

Abstract
Necrotic cell death triggers a range of biological responses including a strong adaptive immune response, yet we know little about the cellular pathways that control necrotic cell death. Inhibitor studies suggest that proteases, and in particular cathepsins, drive necrotic cell death. The cathepsin B-selective inhibitor CA-074-Me blocks all forms of programmed necrosis by an unknown mechanism. We found that cathepsin B deficiency does not prevent induction of pyroptosis and lysosome-mediated necrosis suggesting that CA-074-Me blocks necrotic cell death by targeting cathepsins other than cathepsin B. A single cathepsin, cathepsin C, drives necrotic cell death mediated by the lysosome-destabilizing agent Leu-Leu-OMe (LLOMe). Here we present evidence that cathepsin C-deficiency and CA-074-Me block LLOMe killing in a distinct and cell type-specific fashion. Cathepsin C-deficiency and CA-074-Me block LLOMe killing of all myeloid cells, except for neutrophils. Cathepsin C-deficiency, but not CA-074-Me, blocks LLOMe killing of neutrophils suggesting that CA-074-Me does not target cathepsin C directly, consistent with inhibitor studies using recombinant cathepsin C. Unlike other cathepsins, cathepsin C lacks endoproteolytic activity, and requires activation by other lysosomal proteases, such as cathepsin D. Consistent with this theory, we found that lysosomotropic agents and cathepsin D downregulation by siRNA block LLOMe-mediated necrosis. Our findings indicate that a proteolytic cascade, involving cathepsins C and D, controls LLOMe-mediated necrosis. In contrast, cathepsins C and D were not required for pyroptotic cell death suggesting that distinct cathepsins control pyroptosis and lysosome-mediated necrosis.
Keywords: Caspase-1, inflammasome, lysosome rupture, necrosis, pyroptosis
Abbreviations
- LLOMe
L-leucyl-L-leucine methyl ester
- LMN
lysosome-mediated necrosis, IL, interleukin
- LPS
lipopolysaccharide
- NLR
nucleotide-binding domain and leucine rich repeat
Introduction
Cell death is a highly regulated process that contributes to multiple biologic processes. For example, necrotic cell death has been linked to microbial pathogenesis, ischemic stroke, and induction of adaptive immune responses.20,27,30,33,46,52 While most attention has focused on apoptotic cell death, recent evidence suggests that necrotic, like apoptotic cell death is a highly regulated process.3,7,13,18,22,34 Unlike apoptotic cells, necrotic cells have a compromised plasma membrane, which leads to the leakage of cellular components, such as uric acid, HMGB1, DNA and ATP, into the extracellular media.31,36,37,39,48 This necrotic release of cellular factors has been implicated in the Th2-biased immune response mediated by lysosome-destabilizing adjuvants, such as alum and LLOMe.28,31 Cellular factors released from necrotic cells have also been shown to enhance the efficiency of anti-cancer therapies.2,11,32 While these studies highlight the importance of necrotic cell death, we know little about the cellular pathways that control these necrotic processes.
Necrotic cell death was originally believed to be a non-regulated process caused by trauma or environmental insults.17 However, recent studies indicate that necrotic cell death is, like apoptosis, controlled by inducer-specific cellular factors and susceptible to distinct inhibitors.5,8,28,41,55 In contrast to a multistep cascade driving apoptotic cell death, only single, inducer-specific events have been associated with necrotic cell death. For example, caspase-1 activation has been shown to drive pyroptosis, the best-studied form of necrotic cell death.8,19 Extended caspase-1 activation is highly toxic to target cells, and requires the formation of a multiprotein complex called the inflammasome.8,19,36 The inflammasome complex generally consists of a Nod-like receptor (NLR), a scaffolding protein (Asc) and a catalytic component (caspase-1).38 Agents that block caspase-1 activation, such as high potassium concentrations, caspase-1 inhibitors, and proteasome inhibitors (restricted to the NLR Nlrp1b), generally prevent pyroptotic cell death.29,44,49 Necroptosis, a second form of necrotic cell death, is dependent on RIP-1 signaling.12,15 We have recently described a third form of necrotic cell death, designated as lysosome-mediated necrosis (LMN), that is characterized by lysosome rupture and broad proteolysis of cytosolic proteins.28,35 Intriguingly, the cathepsin B-selective inhibitor, CA-074-Me, blocks not only all known forms of necrotic cell death, but also apoptotic cell death4,21 The mechanism by which CA-074-Me blocks necrotic cell death is unclear and a major focus of this study.
Here we demonstrate that cathepsin B is dispensable for the CA-074-Me inhibition of pyroptotic cell death and lysosome-mediated necrosis. We have previously shown that a single cysteine cathepsin (cathepsin C) controls not only necrotic cell death, but also the adaptive immune response mediated by the dipeptide methyl-ester, LLOMe.28 Here we show that both cathepsin C deficiency and CA-074-Me block LLOMe-induced necrotic cell death in a cell type-specific fashion. Cathepsin C deficiency and CA-074-Me prevented LLOMe killing by all myeloid cells tested, except for neutrophils. LLOMe-mediated killing of neutrophils was blocked by cathepsin C-deficiency, but not by CA-074-Me. These findings suggested that CA-074-Me act does not block cathepsin C directly, consistent with our observation that the inhibitor does not block recombinant cathepsin C. We further found that LLOMe killing is efficiently blocked by lysosomotropic agents and controlled by cathepsin D. Together our findings indicate that multiple cathepsins are part of a proteolytic cascade driving lysosome-mediated cell death.
Results
CA-074-Me and lysosomotropic agents control necrotic cell death in an inducer-specific fashion
While recent studies suggest that necrotic cell death is a highly controlled process, we know little about the regulators and checkpoints guiding necrotic cell death. Inhibitor studies point towards involvement of proteolytic enzymes in necrotic cell death. To determine the role of proteolytic enzymes, particularly lysosomal cathepsins, in necrosis, we used cathepsin inhibitors and lysosomotropic agents. Initially, we used the cathepsin B inhibitor CA-074-Me, as it blocks a wide range of programmed cell death, including all forms of necrotic cell death by an unknown mechanism. To analyze the CA-074-Me block of necrotic cell death, we challenged primary murine macrophages with lysosome-destabilizing agents and pyroptosis inducers in the presence of increasing CA-074-Me concentrations. As a model agent for lysosome-mediated necrosis we used the soluble dipeptide methyl ester LLOMe,28,50,51 and as prototypical pyroptosis inducers we used anthrax lethal toxin (LT) and LPS/nigericin.8,9,25,40 We found that 10 μM CA-074-Me was sufficient to block LLOMe killing, while 50 μM CA-074-Me was required to block pyroptotic cell death mediated by LPS/nigericin and LT (Fig. 1A) indicating inducer-specific CA-074-Me profiles. Inducer-specific inhibitor effects were more pronounced when we used the cathepsin D inhibitor, pepstatin A. Pepstatin A blocked LLOMe-induced cell death, while it had no impact on pyroptotic cell death mediated by LPS/nigericin and LT (Fig. 1B).
