

Abstract
Statins are largely used in clinics in the treatment of patients with cardiovascular diseases for their effect on lowering circulating cholesterol. Lectin-like oxidized low-density lipoprotein (LOX-1), the primary receptor for ox-LDL, plays a central role in the pathogenesis of atherosclerosis and cardiovascular disorders. We have recently shown that chronic exposure of cells to lovastatin disrupts LOX-1 receptor cluster distribution in plasma membranes, leading to a marked loss of LOX-1 function. Here we investigated the molecular mechanism of statin-mediated LOX-1 inhibition and we demonstrate that all tested statins are able to displace the binding of fluorescent ox-LDL to LOX-1 by a direct interaction with LOX-1 receptors in a cell-based binding assay. Molecular docking simulations confirm the interaction and indicate that statins completely fill the hydrophobic tunnel that crosses the C-type lectin-like (CTLD) recognition domain of LOX-1. Classical molecular dynamics simulation technique applied to the LOX-1 CTLD, considered in the entire receptor structure with or without a statin ligand inside the tunnel, indicates that the presence of a ligand largely increases the dimer stability. Electrophoretic separation and western blot confirm that different statins binding stabilize the dimer assembly of LOX-1 receptors in vivo. The simulative and experimental results allow us to propose a CTLD clamp motion, which enables the receptor-substrate coupling. These findings reveal a novel and significant functional effect of statins.
Keywords: LOX-1 receptor, molecular docking, molecular dynamics simulation, monomer-dimer ratio, statin, substrate recognition
Abbreviations
- Ato
atorvastatin
- Cav-1
caveolin-1
- CTLD
C-type lectin-like domain
- DiI
1,1′-dioctadecyl-3,3,3′,3′-tetramethyllindocarbocyanine perchlorate
- DMEM
Dulbecco's modified Eagle's medium
- Flu
fluvastatin
- HEK
human embryonic kidney
- HMG-CoA
3-hydroxy-3-methylglutaryl coenzyme A
- LDL
low-density lipoprotein
- LDL-C
low-density lipoprotein-cholesterol
- Lov
lovastatin
- LOX-1
lectin-like oxidized low-density lipoprotein receptor-1
- Mab
monoclonal antibody
- ox-LDL
oxidized low-density lipoprotein
- Pra
pravastatin
Introduction
Statins are inhibitors of the 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, the major rate-limiting enzyme in cholesterol biosynthesis.1 For their effect on lowering total circulating and LDL-cholesterol (LDL-C), statins are used in clinics in the treatment of patients with cardiovascular diseases. Statins can also exert cholesterol-independent responses, due to the inhibition of the synthesis of the intermediate products of the mevanolate pathway, the isoprenoids, which are important lipid attachments for post-translational modifications of many proteins, such as Ras, Rho, Rac and nuclear lamina.2 Critical cellular functions such as membrane integrity, cell signaling, protein synthesis and cell-cycle progression can be altered by statins.2,3 The discrimination of the circulating LDL-C lowering effect of statins from other pleiotropic effects may be more evident in the early phase of treatment of patients with cardiovascular diseases. Thus, in clinical use, only 10% reduction of LDL-C level is detectable after 24 hours and at least 6–7 d are necessary to lower it significantly.4 Interestingly, a short term statin pre-treatment prevents myocardial damage in patients with acute coronary syndromes (ACS).5 The molecular mechanisms responsible for these effects, however, are still unknown.
Lectin-like oxidized low-density lipoprotein (LOX-1) is the primary receptor for ox-LDL in endothelial cells. This type II membrane glycoprotein is a disulfide-linked homodimer belonging to the C-type lectin-like receptors family. It is composed by 273 residues and 4 domains: a short N-terminal cytoplasmic domain (34 aa), a single transmembrane domain (26 aa), an extracellular region consisting of a coiled-coil domain named NECK (82 aa), and a C-type lectin-like domain named CTLD (131 aa) at the C-terminus6-8 (Fig. 1).
Figure 1.

