected with siRNAs targeting CHK2, DYNHC1 or EB1. The dot plots show SPDF values (mean ± s.e.m.; t-test; 3 independent experiments with a total of 116–123 cells).
Potential application for large-scale screens
Our results strongly nominate the monopolar spindle assay as an approach to systematically identify novel regulators of microtubule plus end assembly. To allow large-scale or even genome-wide screens in the future, we reasoned to simplify the evaluation step of the assay approach. Since asymmetric spindle formation is accompanied with a spindle pole displacement from the center of the cell, the relative position of the spindle pole with its 2 centrosomes can serve as a parameter for asymmetric spindle formation and thus, for increased microtubule assembly rates (Fig. 4C). Therefore, we visualized both, the monopolar spindle (by detecting α-tubulin) and the 2 centrosomes (by detecting γ-tubulin) by immunofluorescence microscopy and calculated a "spindle pole displacement factor (SPDF)" (Fig. 4C). To test the robustness of this approach, we used cells transfected with siRNAs targeting luciferase (control), CHK2, DYNHC1 or EB1 and calculated the SPDF from more than 100 cells. In fact, in full accordance with the results form the evaluation of the asymmetric spindle structures (Fig. 4B), we found a significant, approximately fold3- increase of the SPDF upon depletion of CHK2 or DYNHC1, but not after EB1 repression (Fig. 4D). Thus, calculating the relative position of the spindle pole/centrosomes after treatment with Eg5 inhibitors provides a reliable readout for asymmetric monopolar spindle assembly and hence, for increased microtubule assembly rates. Therefore, we propose that automated calculation of the SPDF might be a useful tool for future large-scale screens to identify novel regulators of microtubule plus end polymerization.
Determination of the role of microtubule plus end assembly rates for the regulation of mitotic spindle orientation
Interestingly, most of the candidates identified in our siRNA screen (Fig. 4B) have been previously implicated in the regulation of spindle orientation, which might indicate a role of microtubule plus end dynamics for proper spindle positioning during mitosis. To test this hypothesis, we treated siRNA transfected cells with low doses of Taxol® to restore normal microtubule assembly rates and assessed spindle orientation in metaphase cells. As controls, we used siRNAs targeting APC, EB1 and TPX2, which are known to result in spindle misorientation, but neither affected microtubule plus end growth rates nor symmetric monopolar spindle formation in the absence or presence of Taxol® (Fig. 5A and B). As expected, all siRNAs that were hits in our monopolar spindle assay also caused increased microtubule assembly rates and both phenotypes were efficiently suppressed upon Taxol® treatment (Fig. 5A and B). Importantly, all siRNAs caused spindle orientation defects and, interestingly, in the cases of CEP170, CLIP170 and NuMA these defects were suppressed upon restoration of normal microtubule polymerization (Figs. 5C and S5) indicating that these genes regulate spindle orientation indeed through their role in microtubule plus end assembly. Thus, we conclude that the monopolar spindle assay can efficiently identify microtubule plus end regulators that can also function in the regulation of spindle orientation through their role in microtubule plus end polymerization.
Figure 5.

Determination of the role of microtubule plus end assembly rates for the regulation of mitotic spindle orientation. (A) Monopolar spindle assay of HCT116 cells transfected with the indicated siRNAs and treated with or without 2 nM Taxol® for 3.5 hrs. The proportion of cells exhibiting asymmetric monopolar mitotic spindles was quantified (mean values ± s.e.m., t-test; 3–7 independent experiments with a total of 1500–3500 mitotic cells). (B) Determination of microtubule plus end assembly rates in mitotic HCT116 cells transfected with siRNAs as in (A) and treated with or without 0.2 nM Taxol® for 2 h. Scatter dot plots show average assembly rates (20 microtubules per cell, mean ± s.e.m., t-test, n = 20–30 cells from 3 independent experiments). (C) Determination of angles of the spindle axes in HCT116 cells synchronized in metaphase and transfected with siRNAs as in (A) in the presence or absence of 0.2 nM Taxol®. The box and whisker plots show the range, mean and quartile of the measurements (t-test, n = 40–151 cells from 2–7 independent experiments).