Figure 1.
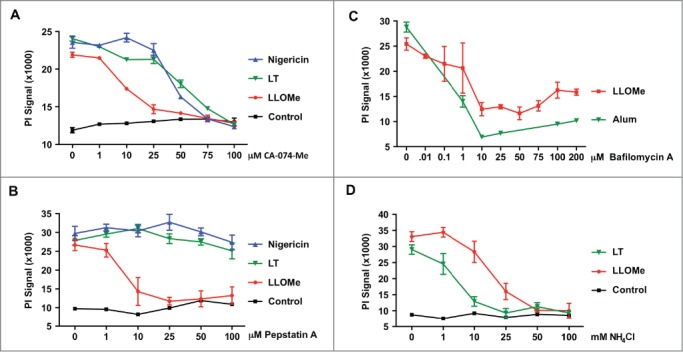
Pyroptotic and lysosome-mediated cell death exhibit distinct protease inhibitor susceptibilities. (A) BALB/c-derived macrophages were primed with LPS, and subsequently challenged with 1.5 mM LLOMe, 15 μM Nigericin or anthrax lethal toxin (LT) (500 ng/ml PA and 250 ng/ml LF) for 3 hours in the presence of increasing doses of CA-074-Me. (B) BALB/c-derived macrophages were primed with 250 ng/ml LPS for 2 hours, and then exposed to 10 (M nigericin, 2 mM LLOMe, or anthrax lethal toxin (LT) (500 ng/ml PA and 250 ng/ml LF) for 2 hours in the presence of increasing concentrations of the cathepsin D inhibitor pepstatin A. Cell death was determined by PI exclusion assays. Representative experiment is shown, and PI measurements were performed in triplicates. Pyroptotic and lysosome-mediated cell death exhibit distinct protease inhibitor susceptibilities. (C) C57BL/6-derived macrophages were exposed to 2 mM LLOMe for 2 hours or to alum (150 (g/ml) for 6 hours in the presence of increasing concentrations of bafilomycin A. (D) C57BL/6-derived macrophages were exposed to 2 mM LLOMe or anthrax lethal toxin (LT) (500 ng/ml PA and 250 ng/ml LF) for 2 hours in the presence of increasing concentrations of NH4Cl. Cell death was determined by PI exclusion assays. Control cells (CT) received NH4Cl only. Cell death was determined by PI exclusion assays. Data shows representative experiments performed in triplicate.
To further characterize the proteolytic processes in necrotic cell death, we tested the pH-dependence of pyroptotic and lysosome-mediated cell death. Towards this, we challenged primary macrophages with pyroptosis inducers and lysosome-destabilizing agents in the presence of lysosomotropic agents. We found that the lysosomotropic agent NH4Cl blocked necrotic cell death mediated by LLOMe and the pyroptosis inducer LT, yet with distinct efficiencies (Fig. 1C). Significantly lower NH4Cl concentrations were required to block LT-induced cell death compared to LLOMe-induced cell death (Fig. 1C). Consistent with pH dependence, bafilomycin A also blocked necrotic cell death mediated by the lysosome-destabilizing agents LLOMe and alum (Fig. 1D). Taken together our findings suggest that pH-dependent proteases control necrotic cell death, consistent with cathepsins in this process.
Major cathepsins are not required for pyroptotic cell death
Cathepsins B and C have been implicated in lysosome-mediated cell death,26,28 and the cathepsin B inhibitor, CA-074-Me blocks pyroptotic cell death (Fig. 1B). To identify cathepsins required for pyroptotic cell death, we challenged a panel of murine macrophages deficient in the predominant cathepsins with pyroptosis inducers. While the cathepsin B inhibitor CA-074-Me blocked pyroptotic cell death (Fig. 1B), cathepsin B-deficiency did not block cell death mediated by the pyroptosis inducers nigericin and ATP (Fig. 2). These findings are consistent with studies using the soluble dipeptide methyl ester LLOMe, which triggers CA-074-Me-dependent (Fig. 1A), but cathepsin B-independent cell death,28 None of the other major cathepsins, including cathepsins C, L and S, were required for nigericin and ATP-mediated cell death (Fig. 2). Taken together, our findings indicated that CA-074-Me blocks pyroptotic cell death by targeting proteases other than the predominant cathepsins.
Figure 2.
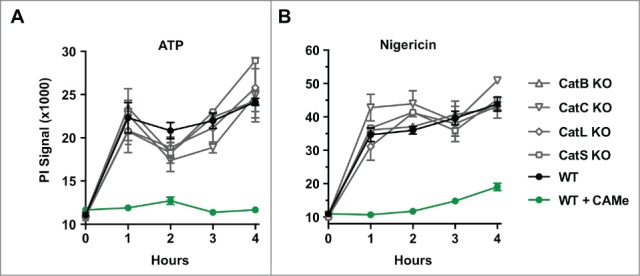
Cathepsin-dependence of pyroptotic cell death. Wild-type, cathepsin B-, cathepsins C-, cathepsin L-, and cathepsin S-deficient C57BL/6 macrophages BALB/c-derived macrophages were primed with LPS for 2 hours, and subsequently challenged with 5 mM ATP (A) or 15 μM nigericin (B) for 4 hours. As a control, wild-type cells were treated with 100 μM CA-074-Me (CAMe). Cell death was measured by PI exclusion. Data show represent a representative experiments performed in triplicate.
The pan-necrosis inhibitor CA-074-Me blocks LLOMe killing in a cell type-specific fashion
We have previously demonstrated that LLOMe-mediated necrotic cell death is blocked by CA-074-Me and by cathepsin C-deficiency,26,28 As LLOMe susceptibility is restricted to specific cell types,26,28 we tested whether the CA-074-Me block of necrotic cell death is also cell type-specific. Towards this, we challenged a range of murine myeloid cell types, such as monocytes, dendritic cells and neutrophils, with LLOMe in the absence and presence of CA-074-Me. While LLOMe killed murine monocytes, dendritic cells and neutrophils with high efficiency, CA-074-Me blocked only LLOMe killing of monocytes and dendritic cells, but not LLOMe killing of neutrophils (Fig. 3A and B). As controls, we challenged cathepsin B and C-deficient myeloid cells with LLOMe. Consistent with earlier studies (26,28), cathepsin C-deficiency blocked LLOMe killing of all myeloid cells tested, including neutrophils (Fig. 3A and B). As expected, cathepsin B-deficiency did not block LLOMe killing of the myeloid cells tested (Fig. 3A and B). LLOMe-induced neutrophil killing was cathepsin C-dependent, but not blocked by CA-074-Me suggesting that CA-074-Me blocks LLOMe killing by targeting proteins other than cathepsin C.
Figure 3.