LOX-1 receptor model. Red cartoon and transparent surface representation of the protein with the double layer phospholipid membrane in spacefill. This picture was produced by using the program Chimera.46
The CTLD domain forms a disulfide-linked heart-shaped homodimer, which assembles in larger functional oligomers through non covalent interactions.7-12 3D structure7,8 and classical molecular dynamics (MD) simulations of the human LOX-1 receptor and of some mutants10,13 have evidenced that a basic spine, located on the CTLD and crossing the entire dimer, as well as a long hydrophobic tunnel, are important elements in the ox-LDL recognition. The tunnel access is surrounded by a quasi-conical surface where hydrophilic and hydrophobic patches are scattered.7,8 Mutations of residues or selective phospholipids that interact with the residues in the tunnel, significantly prevent the binding ability of LOX-1 receptor.14,15 The NECK domain appears as a dimer consisting of 2 α-helices wound in a parallel coiled-coil structure and displays specific residues, which functionally modulate the flexibility of this region.16 During the process of ox-LDL recognition, a flexible NECK domain structure has been proposed to facilitate the required interactions.16 Moreover, molecular modeling and MD simulation have elucidated the NECK structural model able to explain the observed cleavage site that generates the soluble form of LOX-1.17
LOX-1 is up-regulated in atherosclerosis lesions and during atherogenesis18,19 and its activation triggers oxidative stress response causing plaque vulnerability and potential rupture, which leads to acute atherothrombotic vascular occlusion and tissue infarction. LOX-1 is over-expressed during myocardial ischemia reperfusion and appears to be associated with apoptosis, necrosis and left ventricular functional deterioration.20 Deletion of ORL1, the gene which encodes LOX-1 receptor, in Ldlr knockout mice results in much smaller atherosclerosis lesions, drastic reduction of inflammation in aortic wall and of the extent of ischemia/reperfusion injury.21,22 These findings suggest that therapies for the inhibition of LOX-1 receptors may be effective in reducing the rate of atherosclerotic and inflammation processes, provided within 24 hours before stenting during the intervention for myocardial infarction. Specific LOX-1 receptor inhibitors are not yet available and urgently necessary. Exposure to statins disrupts LOX-1 receptor cluster distribution in plasma membranes and blocks LOX-1-mediated ox-LDL binding and internalization, suggesting that statins may protect vascular endothelium against the adverse effects of ox-LDL by disruption of LOX-1 receptor function.23
In this paper, we investigated the statin-mediated LOX-1 inhibition mechanism and demonstrate that statins directly bind and inhibit human LOX-1 receptors in a cell-based assay. The molecular mechanism of inhibition has been elucidated in silico by molecular docking analysis and MD simulation. The simulative and experimental results obtained on the LOX-1 recognition domain and on the dimer-monomer transition, in absence and presence of different statins, give relevant insights about the functional role assumed by the CTLD domain and the substrate in the active cell receptor.
Results
Long-term exposure with statins impairs LOX-1 receptor activity
We have recently demonstrated that LOX-1 receptors are distributed within lipid rafts in the plasma membranes and chronic exposure of LOX-1 expressing cells with lovastatin for 24 and 48 hours leads to a marked reduction of LOX-1-mediated ox-LDL binding and uptake. The reduction is not related to variation on the number of exposed receptors, but to the impairment of their function.23 To further study the phenomenon we analyzed whether different statins, relatively lipofilic (atorvastatin, fluvastatin, lovastatin) or water soluble (pravastatin), are similarly effective on LOX-1 intracellular distribution in lipid rafts and on inhibiting LOX-1 function. We used 2 clones of HEK (Human Embryonic Kidney) stably transfected with human LOX-1-V5 (clone 13 or clone 19), expressing high level of human LOX-1 receptors. From these clones, we isolated caveolin-rich domains by the detergent-free procedure and sucrose gradient flotation centrifugation and we analyzed the fractions by Western blot.23,24 Figure 2A shows that about 50% of LOX-1 band is found in fractions 5 (left panel, Ctrl), composed of lipid rafts, as confirmed by the presence of caveolin-1 (Fig. 2A, right panel, Ctrl). The remaining amount of LOX-1 is found in heavier membrane fractions (Fig. 2A, lines 10, 11, 12). When cells are treated with different concentration of lovastatin, atorvastatin, pravastatin and fluvastatin for 3 d, the LOX-1 band intensity in caveolin-enriched membranes (fraction 5) markedly decreases (Fig. 2A). Densitometric quantification of the fold change in LOX-1 band in fraction 5 in control and cells incubated with statins at 2.5 μM concentration is shown in Fig. 2B. It is worth noting that caveolin-1 band intensity is decreased in lovastatin-treated cells (left panel, Fig. 2A) and in cells treated with atorvastatin, fluvastatin and pravastatin (not shown). No cytotoxicity was observed when cells were incubated in the presence of different doses (ranging from 2 to 20 μM) of atorvastatin and pravastatin for at least 5 d, as measured by the sulforhodamine assay (SRB).25 At concentration higher than 5 μM for lovastatin and 3 μM for fluvastatin a moderate toxicity was observed after 3 d of incubation (not shown).