Discussion
Here, we introduced a simple, cost effective and reliable phenotypic assay for the detection of increased microtubule plus end assembly rates during mitosis, which reflects a key mitotic phenotype strongly associated with whole chromosome instability (CIN) in human cancer cells.5 In fact, our recent work has revealed that an increase in microtubule plus end polymerization is involved in the generation of so-called lagging chromosomes that represent a frequent pre-stage of chromosome missegregation leading to aneuploidy in human cancer cells.5,20 Therefore, it is of utmost interest to identify the genetic lesions that are responsible for the induction of this intriguing cancer-associated phenotype. The assay system we present here now provides the opportunity to systematically identify regulators that restrain microtubule polymerization and thus, ensure proper microtubule plus end assembly rates. Since CIN can contribute to tumorigenesis and tumor progression, those regulators might represent important tumor suppressor genes.
Our assay relies on the inhibition of Eg5/Kif11 to generate monopolar spindles, which can easily be assessed for their symmetry. Although we found a robust causal relationship between increased mitotic microtubule polymerization rates and asymmetric monopolar spindle formation, it is unclear why monopolar spindles become asymmetric when plus end assembly rates increase. In live cell analyses we observed a rapid re-formation of symmetric monopolar spindles upon restoration of normal plus end assembly rates suggesting that the spindle asymmetry is not caused by centrosome mis-positioning before cells enter mitosis. Instead, these analyses reinforces a causal role of abnormally elevated microtubule polymerization rates during mitosis that determine the symmetry status of monopolar spindles.
Interestingly, recent work has revealed that the human kinesin-12 Kif15 can mediate centrosome separation and bipolar spindle assembly when Kif11/Eg5 is absent.21,22 This Kif15 dependent mechanism involves the formation of monopolar spindle intermediates, which resembles the asymmetric monopolar spindles we observe in our assay using Kif11/Eg5 inhibitors.21 Thus, it might be possible that the formation of asymmetric monopolar spindles in the absence of Kif11/Eg5 not only involves increased microtubule plus end assembly, but also the function of Kif15. Alternatively, one might even speculate that Kif15 itself might support microtubule plus end assembly. Thereby, Kif15 might mediate asymmetric monopolar spindle assembly in the absence of Kif11/Eg5 and might also contribute to misorientation of bipolar spindles.
The results we obtained using the novel monopolar spindle assay were verified by direct measurements of microtubule plus end polymerization rates, which is routinely performed by tracking GFP-tagged "end binding" proteins (EBs) in single living cells.5,9 However, systematically screening for increased microtubule assembly rates using a live cell approach is technically difficult and usually restricted to the analyses of a few cells. However, a recent report indicated that EB3-GFP tracking might be performed in an automated manner.10 Since EB3-GFP tracks in mitosis are very complex, automated tracking faces the risk to follow individual microtubules with diminished accuracy. This might be the reason why automatically calculated microtubule polymerization rates derived from automatically recorded EB3-GFP tracks show a rather high mean variation.10 Nevertheless, automated tracking of EB3-GFP in living cells might represent a promising methodology for future systematic microtubule dynamics measurements, in particular in interphase cells where the complexity of individual microtubules is lower than in mitotic cells.
Our phenotypic assay specifically detects increased microtubule plus end assembly rates, which is a highly cancer-relevant phenotype underlying chromosome missegregation in cancer cells.5 Therefore, the most important result form our study is that we have now a simple detection assay available that can be used to systematically identify genes that contribute to CIN and aneuploidy in human cancer cells.