CA-074-Me blocks LLOMe killing in a cell type-specific fashion. (A) Wild type and cathepsin B and C-deficient splenocytes (monocytes, dendritic cells, B cells, or neutrophils) were exposed to 2 mM LLOMe, and cell survival was analyzed by Live/Dead stain and flow cytometry 2 hours post-LLOMe exposure. Wild type macrophages treated with 100 μM CA-074-Me (CAMe) served as controls. Immune cell specificity was determined using cell type-specific antibodies. (B) Wild type and cathepsin B- and C-deficient monocytes, and neutrophils were exposed to increasing concentrations of LLOMe, and cytopathic effects were analyzed by Live/Dead stain and flow cytometry 2 hours post-LLOMe exposure. Wild type and cathepsin B-deficient macrophages were also treated with 100 μM CA-074-Me. Immune cell specificity was determined using cell type-specific antibodies and flow cytometry. Data show represent a representative experiments performed in triplicate. ns, not significant; ***P< 0.001.
We next wanted to know whether the inability of CA-074-Me to block LLOMe killing of neutrophils might stem from low esterase levels in this cell type. Cellular esterases are required to hydrolyze the methyl ester domain of CA-074-Me in order to generate the active inhibitor,10 To test whether CA-074-Me was still able to block neutrophil killing mediated by other cell death inducers, neutrophils were exposed to the pyroptosis inducer nigericin in the presence of CA-074-Me. While CA-074-Me failed to prevent LLOMe killing of neutrophils (Figs. 3 and4A), the inhibitor efficiently blocked nigericin-mediated neutrophil death (Fig. 4B) indicating that CA-074-Me is active in these cell types.
Figure 4.
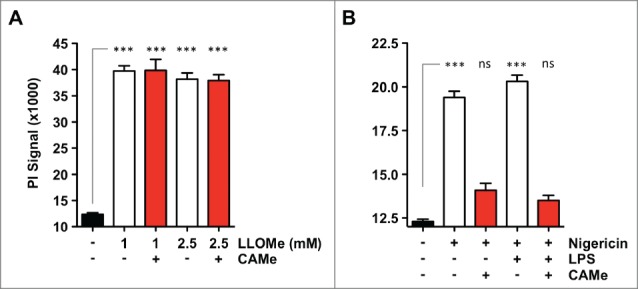
CA-074-Me is active in neutrophils. (A) C57BL/6-derived neutrophils were challenged with 2 mM LLOMe for 2 hours in the absence or presence of 100 μM CA-074-Me (CAMe). (B) C57BL/6-derived neutrophils were primed with 250 ng/ml LPS for 2 hours, and then challenged with 10 μM nigericin for 2 hours in the absence or presence of 100 μM CA-074-Me. Cell death was determined by PI exclusion assays. Data show represent a representative experiments performed in triplicate. ns, not significant; ***P< 0.001.
The cathepsin inhibitor CA-074-Me does not block cathepsin C
We have previously demonstrated that a single protein, cathepsin C, is critical for LLOMe-induced cell death and immune responses,28 We next wanted to know whether cathepsin C is also responsible for the CA-074-Me block of LLOMe-mediated cell death, as cathepsin B is not required for this process.28 CA-074-Me requires prior cleavage by cellular esterases, and is therefore inactive in cell-free suspensions (42). We therefore performed in vitro assays with recombinant cathepsins in the presence of CA-074, which is active in vitro, as it does not require prior cleavage of the methyl-ester group for activation. Consistent with our inhibitor and knockout studies, CA-074 did not block the activity of recombinant cathepsin C even at exceeding concentrations (Fig. 5). As positive controls we used the cathepsin C inhibitor, Gly-Phe-diazomethylketone (GF-DMK), which blocked recombinant cathepsin C in the nanomolar range (Fig. 5). As a control for CA-074, we tested the activity of recombinant cathepsin B in the presence of the inhibitor. As expected, CA-074 blocked recombinant cathepsin B in the nanomolar range, while the cathepsin C inhibitor, GF-DMK, had no impact on cathepsin B activity (Fig. 5). Taken together, these findings suggested that the critical protease for LLOMe killing, cathepsin C, is not involved in the CA-074-Me block of LLOMe killing.
Figure 5.
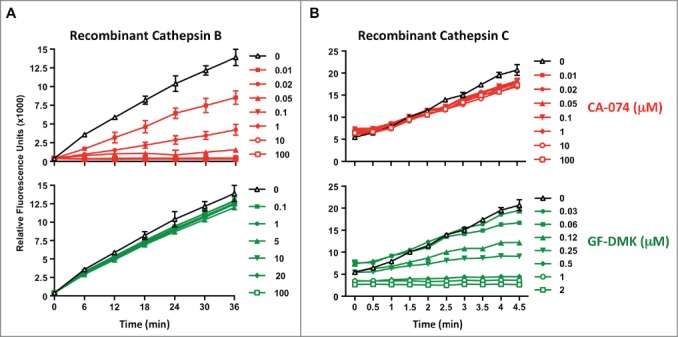
CA-074 does not inhibit recombinant cathepsin C. The in vitro assay was performed in the presence of recombinant cathepsin B (A) or cathepsin C (B), the corresponding cathepsin B and C substrates, and increasing concentrations of the cathepsin B inhibitor CA-074, or the cathepsin C inhibitor GF-DMK. Cathepsin B and C activity was measured by analyzing Gly-Arg-AMC and Arg-Arg-AMC cleavage at 460 nm, respectively. Experiment was performed in triplicate.
CA-074-Me blocks processing of inflammatory proteins in pyroptotic and lysosome-mediated cell death
Caspase-1 activation/Nlrp3 signaling is the central event in pyroptosis, and perpetual caspase-1 activation is the driving force in pyroptotic cell death.8,19 To determine whether this critical step in pyroptotic cell death is targeted by CA-074-Me, we tested whether the inhibitor blocks IL-1β processing, as an indicator for caspase-1 activation, in cells challenged with the pyroptosis inducer nigericin. We found that CA-074-Me blocks IL-1β processing and at concentrations that also prevented cell death in nigericin-treated cells (Fig. 6A). While LLOMe killing is independent of caspase-1 activation23,28,35,45 processing of IL-1β also occurs in LLOMe-treated cells (Fig. 6B). Intriguingly, CA-074-Me also blocked proteolysis of pro-IL-1β in LLOMe-treated cells at concentrations that prevented LLOMe killing (Fig. 6B). As expected, cathepsin C-deficiency blocked cell death and pro-IL-1β proteolysis mediated by LLOMe, but not by the pyroptosis inducer nigericin (Fig. S1). As CA-074-Me prevents the activation of caspase-1 (Fig. 6A) or cathepsin C (Fig. 4) without targeting these proteases directly (42), it is reasonable to assume that CA-074-Me blocks an upstream event in necrotic cell death, preceding activation of caspase-1 and cathepsin C.
Figure 6.