Figure 2.

Detergent-free purification of caveolin-rich membrane fractions by sucrose gradient. (A) HEK-293 stably expressing LOX-1-V5 (clone 19) were treated or not with different statins, as indicated, for 3 d. Lysates were subjected to sucrose gradient centrifugation to isolate caveolin-enriched lipid rafts and immunoblotted with anti-V5 (α LOX-1)(left panel) and anti-caveolin (α Cav-1)(right panel). (B) Densitometric measurements of LOX-1 subunit band in fraction 5 derived from control or treated cells with 2.5 μM drug concentration. The data represent the average ± standard deviation (SD) of 3 separate experiments.
LOX-1-ox-LDL binding was monitored in COS fibroblasts transiently transfected with full length human LOX-1-V5 DNA by using ox-LDL labeled with the highly fluorescent lipophilic dye DiI.23,26 This well-established cell-based binding assay9,13 allows a high transfection efficiency (about 40% of transfected cells), a very strong expression level of LOX-1 receptors and very high ox-LDL binding and uptake. Since COS cells do not express endogenous LOX-1, only transfected cells specifically bind labeled ox-LDL. The binding specificity was confirmed by incubating with 100-fold excess of cold ox-LDL, which completely abolishes the signal of DiI-ox-LDL bound to LOX-1.9 Incubation of cells with different statins (lovastatin, atorvastatin, fluvastatin and pravastatin), at different concentration for 48 hours, leads to a marked reduction of the intensity of the signal of DiI-ox-LDL bound to LOX-1 compared to the control value (Fig. S1, supplementary data). As mentioned, fluvastatin could not be used at concentration higher than 2 μM for its moderate cytotoxicity.
Statins inhibit LOX-1 function by direct interaction
The binding experiments were carried out at 4°C and for a shorter time of incubation (60 min), in order to avoid any cholesterol-lowering effect. We analyzed ox-LDL binding by counting positive DiI-ox-LDL fluorescent cells (red fluorescence) in the absence and in the presence of the indicated statins. Figure 3A shows representative images of untreated cells (Ctrl) and cells treated with Lovastatin, Fluvastatin and Atorvastatin. About 40% of control cells (Fig. 3A, panel Ctrl) are expressing LOX-1 receptors and bind the fluorescently labeled DiI-ox-LDL. When lovastatin, atorvastatin, fluvastatin and pravastatin are present in the binding assay a marked reduction in the number of red fluorescent positive cells is observed. Red fluorescent cells were counted and the percentage of positive cells with respect to all Hoechst-positive nuclei was calculated. Lovastatin induces a reduction of DiI-ox-LDL positive cells of 61 ± 2% vs controls (100%) and atorvastatin, fluvastatin and pravastatin of 47 ± 4%, 45 ± 3% and 33 ± 5% vs controls, respectively. Remarkably these values are very similar to those obtained by incubating transfected cells with DiI-ox-LDL at 37°C for 1 hour to measure the rate of DiI-ox-LDL internalization in the presence of statins (Fig. 3B, uptake).