In fact, with this study we have already embarked on systematically identifying those genes. By using pre-selected candidate siRNAs we identified genes that have not been previously implicated in CIN in human cancer. For instance, loss of +TIPs such as CLASPs and CLIP-170 and also components of the dynein-dynactin complex (dynein, p150GLUED, Lis1)7 significantly increases microtubule plus end polymerization rates suggesting a restraining role of these proteins for microtubule polymerization. Interestingly, also centrosomal proteins such as PCM-1 and Cep170 were found to be involved in dampening microtubule plus end polymerization, which is in agreement with our previous findings showing that loss of centrosomal Chk2 or elevated levels of centrosomal Aurora-A kinase also contributes to increased microtubule plus end assembly.5 It is currently unknown how these centrosomal proteins affect microtubule plus ends, but it is interesting to note that many +TIPs are also localized at centrosomes where they might also contribute to the regulation of microtubule plus end behavior.23
Based on the identification of the first candidates that restrain microtubule plus end assembly it will be also of great interest to investigate whether those genes are downregulated in CIN cancer cells that are characterized by increased plus end assembly rates. For this, comparative gene expression profiling of cell lines with or without abnormal microtubule assembly would be a valuable and complementary approach to identify novel regulators of microtubule plus end assembly.
It is intriguing that many microtubule plus end regulators we identified and validated in our screen are also required for proper orientation of a bipolar mitotic spindle. This suggests that microtubule plus end assembly might influence spindle orientation. Indeed, restoration of proper microtubule assembly rates by low dose Taxol® treatment showed that a subset of spindle regulators such as Cep170, Clip170 and NuMA contribute to proper spindle orientation by restraining proper microtubule plus end dynamics. However, other spindle orientation regulators that are also required for proper microtubule plus end assembly act on spindle positioning independent of microtubule plus end polymerization, which suggests that these regulators might have dual functions, in spindle orientation and in the regulation of microtubule plus end polymerization.
In sum, our work provides an easy and efficient phenotypic assay for the identification of microtubule plus end assembly regulators. This will not only provide novel insights into a key process in mitosis, but is also likely to identify novel genes that contribute to chromosomal instability in human cancer.
Materials and Methods
Cell culture
HCT116 and isogenic CHK2-deficient cells24 were a gift from Bert Vogelstein (Baltimore, USA); colorectal cancer cell lines were from ATCC. All cell lines were cultured at 37°C with 5% CO2 in RPMI1640 containing 10% fetal calf serum, 1% Glutamine, 100 μg/ml streptomycin and 100 U/ml penicillin (Invitrogen).
Cell treatments
To restore normal microtubule plus end assembly rates, cells were either treated with 0.2 - 2 nM of Taxol® (Sigma)5 or with 0.5 μM of the Aurora-A inhibitor MLN8054 (Selleck Chemicals).17 To increase microtubule plus end assembly rates, cells were treated with 0.125 - 2.5 nM of nocodazole (Sigma).5
cDNAs and plasmid transfection
pcDNA3-AURKA plasmid was a gift from Martin Eilers (Würzburg, Germany). Stable HCT116 cell lines overexpressing AURKA were described previously.5 For live-cell microscopy, pEGFP-EB3 (kindly provided by Linda Wordeman, Seattle, USA) was used to track microtubule plus tips. pCR3.1-Cep192-GFP and pEYFP-TPX2 were kind gifts from David J. Sharp (New York, USA) and Oliver Gruss (Heidelberg, Germany), respectively. All DNA transfections were carried out by electroporation using an electroporator (BioRad) at 300 V and 500 μF.