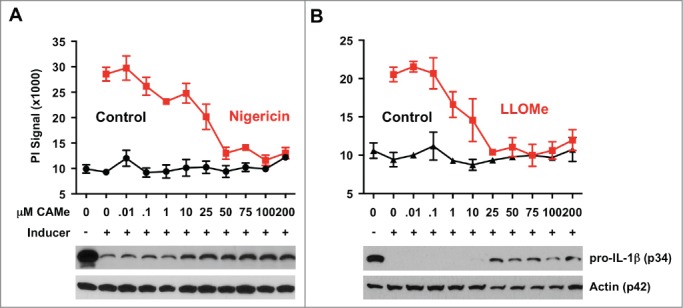
CA-074-Me blocks IL-1β processing in pyroptotic and lysosome-mediated cell death. CA-074-Me response of macrophages exposed to the lysosome-destabilizing agent LLOMe and the pyroptosis inducer nigericin. C57BL/6-derived macrophages were primed with 250 ng/ml LPS for 2 hours and then exposed to 10 (M nigericin (A) or to 2 mM LLOMe (B) for 2 hours in the presence of increasing concentrations of CA-074-Me (CAMe). Control cells (control) received CA-074-Me only. Cell death was determined by propidium iodide (PI) exclusion assays. Levels of pro-IL-1β or actin (control) were determined from lysates of LPS or nigericin-treated macrophages by immunoblotting (lower panel). Cell death assay was performed in triplicate.
LLOMe-mediated cell death is controlled by cathepsin D
Our findings indicated that CA-074-Me blocks LLOMe-induced lysosome rupture and cell death without targeting cathepsin C directly. Among the macrophage-associated cathepsins, cathepsin C is unique in that it is incapable of autocatalytic activation, as it lacks an endoproteolytic activity.14 Cathepsin C is activated early in the endocytic pathway by other lysosomal enzymes, such as cathepsin D.14,16,47 We have shown that the cathepsin D inhibitor, pepstatin b, specifically blocks LLOMe-induced cell death, but not pyroptotic cell death (Fig. 1B). To test whether cathepsin D is required for LLOMe-mediated cell death, we targeted cathepsin D expression in RAW264.7 macrophages by using small interfering RNA (siRNA). Initially we tested available transfection agents for their efficiency in downregulating gene expression by using siRNA in RAW264.7 cells. We found that siQuest most efficiently down-regulates a control protein (GAPDH) using siRNA in RAW264.7 cells (Fig. 7A). Transfection of anti-cathepsin D siRNA by siQuest significantly reduced cathepsin D expression in RAW264.7 cells and LLOMe-induced cell death (Fig. 7B and C) suggesting that cathepsin D is required for LLOMe-induced necrotic cell death.
Figure 7.
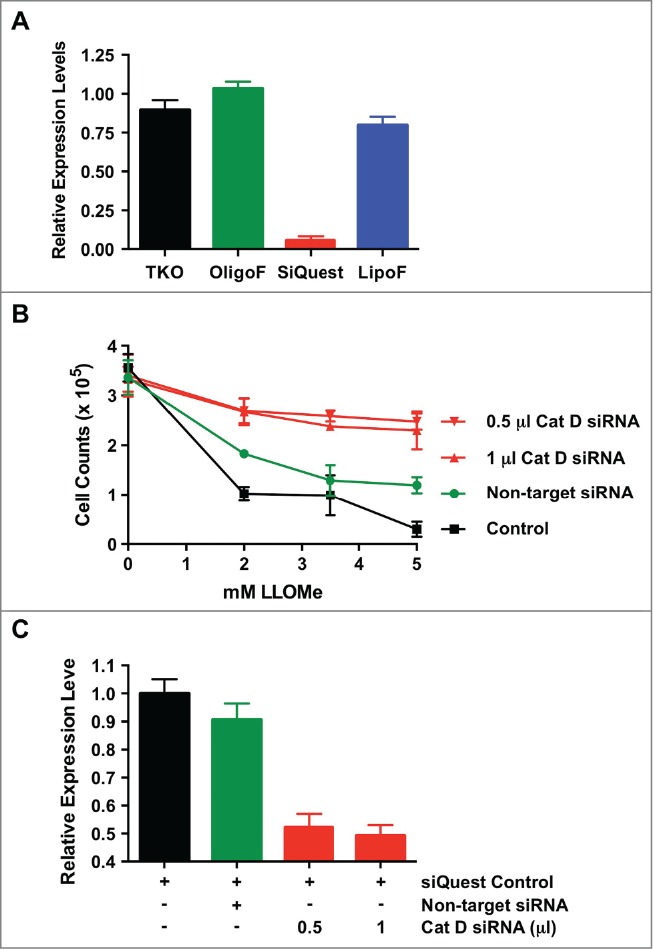
Cathepsin D controls LLOMe-mediated cell death. (A) Knock-down control assays in RAW264.7 macrophages. RAW264.7 macrophages were transfected with anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) siRNA in the presence of 2 μl of the transfection reagents, TKO, siQuest, Oligofectamine (OligoF) and Lipofectamine2000 (LipoF). Expression levels of GAPDH were determined by quantitative PCR one day post-transfection. (B) RAW264.7 macrophages were transfected using siQuest with increasing amount of anti-cathepsin D siRNA, non-target siRNA and siQuest only, as a negative control, and cathepsin D expression were determined by quantitative PCR one day post transfection. (C) Cell survival was determined by analyzing cell counts of RAW264.7 macrophages transfected with anti-cathepsin D siRNA, scrambled siRNA and siQuest following exposure to increasing amount of LLOMe (mM) only one day post LLOMe exposure.
In summary, we provide evidence that the cathepsin inhibitor CA-074-Me blocks an early event in necrotic cell death mediated by lysosome-destabilizing agents and pyroptosis inducers. Our data suggests that CA-074-Me targets an event upstream of caspase-1 and cathepsin C activation in pyroptotic and lysosome-mediated cell death, respectively. Using CA-074-Me, cathepsin-deficient macrophages and recombinant cathepsins allowed us to demonstrate that the CA-074-Me block of necrotic cell death is independent of cathepsins B and C. Inhibition of lysosome-mediated necrosis by lysosomotropic agents and cathepsin D knockdown assays suggest that pH-dependent proteases and cathepsin D control early processes in lysosome-mediated necrosis. In summary, our findings indicate that a cascade of proteolytic events control necrotic cell death and the ensuing adaptive immune response mediated by lysosome-destabilizing adjuvants
Discussion
The cathepsin B inhibitor CA-074-Me blocks all forms of necrotic cell death, including pyroptosis, necroptosis and lysosome-mediated necrosis4,25,28,35 by an unknown mechanism. Using knockout approaches and recombinant cathepsins we demonstrate that cathepsin B is not required for pyroptotic cell death and lysosome-mediated necrosis. These results suggested that CA-074-Me blocks necrotic cell death by targeting cathepsins/proteases other than cathepsin B. We have previously shown that a single cathepsin, cathepsin C controls lysosome-mediated necrosis and the adaptive immune response triggered by the lysosome-disrupting agent LLOMe.28 However, CA-074-Me and cathepsin C-deficiency blocked LLOMe killing of a range of susceptible immune cells, with the exception of neutrophils. LLOMe killing of neutrophils was only blocked by cathepsin C-deficiency, but not by CA-074-Me. These findings suggested that CA-074-Me blocks LLOMe killing in susceptible immune cells by targeting a cellular protease that is lacking in neutrophils. It is also conceivable that multiple proteases act redundantly in neutrophils and one of these cathepsins is not CA074Me-sensitive. As cathepsin C-deficiency still blocks LLOMe killing of neutrophils, it is reasonable to assume that the cellular target for CA-074-Me is not cathepsin C, consistent with the failure of the inhibitor to block recombinant cathepsin C.