Figure 3.

Effects of short exposure of statins on LOX-1 mediated ox-LDL binding. (A) COS cells transiently transfected with LOX-1-V5 were incubated with 10 μg/ml DiI-ox-LDL (red fluorescence) for 1 hour at 4°C in the absence (Ctrl) or in the presence of 2 μM lovastatin, fluvastatin or 4 μM atorvastatin. Nuclei are blue stained with Hoechst 33342. Scale bar 20 μm. (B) Histogram shows the percentage of red positive cells with respect of all Hoechst-positive nuclei incubated with 10 μg/ml DiI-ox-LDL without (Ctrl) or with 2 μM lovastatin, atorvastatin, fluvastatin and pravastatin for 1h at 37°C to measure ox-LDL internalization (uptake). (C) Percentage of DiI-ox-LDL red positive cells pre-treated (dashed line) or not (continuous line) with different concentration of lovastatin before the binding assay done in the presence of lovastatin (1h at 4°C). (D) Quantification of DiI-ox-LDL binding by spectrofluorometer. The data represent the mean ± standard deviation calculated from 4 separate experiments. A P value < 0.01 was considered statistically significant.
We asked whether incubation of cells with statins for 24 hours could improve the inhibitory effect obtained with statins present only during the binding assay. Figure 3C shows that, although cholesterol reduction may have impaired lipid rafts integrity and LOX-1 receptors distribution, a longer incubation time only moderately improves the inhibitory effect of lovastatin (difference ≈ 8 ± 4% at 2.5 μM).
Quantification of bound fluorescent DiI-ox-LDL was measured by its extraction from stained cells with isopropanol and spectrofluorometric analysis. As shown in Figure 3D, incubation with 2 μM lovastatin for 60 min at 4°C inhibits the DiI-ox-LDL binding to LOX-1 of 62 ± 8% compared to the control value (100%) (P < 0.01) and 2 μM atorvastatin, pravastatin or fluvastatin reduce fluorescence signal of 45 ± 9%, 46 ± 9% and 41 ± 8% respectively (P < 0.01). A similar rate of inhibition (data not shown) was found when cells were incubated with DiI-ox-LDL at 37°C for 60 minutes, to measure ox-LDL internalization.
Statin mechanism of action: in silico analysis of statins as CTLD ligands
The molecular mechanism of statin-mediated human LOX-1 inhibition has been investigated by docking analysis and MD simulations. From a structural point of view, statins can be broadly divided into 3 parts: (a) an analog of the target enzyme substrate HMG-CoA reductase composed by an hydrophilic branch generally containing one carboxyl and 2 hydroxyl groups (Fig. S2, supplementary data, evidenced by the black line); (b) a complex central hydrophobic ring structure, covalently linked to the substrate analog, involved in the binding to the reductase (Fig. S2, supplementary data, evidenced by the dark gray line), and (c) a variety of side groups on the rings that define the solubility properties of the drugs and therefore many of their pharmacokinetic properties (Fig. S2, supplementary data, evidenced by the light gray lines).27
Docking simulations indicate that statins are able to completely fill the hydrophobic tunnel (Fig. 4) using a common strategy, characterized by small differences, depending on the chemical structure of each drug. In all the docked compounds (Fig. 4) the substrate analog for the HMG-CoA reductase, portion (a), is always protruding toward the solvent and is involved in hydrogen bonds with residues located at the border of the tunnel. Four hydrophobic residues (Ile149, Leu157, Phe158 and Ala194), forming the tunnel, are involved in contacts with the central ring of statin, portion (b), and with the hydrophobic side groups protruding from the ring, portion (c) (Fig. 4). In detail, the aromatic residue Phe158 is often involved in stacking interactions with the drug rings, while Tyr197, due to the side chain hydroxyl, is involved in hydrogen bonds (Fig. 4). This binding mode is facilitated by small cavities adjacent to the hydrophobic tunnel that are filled with portions of the drugs. The range of binding energies (from about −10 to −12 kcal/mol), shown in Table 1, indicate that statins are strong, direct inhibitors of the human LOX-1 and hence of the LOX-1-ox-LDL recognition.