siRNA transfection
siRNA transfections were carried out using Interferin transfection reagent (Polyplus) according to the manufacturer′s instructions and the following siRNAs (Sigma) were used:
LUCIFERASE (control): 5′-CUUACGCUGAGUACUUCGAUU-3′
APC: 5′-AAGACGUUGCGAGAAGUUGGA-3′
AURKA: 5′-GGCAACCAGUGUACCUCAU-3′
CEP170: 5′-GAAGGAAUCCUCCAAGUCA-3′
CEP192: 5′-AGCAGCUAUUGUUUAUGUUGAAAA-3′
CHK2 #1: 5′-CCUUCAGGAUGGAUUUGCCAAUC-3′
CHK2 #2: 5′-AAUGUGUGAAUGACAACUACU-3′
CH-TOG: 5′-GAGCCCAGAGUGGUCCAAA-3′
CLASP1: 5′-GGAUGAUUUACAAGACUGG-3′
CLASP2: 5′-GACAUACAUGGGUCUUAGA-3′
CLIP170: 5′-GCACAGCUCUGAAGACACC-3′
DYNHC1: 5′-GCCAAAAGUUACAGACUUU-3′
EB1: 5′-AUUCCAAGCUAAGCUAGAA-3′
EB3: 5′-ACUAUGAUGGAAAGGAUUAC-3′
Gα1/3: 5′-CCGAAUGCAUGAAAGCAUG-3′
Gα2: 5′-CUUGAGCGCCUAUGACUUG-3′
KIF18B: 5′-CGUACAACACCCUCAAAUA-3′
LIS1: 5′-AAGAGUUGUGCUGAUGACAAG-3′
MCAK: 5′-GCAGGCUAGCAGACAAAU-3′
NuMA: 5′-CAGCGCCAACUCAUCGUUCUA-3′
PCM1: 5′-GUAUCACAUCUGAACUAAA-3′
TP53: 5′-GUAAUCUACUGGGACGGAA-3′
P150GLUED: 5′-GACUUCACCCCUUGAUUAA-3′
RHAMM: 5′-GGUGCUUAUGAUGUUAAA-3′
SLAIN2: 5′-CUCUAUAGAUAGUGAGUUA-3′
TACC3: 5′-GUGGAUUACCUGGAGCAGU-3′
TPX2: 5′-GAAUGGAACUGGAGGGCUU-3′.
Immunofluorescence microscopy
To evaluate monoastral mitotic spindles, cells were treated with Eg5/Kif11 inhibitors (68 μM monastrol (Sigma)12 or 2 μM of the Dimethylenastron (DME; Calbiochem)13) for 3 hours and then fixed with 2% p-formaldehyde in PHEM (60 mM Pipes, 27 mM Hepes, 10 mM EGTA, 4 mM MgSO4, pH 7.0) followed by treatment with methanol at –20°C for 5 minutes. The following antibodies and dilutions were used for immunofluorescence microscopy experiments: anti-α-tubulin (1:700, B-5–1–2, Santa Cruz), anti-γ-tubulin (1:600, Sigma, #T3559), anti-Centromere (CREST, 1:1000, Europa Bioproducts, #90C-CS1058). Secondary antibodies conjugated to Alexa Fluor-488/-594 (1:1000, Molecular Probes, A11029/A11032) were used. DNA was stained with Hoechst33342 (Sigma). Microscopy of fixed cells was performed on a Leica DM6000B microscope (Leica) equipped with a charged-coupled device Orca-II-ERA camera (Hamamatsu). Images were recorded with a Z-optical spacing of 0.2 μm, deconvolved and analyzed using the Leica LAS-AF software (Leica).
Live cell microscopy
Live cell time-lapse microscopy was performed as described.5 Cells expressing GFP-H2B and mCherry-tubulin were seeded onto glass-bottom dishes (Ibidi), treated with 2 μM DME for 2 hours before 15 z-stack images were recorded every 30–90 seconds using a DeltaVision Elite microscope (GE Healthcare) with an Olympus 60X or 100X, 1.42 NA objective at 37°C and 5% CO2 atmosphere. Images were deconvolved and maximal projections were used to construct videos.
To follow the re-formation of symmetric monopolar spindles in response to Taxol® or MLN8084 treatment in living cells, cells expressing GFP-tagged H2B and mCherry-tagged α-tubulin were treated with 2 μM DME for 2 hrs before time-lapse microscopy was started. After one minute of recording the growth medium was replaced with medium containing either 2 nM Taxol® or 0.5 μM MLN8054 in the continued presence of DME. Images were recorded with 15 z-stacks every 30–90 seconds.