We have previously shown that CA-074-Me prevents lysosome rupture mediated by the lysosome-destabilizing agents, LLOMe, alum and silica.28 Pharmacological studies have demonstrated that the carboxypeptidase activity of cathepsin C converts the LLOMe dipeptide into a lysosome-disrupting polymer.28,50,51 Lysosome rupture is a highly toxic event that ultimately leads to necrotic cell death.4,25,28,35 As CA-074-Me blocks lysosome rupture and cell death, it is reasonable to assume that the inhibitor interferes with an event upstream of cathepsin C-mediated polymer formation. CA-074-Me also prevents the critical step in pyroptotic cell death, namely caspase-1 activation.1,25,42 As CA-074-Me does not target caspase-1 directly,25,42 CA-074-Me appears to block an event upstream of caspase-1 activation in pyroptotic cell death. Unlike the cathepsin C and D dependence of LLOMe-induced necrotic cell death, the predominant macrophage-associated cathepsin, such as cathepsin C, were not required for pyroptotic cell death.
While CA-074-Me appears to target an early event pyroptosis and lysosome-mediated necrosis, our studies in neutrophils suggest that the inhibitor does not block a common upstream event in the necrotic process, but two distinct cellular events. We found that CA-074-Me efficiently blocked cell death mediated by pyroptosis inducers, while the inhibitor had no impact on LLOMe-mediated necrosis of neutrophils. These results suggested that CA-074-Me block of pyroptotic cell death of neutrophils by targeting a neutrophil-specific protein that is not required for lysosome-mediated cell death. Taken together, these findings indicate that CA-074-Me blocks pyroptosis and lysosome-mediated necrosis by targeting different host proteins. Future studies are required to determine the identity of the CA-074-Me-targeted proteases controlling pyroptotic and lysosome-mediated cell death.
Our findings indicate that CA-074-Me blocks an upstream event of caspase-1 or cathepsin C activation in pyroptosis and lysosome-mediated cell death. We found that lysosomotropic agents block pyroptotic and lysosome-mediated necrosis suggesting involvement of pH-dependent proteases in necrotic cell death. As lysosomotropic agents prevent the acidification of lysosomal compartments, the most likely explanation is that lysosomal pH changes neutralize or prevent the autoactivation of lysosomal proteases critical for necrosis induction. Cathepsin C, the critical protease for LLOMe-mediated necrotic cell death,28 lacks endoproteolytic activity, and is unable to undergo auto-proteolysis.14 We therefore tested the requirement for other proteases, such as cathepsin D, in the activation of cathepsin C54. We found that the cathepsin D inhibitor, pepstatin A, and downregulation of cathepsin D block necrotic cell death mediated by LLOMe, but not by pyroptosis inducers. It is conceivable that cathepsin D acts upstream of cathepsin C, and activates this critical cathepsin in LLOMe-mediated cell death. However, this proteolytic cascade seems to be specific to lysosome-mediated cell death, as cathepsins C and D are dispensable for pyroptotic cell death. Taken together, our studies suggest that distinct proteolytic cascades control pyroptotic and lysosome-mediated cell death.
Cathepsins have also been linked to apoptotic pathways mediated by staurosporine, p53, oxidative stress and TNF-α.6,24,53 For example, CA-074-Me and cathepsin B-deficiency have been shown to interfere with TNFα-induced apoptotic cell death24 suggesting a role of cathepsin B in apoptotic cell death. However, cathepsin B involvement in apoptotic cell death might be inducer-specific. For example, cathepsin B-deficiency does not prevent apoptosis mediated by etoposide or IL-3 deprivation, while cathepsin B it is required for processing of the proapoptotic proteins, Bid and caspase-343. Consistent with our findings in pyroptosis and lysosome-mediated necrosis, CA-074-Me blocks activation of apoptotic caspases without targeting these caspases directly.24 Taken together, these studies suggest that CA-074-Me blocks an upstream event in apoptotic and necrotic cell death.
In summary, we demonstrated that the cathepsin B inhibitor CA-074-Me prevents lysosome-mediated cell death without targeting cathepsins B or C directly. Here, we provide evidence that CA-074-Me blocks an early event in pyroptosis and lysosome-mediated cell death. We further demonstrate that acid pH-dependent proteases and cathepsin D control early events in necrotic cell death mediated by lysosome-disrupting adjuvants. Taken together, our studies suggest that a proteolytic cascade drives pyroptosis and lysosome-mediated cell death. As these cell death-inducing agents activate a strong adaptive immune response, our findings indicate that multiple proteases control early events in necrotic cell death and immune responses mediated by cell death-inducing adjuvants. As recent findings highlight the importance of necrotic cell death in many biological processes, identification of cellular factors driving necrotic cell death will help us harness these processes and understand how they become dysregulated in disease.
Material and Methods
Chemicals and reagents
Bafilomycin A was purchased from LC Laboratories (Woburn, MA). Imject alum was purchased from Thermo Scientific (Rockford IL). Nigericin was purchased from Calbiochem (San Diego, CA). NH4Cl was purchased from Fisher Scientific (Pittsburgh, PA). Propidium Iodide was purchased from Sigma (St. Louis, MO). CA-074-Me was purchased from Peptides International (Louisville, KY). MG115 was purchased from Calbiochem (San Diego, CA).
Cell culture
Wild type C57BL/6 and BALB/c mice were purchased from Jackson Labs (Bar Harbor, MN). Bone marrow-derived macrophages were generated as described previously.28 Briefly, bone marrow from femurs and tibias of wild type, and cathepsin-deficient mice was isolated and grown for a week in DMEM, containing 10% FCS, 20% L929 preconditioned media, 1% HEPES, 1% MEM nonessential amino acids, and 0.1% BME. For the preparation of primary dendritic cells, cells were conditioned in RPMI with 10% FCS, 1% HEPES, 1% MEM nonessential amino acids, 0.1% BME, and 20 ng/ml GM-CSF.