Figure 4.

Schematic view of the interactions between statins and CTLD. The 2D depictions show hydrogen bonds as black dashed lines between the interaction partners on either side. Hydrophobic interactions are illustrated as smooth green contour lines between the respective amino acids and the ligand. Stacking interactions are indicated by green dashed lines between filled green circles. The ligand itself is drawn utilizing the 2D draw engine according to chemical drawing conventions. This image has been produced using the PoseView software.64
Table 1.
Results of the CTLD – statins molecular docking analysis
| Statin | Binding Energy (kcal/mol) |
|---|---|
| Atorvastatin | −11.7 |
| Fluvastatin | −11.4 |
| Lovastatin | −11.6 |
| Pravastatin | −10.2 |
Molecular dynamics simulations and stability of the structures
MD simulation technique has been applied to characterize the structural and dynamical properties of LOX-1 recognition domain in the presence of NECK domain, including or not a statin which fills the tunnel. Among the different statins we have selected atorvastatin since forms the most stable complex in the docking simulation (Table 1). Two different systems have been simulated and compared: the full LOX-1 receptor model (i.e. CTLD, NECK, transmembrane domain) named LOX-1.apo and the full LOX-1 receptor model in complex with atorvastatin, named LOX-1.lig.
The PROCHECK28 and MolProbity29 validation methods have been applied to LOX-1.apo and LOX-1.lig structures at 3 different stages: (I) the initial model, (II) after minimization (before starting simulation) and (III) after first 25 ns of simulation. Comparison of the validation methods confirm that both models are reliable after 25 ns of simulation, the time from which the structural analyses were started (Table S2, supplementary data). Moreover, the secondary structure analysis shows that the CTLDs subunits maintain the secondary structure pattern observed in the LOX-1 CTLD X-ray structure7,8 (Fig. S3 A,B,C, supplementary data), indicating that CTLDs are sampling reliable, natively folded conformations.
The LOX-1.apo main chain RMSD values (Fig. 5A, black line), calculated using the MD starting structure as a reference, indicate growing deviation values in the first 60 ns of simulation (up to 8 Å) and a large increase in the last 40 ns, reaching a value of 18Å. The same parameter evaluated for LOX-1.lig simulation (Fig. 5A, red line) indicates a relatively rapid increase in the first 35 ns of simulation, followed by a slight decrease in the last 65 ns that reaches a plateau at a value of about 10 Å. The RMSDs of only CTLD in LOX-1.apo and in LOX-1.lig, plotted without considering the NECK contributions in the geometrical analysis (Fig. 5B), indicate that CTLD from LOX-1.apo (black line) oscillates in the first 65 ns of trajectory and then stabilize at a value of about 2 Å while the CTLD from the LOX-1.lig simulation (red line) is stable along all the trajectory. The absence of large displacements in the RMSD of only the CTLD (Fig. 5B) indicates that the deviations previously observed in the full receptor (Fig. 5A) are due to the presence of the NECK domain. In fact, in LOX-1.apo and LOX-1.lig the motion is wide and mainly dominated by the CTLD bend due to the NECK junction flexibility represented by the region in proximity of the inter-subunits Cys140 disulfide bridge.
Figure 5.

RMSD analysis. (A) RMSD time dependence for the full LOX-1.apo (black line) and LOX-1.lig models (red line) simulations. (B) RMSD time dependence for the CTLD structures only. The black line indicates the CTLD in LOX-1.apo simulation while the red line shows the CTLD in LOX-1.lig simulation.
Other CTLD motions in the 2 systems can be also appreciated from the histogram, reporting the distance distribution between the monomers mass centers that monitors the subunits association during the trajectories (Fig. 6). In the LOX-1.apo system the distribution is spread over a large range of distances while in LOX-1.lig is peaked in a delimited range, indicating that the statin ligand blocks the fluctuations and the separation of the monomers.
Figure 6.