Determination of microtubule plus end assembly rates
Microtubule assembly rates were measured as described.5 Briefly, cells were transfected with plasmids expressing GFP- or mCherry tagged EB3 and treated with 2 μM of DME for one hour before z-stack images were recorded every 2 seconds. Average assembly rates (μm/min) per cell were calculated from 20 individual microtubules per cell.
Determination of spindle orientation
Cells were transfected with siRNAs, synchronized at G1/S by a double thymidine block and seeded onto Fibronectin coated cover slips (BD Biosciences, Germany, #354088) in the presence or absence of 0.2 nM Taxol®. Eight hours after release form the synchronization block when the proportion of metaphase cells was maximal cells were fixed in ice cold methanol for 6 min. Fixed cells were stained for α-tubulin, γ-tubulin and with Hoechst33342 to visualize metaphase spindles, centrosomes and chromosomes, respectively. Microscopy was performed on a DeltaVision-ELITE microscope (GE Healthcare) equipped with a PCO Edge® sCMOS camera (PCO, Germany). Images were recorded with a Z-optical spacing of 0.4 μm, deconvolved and analyzed using the SoftWorx 5.0 (Applied Precision) software. Spindle orientation was determined by measuring the angle between the centrosome axis and the growth surface (substratum) following the mathematic formula α = (sinα × 180°)/π.
Western blotting
Western blotting was performed as described previously,25 using the following antibodies and dilutions: anti-Aurora-A (1:2000, H130, Santa Cruz), anti- Cep192 (1:1000; Bethyl, A302–324A), anti-Chk2 (1:800, DCS-270, Santa Cruz), anti-ch-TOG (1:1000; H4, Santa Cruz), anti-Clasp2 (1:1000; F-3, Santa Cruz), anti-Clip170 (1:800, Cell Signaling, #8977), anti-EB1 (1:1000; F-7, Santa Cruz), anti-EB3 (1:1000; 7, Santa Cruz), anti-MCAK (1:250; kindly provided by Linda Wordeman, Seattle, USA), anti-NuMA (1:400, A73-B/D12, Santa Cruz), anti-PCM1 (1:1000; kindly provided by Oliver Gruss, Heidelberg, Germany), anti-p53 (1:1000; DO-1, Santa Cruz), anti-p150glued (1:800; BD Biosciences), anti-RHAMM (1:6000, kindly provided by V. Assmann, Hamburg, Germany), anti-TACC3 (1:1000; H300, Santa Cruz), anti-Tpx2 (1:1000; 18D5, Santa Cruz), anti-β-actin (1:40000, AC-15, Sigma). Secondary antibodies conjugated to horseradish peroxidase (1:10000, Jackson ImmunoResearch) were used and proteins were detected by enhanced chemoluminescence.
RNA Isolation and RT-PCR
When antibodies were not available, knockdown efficiencies of the siRNA transfections were determined by RT-PCR. Total RNA was isolated from HCT116 cells after siRNA transfections using the RNeasy® Mini Kit (Qiagen) according to the manufacturer`s protocol. cDNA was synthesized from 1μg of isolated RNA using Maxima reverse transcriptase (Thermo Scientific) and used for real-time PCR analysis using DreamTaq DNA-Polymerase (Thermo Scientific). GAPDH was used as reference gene. The following primer pairs were used for PCR analysis:
APC forward, 5′-AGGAATTTGTCTTGGCG-3′, and reverse, 5′-GCAGCACTTCCCATAGC-3′;
CEP170 forward, 5'-TGGAAGCAAACGTTGGG-3', and reverse, 5'-CACTGAGCCTCCTCCC-3'
CLASP1 forward, 5′-GGGCCTGCAGAACTTAC-3′, and reverse, 5′-GCTGCCTTTCTCACGTC-3′;
DYNHC1 forward, 5′-GTTAACCGCTGGATCC-3′, and reverse, 5′-CTTCAAAGCATGCTACC-3′;
KIF18B forward, 5′-CAGTGATGATCGCTGCC-3′, and reverse, 5′-CTCTGGCAGTGCATGTG-3′;
LIS1 forward, 5′-GGCCAAATCAAGATGGC-3′, and reverse, 5′-GGCACTCCCACACTTTT-3′;
SLAIN2 forward, 5′-TTCTAGCCCGGATGCAG-3′, and reverse, 5′-AAGGTATGGCTGAAGCC-3′.