Generation of splenic cell suspensions
Splenic cell suspensions were produced as previously described.28 Briefly, excised spleens from age-matched C57BL/6 and cathepsin B or C-deficient mice were incubated in RPMI medium containing 2 mg/ml collagenase and 30 μg/ml DNase at 37°C for 30 min. Following drug treatment splenic cell suspensions were washed with PBS and stained with the Blue LIVE/DEAD viability dye (BluVID; Invitrogen) for 20 minutes on ice. Cells were subsequently washed in FACS buffer (PBS, 2% Fetal Bovine Serum, 0.05% NaN3 [wt/vol]), and stained with monoclonal antibodies against cell type-specific markers (BD Pharmingen and eBioscience). Monocytes, and neutrophils were CD45/B220neg CD3eneg, and divided further into CD11bpos and CD11bneg subsets. Neutrophils were CD11bhigh and Ly6Gpos, and monocytes were CD11bhigh/intermediate CD11cneg Ly6Gneg and F4/80neg. Frequency of live cells from each subpopulation was determined by gating on the BluVIDneg cells. Cells were fixed in 1% paraformaldehyde, and analyzed using a BD LSRII flow cytometer.
Cell death assays
Macrophages were plated in a 96-well plate at 1 × 106 cells/ml, and cell death was determined by measuring membrane impairment by analysis of propidium iodide exclusion, or by measuring LDH activity, at specified time points. Propidium iodide was added to a final concentration of 30 μM 10 min prior to analysis, and LDH activity was measured using the CytotoxOne kit (Promega, Madison, WI) according to the manufacturer's description.
Western blotting
Cell lysates were collected and spun at 13,000 rpm for 10 min at 4°C. Lysates were placed in water bath at 100°C for three min, and run on 12% Tris-HCl gels (Bio-Rad). Gels for westerns were then blotted onto PVDF membranes with a semi-dry transfer (Bio-Rad). Membranes were probed with anti-mouse antibodies directed against pro-IL-1β (R&D Systems, Kingstown, RI), pro-caspase-1 (sc-514; Santa Cruz Biotechnologies, Santa Cruz, CA), and actin (AC-40 Sigma-Aldrich, St. Louis, MO). Antibodies against goat were obtained from Santa Cruz.
Cathepsin cleavage assay
Recombinant active murine cathepsin C was purchased form R&D Systems (Minneapolis, MN), and the in vitro assay was performed according to the manufacturer's description. The cathepsin B substrate Arg-Arg-AMC and the cathepsin C substrate Gly-Phe-AMC were purchased from Bachem (Torrance, CA), and the cathepsin C inhibitor, Gly-Phe-diazomethylketone (Gly-Phe-DMK), was from MP Biomedicals (Solon, OH). The cathepsin B assay was performed with 5 μg of recombinant active murine cathepsin B in the presence of 200 μM of the cathepsin B substrate Arg-Arg-AMC and various inhibitors. Cathepsin B was activated with 1 mM dithiothreitol and 0.5% Triton X-100 for 1 hour. The cathepsin C assay was performed with 1 μg of recombinant active murine cathepsin C in the presence of 200 μM of the cathepsin C substrate Gly-Phe-AMC and various inhibitors. The generation of free AMC was determined by measuring excitation and emission wavelengths of 380 nm and 460 nm for 10 min at 30°C using a Victor 3 plate-reader (Perkin-Elmer Life, Mountain View, CA). Cathepsin B and C activity was determined by measuring the slope of the increase in AMC fluorescence over time.
SIRNA knockdown assay
RAW264.7 cells were transfected with 50-200 nM siRNAs specific for cathepsin D or non-target control. siRNA pools (cathepsin D and non-target) were purchased from Dharmacon. Transfection efficiency of RAW264.7 cells was tested with the transfection reagents, siQuest and TKO (Mirus Bio Corporation, Madison, WI), Lipofectamine2000 and Oligofectamine (Life Technology). siRNAs were complexed with the lipid siQuest and cells incubated with the complexed siRNAs in 1 mL complete medium for 24 hrs. At that time knockdown of cathepsin D was determined by quantitative (q)PCR (Biorad). Cathepsin D reduction of 50% or greater was routinely observed with siQuest (by qPCR). Cells treated with lipid-complexed non-target siRNAs or siQuest only showed no significant knockdown.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported in part by the US National Institutes of Health grants (1R56AI092497-01A1 to J.B, H.L and L.J., and by the Martin Turkish and the M-O’Connor-Foundation; and 5R01AI088027 to K.C., and U54-AI057158 (Northeast Biodefense Center-Lipkin) to D.G.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Averette KM, Pratt MR, Yang Y, Bassilian S, Whitelegge JP, Loo JA, Muir TW, Bradley KA. 2009. Anthrax lethal toxin induced lysosomal membrane permeabilization and cytosolic cathepsin release is Nlrp1b/Nalp1b-dependent . Plos One 4:e7913; PMID:19924255; http://dx.doi.org/ 10.1371/journal.pone.0007913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aymeric L, Apetoh L, Ghiringhelli F, Tesniere A, Martins I, Kroemer G, Smyth MJ, Zitvogel L. 2010. Tumor cell death and ATP release prime dendritic cells and efficient anticancer immunity. Cancer Res 70:855-8; PMID:20086177; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-3566 [DOI] [PubMed] [Google Scholar]
- 3. Basu S, Binder RJ, Suto R, Anderson K, Srivastava PK. 2000. Necrotic but not apoptiotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF kappa B pathway. Cell Stress Chaperon 5:373-373; PMID:11058573 [DOI] [PubMed] [Google Scholar]
- 4. Berghe TV, Vanlangenakker N, Parthoens E, Deckers W, Devos M, Festjens N, Guerin CJ, Brunk UT, Declercq W, Vandenabeele P. 2010. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ 17:922-30; PMID:20010783; http://dx.doi.org/ 10.1038/cdd.2009.184 [DOI] [PubMed] [Google Scholar]
- 5. Bergsbaken T, Fink SL, Cookson BT. 2009. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 7:99-109; PMID:19148178; http://dx.doi.org/ 10.1038/nrmicro2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bidere N, Lorenzo HK, Carmona S, Laforge M, Harper F, Dumont C, Senik A. 2003. Cathepsin D triggers bax activation, resulting in selective apoptosis-inducing factor (AIF) relocation in T lymphocytes entering the early commitment phase to apoptosis. The Journal of biological chemistry 278:31401-11; PMID:12782632; http://dx.doi.org/ 10.1074/jbc.M301911200 [DOI] [PubMed] [Google Scholar]
- 7. Borner C, Monney L. 1999. Apoptosis without caspases: an inefficient molecular guillotine? Cell Death Differ 6:497-507; PMID:10381652; http://dx.doi.org/ 10.1038/sj.cdd.4400525 [DOI] [PubMed] [Google Scholar]