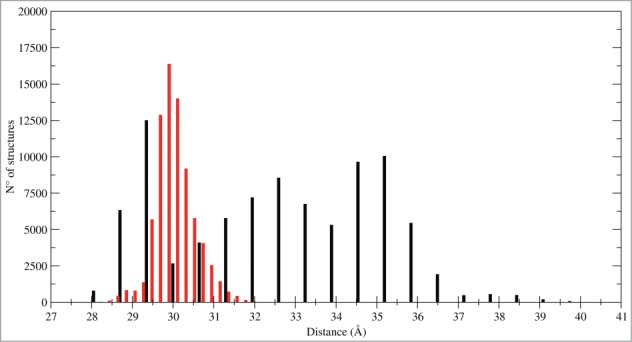
Inter-subunits distances. Histogram of distances between the mass centers of the 2 CTLD monomers: CTLD in LOX-1.apo (black bars) and CTLD in LOX-1.lig (red bars).
Large amplitude motions characterize the LOX-1 dynamical behavior
The principal component analysis (PCA),30,31 applied to highlight the correlation motion on the 268 Cα atoms of CTLD in LOX-1.apo and LOX-1.lig, indicates that, although the motion is dispersed over 828 eigenvectors, about 90% of total motion is included in the first eigenvector (data not shown). This finding is in line with PCA analysis of other systems.32,33 Projections of the motions of CTLD along the first eigenvector of the LOX-1.apo simulation (Fig. 7A) indicate that monomers undergo a large oscillation and move away one from the other generating a wide “clamp motion,” as already shown in Fig. 6. In the CTLD of LOX-1.lig (Fig. 7B), this oscillation is almost absent, the dimer interface is firmly anchored and the principal motion is mainly located in the external loops.
Figure 7.
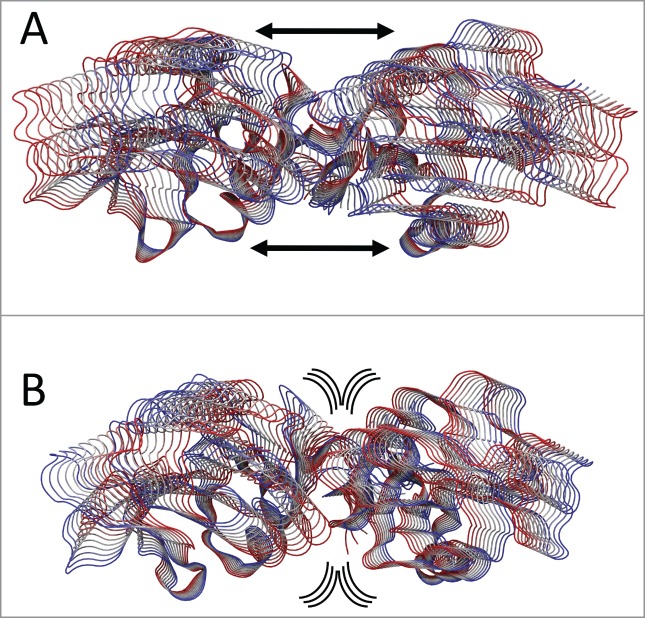
PCA analysis. Tube representation of the motion projections along the first eigenvector for (A) CTLD in LOX-1.apo and (B) CTLD in LOX-1.lig. The direction of the subunits motion is indicated by the black arrows and by the flanked tubes, the vs. being defined from red to blue. Arrows underline the direction of the motion. This picture has been produced using the program VMD.47
Dimers versus monomers ratio in human LOX-1
In order to experimentally verify whether in living cells the filling of the CTLD tunnel is involved in the stabilization of LOX-1 receptor dimer form, cells have been treated with different statins or the natural substrate ox-LDL. The dimer vs. monomer ratio has been evaluated either purifying full-length LOX-1 receptors from cell membranes or collecting the soluble form of LOX-1 from conditioned medium.