Statistical analyses
All data are shown as mean ± standard deviations (SD) or standard error of the mean (SEM). Where indicated students t-tests using the Prizm software package, version 4 (Graphpad) were applied. Quantification of mitotic spindles are based on at least 3 independent experiments, in which at least 1500 mitotic figures were evaluated. Spindle orientation was determined in 40–151 individual cells (see Fig. S5) and spindle angles were calculated according to the formula: α = (sinα × 180°)/π using the Prism software package, version 4 (GraphPad Software).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Linda Wordeman, Bert Vogelstein, David J. Sharp, Oliver Gruss, Martin Eilers and Volker Assmann for materials and Heike Krebber for microscopy support.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft (KFO179) and by a DFG funded Heisenberg professorship awarded to H.B.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Rajagopalan H, Lengauer C. Aneuploidy and cancer. Nature 2004; 432:338-41; PMID:15549096; http://dx.doi.org/ 10.1038/nature03099 [DOI] [PubMed] [Google Scholar]
- 2. McGranahan N, Burrell AR, Endesfelder D, et al. Cancer chromosomal instability: therapeutic and diagnostic challenges. EMBO Rep 2012; 13:528-38; PMID:22595889; http://dx.doi.org/ 10.1038/embor.2012.61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Holland JA, Cleveland WD. Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol 2009; 10:478-87; PMID:19546858; http://dx.doi.org/ 10.1038/nrm2718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thompson LS, Bakhoum FS, Compton AD. Mechanisms of chromosomal instability. Curr Biol 2010; 20:R285-95; PMID:20334839; http://dx.doi.org/ 10.1016/j.cub.2010.01.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ertych N, Stolz A, Stenzinger A., et al. Increased microtubule assembly rates influence chromosomal instability in colorectal cancer. Nature Cell Biology 2014; 16:779-91; PMID:24976383; http://dx.doi.org/ 10.1038/ncb2994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stolz A, Ertych N, Kienitz A., et al. The CHK2-BRCA1 tumour suppressor pathway ensures chromosomal stability in human somatic cells. Nature Cell Biol 2010; 12:492-9; PMID:20364141; http://dx.doi.org/ 10.1038/ncb2051 [DOI] [PubMed] [Google Scholar]
- 7. Akhmanova A, Steinmetz OM. Microtubule +TIPs at a glance. J Cell Sci 2010; 123:3415-9; PMID:20930136; http://dx.doi.org/ 10.1242/jcs.062414 [DOI] [PubMed] [Google Scholar]
- 8. Lu SM, Johnston AC. Molecular pathways regulating mitotic spindle orientation in animal cells. Development 2013; 140:1843-56; PMID:23571210; http://dx.doi.org/ 10.1242/dev.087627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stepanova T, Slemmer J, Hoogenraad CC, et al. Visualization of microtubule growth in cultured neurons via the use of EB3-GFP (end-binding protein 3-green fluorescent protein). J Neurosci 2003; 23:2655-64; PMID:12684451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sironi L, Solon J, Conrad C, et al. Automatic quantification of microtubule dynamics enables RNAi-screening of new mitotic spindle regulators. Cytoskeleton (Hoboken) 2011; 68:266-78; PMID:21491614; http://dx.doi.org/ 10.1002/cm.20510 [DOI] [PubMed] [Google Scholar]
- 11. Kapoor MT, Mayer UT, Coughlin LM, et al. Probing spindle assembly mechanisms with monastrol, a small molecule inhibitor of the mitotic kinesin, Eg5. J Cell Biol 2000; 150:975-88; PMID:10973989; http://dx.doi.org/ 10.1083/jcb.150.5.975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mayer UT, Kapoor MT, Haggarty JS, et al. Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science 1999; 286:971-4; PMID:10542155; http://dx.doi.org/ 10.1126/science.286.5441.971 [DOI] [PubMed] [Google Scholar]