- 8. Brennan MA, Cookson BT. 2000. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol Microbiol 38:31-40; PMID:11029688; http://dx.doi.org/ 10.1046/j.1365-2958.2000.02103.x [DOI] [PubMed] [Google Scholar]
- 9. Brojatsch J, Casadevall A, Goldman DL. 2014. Molecular determinants for a cardiovascular collapse in anthrax. Front Biosci (Elite Ed) 6:139-47; PMID:24389148; http://dx.doi.org/ 10.2741/E697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Buttle DJ, Murata M, Knight CG, Barrett AJ. 1992. CA074 methyl ester: a proinhibitor for intracellular cathepsin B. Arch Biochem Biophys 299:377-80; PMID:1444478; http://dx.doi.org/ 10.1016/0003-9861(92)90290-D [DOI] [PubMed] [Google Scholar]
- 11. Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, Schmitt E, Hamai A, Hervas-Stubbs S, Obeid M, et al. 2005. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med 202:1691-701; PMID:16365148; http://dx.doi.org/ 10.1084/jem.20050915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Christofferson DE, Yuan JY. 2010. Necroptosis as an alternative form of programmed cell death. Curr Opin Cell Biol 22:263-8; PMID:20045303; http://dx.doi.org/ 10.1016/j.ceb.2009.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Creagh EM, O'Neill LA. 2006. TLRs, NLRs and RLRs: a trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol 27:352-7; PMID:16807108; http://dx.doi.org/ 10.1016/j.it.2006.06.003 [DOI] [PubMed] [Google Scholar]
- 14. Dahl SW, Halkier T, Lauritzen C, Dolenc I, Pedersen J, Turk V, Turk B. 2001. Human recombinant pro-dipeptidyl peptidase I (cathepsin C) can be activated by cathepsins L and S but not by autocatalytic processing. Biochemistry-Us 40:1671-8; PMID:11327826; http://dx.doi.org/ 10.1021/bi001693z [DOI] [PubMed] [Google Scholar]
- 15. Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. 2005. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury ( 1, 112, 2005). Nat Chem Biol ; PMID:16408008 [DOI] [PubMed] [Google Scholar]
- 16. Diment S, Leech MS, Stahl PD. 1988. Cathepsin-D is membrane-associated in macrophage endosomes. J Biol Chem 263:6901-7; PMID:3360812 [PubMed] [Google Scholar]
- 17. Edinger AL, Thompson CB. 2004. Death by design: apoptosis, necrosis and autophagy. Curr Opin Biol 16:663-9; PMID:15530778; http://dx.doi.org/ 10.1016/j.ceb.2004.09.011 [DOI] [PubMed] [Google Scholar]
- 18. Festjens N, Vanden Berghe T, Vandenabeele P. 2006. Necrosis, a well-orchestrated form of cell demise: signalling cascades, important mediators and concomitant immune response. Bio Biophysica Acta 1757:1371-87; PMID:16950166; http://dx.doi.org/ 10.1016/j.bbabio.2006.06.014 [DOI] [PubMed] [Google Scholar]
- 19. Fink SL, Cookson BT. 2006. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol 8:1812-25; PMID:16824040; http://dx.doi.org/ 10.1111/j.1462-5822.2006.00751.x [DOI] [PubMed] [Google Scholar]
- 20. Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G. 2009. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol 10:241-7; PMID:19221555; http://dx.doi.org/ 10.1038/ni.1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fujisawa A, Kambe N, Saito M, Nishikomori R, Tanizaki H, Kanazawa N, Adachi S, Heike T, Sagara J, Suda T. 2007. Disease-associated mutations in CIAS1 induce cathepsin B-dependent rapid cell death of human THP-1 monocytic cells. Blood 109:2903-11; PMID:17164343 [DOI] [PubMed] [Google Scholar]
- 22. Galluzzi L, Aaronson SA, Abrams J, Alnemri ES, Andrews DW, Baehrecke EH, Bazan NG, Blagosklonny MV, Blomgren K, Borner C, et al. 2009. Guidelines for the use and interpretation of assays for monitoring cell death in higher eukaryotes. Cell Death Differ 16:1093-107; PMID:19373242; http://dx.doi.org/ 10.1038/cdd.2009.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guicciardi ME, Gores GJ. 2013. Complete lysosomal disruption: a route to necrosis, not to the inflammasome. Cell Cycle 12:1995; PMID:23759574; http://dx.doi.org/ 10.4161/cc.25317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guicciardi ME, Miyoshi H, Bronk SF, Gores GJ. 2001. Cathepsin B knockout mice are resistant to tumor necrosis factor-alpha-mediated hepatocyte apoptosis and liver injury: implications for therapeutic applications. Am J Pathol 159:2045-54; PMID:11733355; http://dx.doi.org/ 10.1016/S0002-9440(10)63056-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hentze H, Lin XY, Choi MS, Porter AG. 2003. Critical role for cathepsin B in mediating caspase-1-dependent interleukin-18 maturation and caspase-1-independent necrosis triggered by the microbial toxin nigericin. Cell Death Differ 10:956-68; PMID:12934070; http://dx.doi.org/ 10.1038/sj.cdd.4401264 [DOI] [PubMed] [Google Scholar]
- 26. Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, Fitzgerald KA, Latz E. 2008. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 9:847-56; PMID:18604214; http://dx.doi.org/ 10.1038/ni.1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Iyer SS, Pulskens WP, Sadler JJ, Butter LM, Teske GJ, Ulland TK, Eisenbarth SC, Florquin S, Flavell RA, Leemans JC, et al. 2009. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. Proc Natl Acad Sci USA 106:20388-93; PMID:19918053; http://dx.doi.org/ 10.1073/pnas.0908698106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jacobson LS, Lima H, Jr, Goldberg MF, Gocheva V, Tsiperson V, Sutterwala FS, Joyce JA, Gapp BV, Blomen VA, Chandran K, et al. 2013. Cathepsin-mediated necrosis controls the adaptive immune response by Th2 (T helper type 2)-associated adjuvants. J Biol Chem 288:7481-91; PMID:23297415; http://dx.doi.org/ 10.1074/jbc.M112.400655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Keller M, Ruegg A, Werner S, Beer HD. 2008. Active caspase-1 is a regulator of unconventional protein secretion. Cell 132:818-31; PMID:18329368; http://dx.doi.org/ 10.1016/j.cell.2007.12.040 [DOI] [PubMed] [Google Scholar]
- 30. Kono H, Rock KL. 2008. How dying cells alert the immune system to danger. Nature reviews. Immunology 8:279-89; PMID:18340345; http://dx.doi.org/ 10.1038/nri2215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kool M, Soullié T, van Nimwegen M, Willart MA, Muskens F, Jung S, Hoogsteden HC, Hammad H, Lambrecht BN. 2008. Alum adjuvant boosts adaptive immunity by inducing uric acid and activating inflammatory dendritic cells. J Exp Med 205:869-82; PMID:18362170; http://dx.doi.org/ 10.1084/jem.20071087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kroemer G, et al. 2005. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ 12:1463-7; PMID:16247491; http://dx.doi.org/ 10.1038/sj.cdd.4401724 [DOI] [PubMed] [Google Scholar]