- 13. Muller C, Gross D, Sarli V, et al. Inhibitors of kinesin Eg5: antiproliferative activity of monastrol analogues against human glioblastoma cells. Cancer Chemother Pharmacol 2007; 59:157-64; PMID:16703323; http://dx.doi.org/ 10.1007/s00280-006-0254-1 [DOI] [PubMed] [Google Scholar]
- 14. Lengauer C, Kinzler WK, Vogelstein B. Genetic instability in colorectal cancers. Nature 1997; 386:623-7; PMID:9121588; http://dx.doi.org/ 10.1038/386623a0 [DOI] [PubMed] [Google Scholar]
- 15. Jordan AM, Wilson L. Microtubules as a target for anticancer drugs. Nat Rev Cancer 2004; 4:253-65; PMID:15057285; http://dx.doi.org/ 10.1038/nrc1317 [DOI] [PubMed] [Google Scholar]
- 16. Brouhard JG, Stear HJ, Noetzel LT, et al. XMAP215 is a processive microtubule polymerase. Cell 2008; 132:79-88; PMID:18191222; http://dx.doi.org/ 10.1016/j.cell.2007.11.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Manfredi GM, Ecsedy AJ, Meetze AK, et al. Antitumor activity of MLN8054, an orally active small-molecule inhibitor of Aurora A kinase. Proc Natl Acad Sci U S A 2007; 104:4106-11; PMID:17360485; http://dx.doi.org/ 10.1073/pnas.0608798104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kufer AT, Sillje HH, Korner R., et al. Human TPX2 is required for targeting Aurora-A kinase to the spindle. J Cell Biol 2002; 158:617-23; PMID:12177045; http://dx.doi.org/ 10.1083/jcb.200204155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Joukov V, Nicolo DA, Rodriguez A, et al. Centrosomal protein of 192 kDa (Cep192) promotes centrosome-driven spindle assembly by engaging in organelle-specific Aurora A activation. Proc Natl Acad Sci U S A 2010; 107:21022-7; PMID:21097701; http://dx.doi.org/ 10.1073/pnas.1014664107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gregan J, Polakova S, Zhang L, et al. Merotelic kinetochore attachment: causes and effects. Trends Cell Biol 2011; 21:374-81; PMID:21306900; http://dx.doi.org/ 10.1016/j.tcb.2011.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sturgill GE, Ohi R. Kinesin-12 differentially affects spindle assembly depending on its microtubule substrate. Curr Biol 2013; 23:1280-90; PMID:23791727; http://dx.doi.org/ 10.1016/j.cub.2013.05.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tanenbaum EM, Macurek L, Janssen A, et al. Kif15 cooperates with eg5 to promote bipolar spindle assembly. Curr Biol 2009; 19:1703-11; PMID:19818618; http://dx.doi.org/ 10.1016/j.cub.2009.08.027 [DOI] [PubMed] [Google Scholar]
- 23. Bettencourt-Dias M, Glover MD. Centrosome biogenesis and function: centrosomics brings new understanding. Nat Rev Mol Cell Biol 2007; 8:451-63; PMID:17505520; http://dx.doi.org/ 10.1038/nrm2180 [DOI] [PubMed] [Google Scholar]
- 24. Jallepalli VP, Lengauer C, Vogelstein B., et al. The Chk2 tumor suppressor is not required for p53 responses in human cancer cells. J Biol Chem 2003; 278:20475-9; PMID:12654917; http://dx.doi.org/ 10.1074/jbc.M213159200 [DOI] [PubMed] [Google Scholar]
- 25. Stolz A, Vogel C, Schneider V, et al. Pharmacologic abrogation of the mitotic spindle checkpoint by an indolocarbazole discovered by cellular screening efficiently kills cancer cells. Cancer Res 2009; 69:3874-83; PMID:19366805; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-3597 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.