- 33. Li HF, Ambade A, Re F. 2009. Cutting Edge: Necrosis Activates the NLRP3 Inflammasome. J Immunol 183:1528-32; PMID:19596994; http://dx.doi.org/ 10.4049/jimmunol.0901080 [DOI] [PubMed] [Google Scholar]
- 34. Liew FY, Xu D, Brint EK, O'Neill LA. 2005. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol 5:446-58; PMID:15928677; http://dx.doi.org/ 10.1038/nri1630 [DOI] [PubMed] [Google Scholar]
- 35. Lima H, Jr., Jacobson LS, Goldberg MF, Chandran K, Diaz-Griffero F, Lisanti MP, Brojatsch J. 2013. Role of lysosome rupture in controlling nlrp3 signaling and necrotic cell death. Cell Cycle 12:1868-78; PMID:23708522; http://dx.doi.org/ 10.4161/cc.24903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. 2006. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440:228-32; PMID:16407890; http://dx.doi.org/ 10.1038/nature04515 [DOI] [PubMed] [Google Scholar]
- 37. Marichal T, Ohata K, Bedoret D, Mesnil C, Sabatel C, Kobiyama K, Lekeux P, Coban C, Akira S, Ishii KJ, et al. 2011. DNA released from dying host cells mediates aluminum adjuvant activity. Nat Med 17:996-1002; PMID:21765404; http://dx.doi.org/ 10.1038/nm.2403 [DOI] [PubMed] [Google Scholar]
- 38. Martinon F, Burns K, Tschopp J. 2002. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 10:417-26; PMID:12191486; http://dx.doi.org/ 10.1016/S1097-2765(02)00599-3 [DOI] [PubMed] [Google Scholar]
- 39. Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. 2006. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440:237-41; PMID:16407889; http://dx.doi.org/ 10.1038/nature04516 [DOI] [PubMed] [Google Scholar]
- 40. Muehlbauer SM, Evering TH, Bonuccelli G, Squires RC, Ashton AW, Porcelli SA, Lisanti MP, Brojatsch J. 2007. Anthrax lethal toxin kills macrophages in a strain-specific manner by apoptosis or caspase-1-mediated necrosis. Cell Cycle 6:758-66; PMID:17374996; http://dx.doi.org/ 10.4161/cc.6.6.3991 [DOI] [PubMed] [Google Scholar]
- 41. Muehlbauer SM, Lima H, Jr, Goldman DL, Jacobson LS, Rivera J, Goldberg MF, Palladino MA, Casadevall A, Brojatsch J. 2010. Proteasome inhibitors prevent caspase-1-mediated disease in rodents challenged with anthrax lethal toxin. Am J Pathol 177:735-43; PMID:20595632; http://dx.doi.org/ 10.2353/ajpath.2010.090828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Newman ZL, Leppla SH, Moayeri M. 2009. CA-074Me protection against anthrax lethal toxin. Infect Immun 77:4327-36; PMID:19635822; http://dx.doi.org/ 10.1128/IAI.00730-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Oberle C, Huai J, Reinheckel T, Tacke M, Rassner M, Ekert PG, Buellesbach J, Borner C. 2010. Lysosomal membrane permeabilization and cathepsin release is a Bax/Bak-dependent, amplifying event of apoptosis in fibroblasts and monocytes. Cell Death Differ 17:1167-78; PMID:20094062; http://dx.doi.org/ 10.1038/cdd.2009.214 [DOI] [PubMed] [Google Scholar]
- 44. Petrilli V, Dostert C, Muruve DA, Tschopp J. 2007. The inflammasome: a danger sensing complex triggering innate immunity. Curr Opin Immunol 19:615-22; PMID:17977705; http://dx.doi.org/ 10.1016/j.coi.2007.09.002 [DOI] [PubMed] [Google Scholar]
- 45. Reinheckel T. 2013. On the road to inflammation: linking lysosome disruption, lysosomal protease release and necrotic death of immune cells. Cell Cycle 12:1994; PMID:23759575; http://dx.doi.org/ 10.4161/cc.25316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rovere-Querini P, Capobianco A, Scaffidi P, Valentinis B, Catalanotti F, Giazzon M, Dumitriu IE, Müller S, Iannacone M, Traversari C, et al. 2004. HMGB1 is an endogenous immune adjuvant released by necrotic cells. EMBO Rep 5:825-30; PMID:15272298; http://dx.doi.org/ 10.1038/sj.embor.7400205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Samarel AM, Ferguson AG, Decker RS, Lesch M. 1989. Effects of cysteine protease inhibitors on rabbit cathepsin-D maturation. Am J Physiol 257:C1069-79; PMID:2610247 [DOI] [PubMed] [Google Scholar]
- 48. Scaffidi P, Misteli T, Bianchi ME. 2002. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418:191-5; PMID:12110890; http://dx.doi.org/ 10.1038/nature00858 [DOI] [PubMed] [Google Scholar]
- 49. Squires RC, Muehlbauer SM, Brojatsch J. 2007. Proteasomes control caspase-1 activation in anthrax lethal toxin-mediated cell killing. J Biol Chem 282:34260-7; PMID:17878154; http://dx.doi.org/ 10.1074/jbc.M705687200 [DOI] [PubMed] [Google Scholar]
- 50. Thiele DL, Lipsky PE. 1990. The action of leucyl-leucine methyl ester on cytotoxic lymphocytes requires uptake by a novel dipeptide-specific facilitated transport system and dipeptidyl peptidase I-mediated conversion to membranolytic products. J Exp Med 172:183-94; PMID:1972727; http://dx.doi.org/ 10.1084/jem.172.1.183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Thiele DL, Lipsky PE. 1990. Mechanism of L-leucyl-L-leucine methyl ester-mediated killing of cytotoxic lymphocytes: dependence on a lysosomal thiol protease, dipeptidyl peptidase I, that is enriched in these cells. Proc Natl Acad Sci U S A 87:83-7; PMID:2296607; http://dx.doi.org/ 10.1073/pnas.87.1.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ting JPY, Willingham SB, Bergstralh DT. 2008. NLRs at the intersection of cell death and immunity. Nature Reviews Immunology 8:372-9; PMID:18362948; http://dx.doi.org/ 10.1038/nri2296 [DOI] [PubMed] [Google Scholar]
- 53. Turk B, Stoka V, Rozman-Pungercar J, Cirman T, Droga-Mazovec G, Oresić K, Turk V. 2002. Apoptotic pathways: involvement of lysosomal proteases. Biol Chem 383:1035-44; PMID:12437086; http://dx.doi.org/ 10.1515/BC.2002.112 [DOI] [PubMed] [Google Scholar]
- 54. Turk V, Turk Turk B. D. 2001. Lysosomal cysteine proteases: facts and opportunities. The EMBO journal 20:4629-33; PMID:11532926; http://dx.doi.org/ 10.1093/emboj/20.17.4629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. 2010. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 11:700-14; PMID:20823910; http://dx.doi.org/ 10.1038/nrm2970 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.