In a first set of experiments caveolin-rich domains of HEK-293 expressing LOX-1-V5, treated or not with different statins for 2 d, have been isolated as described in Figure 2 and analyzed in high reducing conditions by probing the blots with anti-V5 antibodies. Since the results using different statins are similar, Figure 8A shows the experiments obtained with 2 μM lovastatin or 1 μM fluvastatin. In non-treated cells (Ctrl) 90% of LOX-1 runs as a monomer (46 kDa) in the presence of 20 mM DTT, while, when cells have been treated with lovastatin or fluvastatin and samples analyzed with the same high reducing conditions, the dimer/monomer ratio reverses.
Figure 8.
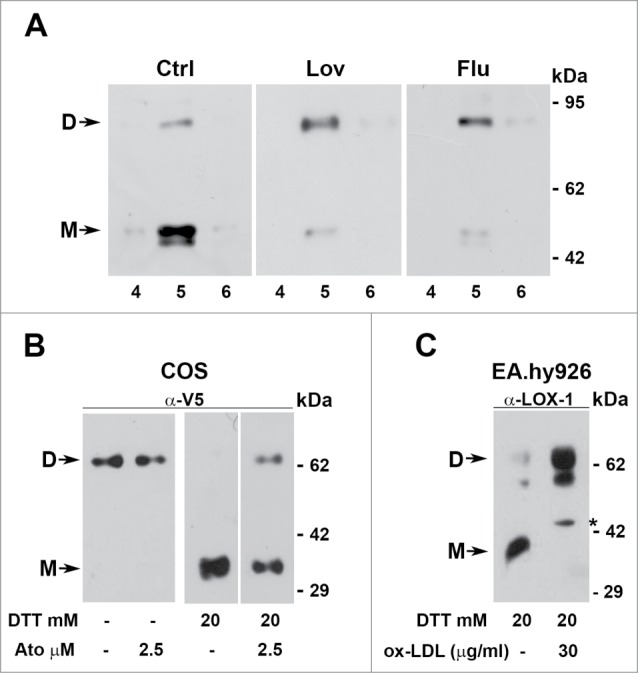
Dimers versus monomers ratio. (A) HEK-293 stably expressing LOX-1-V5 (clone #19) were treated or not with lovastatin and fluvastatin for 2 d. Figure shows the protein gel blot analysis of lipid rafts (fractions 4, 5 and 6) purified by sucrose gradient visualized with Mab anti-V5. (B) Western blot of sLOX-1 released in COS transfected culture medium. Equal volumes of medium derived from cells treated or not with different concentration of atorvastatin for 1 hour was TCA precipitated and analyzed by western blot. Samples were run in the absence or in the presence of 20 mM DTT and immunoreactive bands were visualized with Mab anti-V5. (C) Analysis of conditioned medium derived from human umbilical vein endothelial cells treated or not with ox-LDL. Samples were run in the presence of 20 mM DTT and immunoreactive bands visualized with rabbit anti-LOX-1 antibodies. The asterisk indicates a product of LOX-1 degradation, rarely present. D and M refers to dimers and monomers respectively.
The same behavior is observed analyzing the truncated soluble pool of LOX-1 (sLOX-1).17 When sLOX-1, accumulated in conditioned medium from LOX-1 expressing COS cells, treated or not with statins, is analyzed in non reducing conditions (Fig. 8B, left panel), 100% of the molecules are recovered as dimers. In the presence of 20 mM DTT, able to reduce all disulphide bonds, 100% of sLOX-1 derived from control medium is detected as a monomer, while at least 40% of sLOX-1, which derives from cells treated with statins for 1 hour, runs as a dimer (Fig. 8B, right panel). The figure shows the results obtained incubating cells with atorvastatin, but addition to the cells of other statins, such as lovastatin (2.5 μM), fluvastatin (1 μM) or pravastatin (2.5 μM), gives similar results (not shown).
Dimer versus monomer ratio was also studied in the human umbilical vein endothelial EA.hy926 cell line, which naturally expresses LOX-1 receptors. The truncated soluble LOX-1, released and accumulated in conditioned medium, was analyzed by protein gel blot with polyclonal anti-LOX-1 antibodies, which recognize an epitope in the middle region of the receptor. In cells treated with 30 μg/ml ox-LDL, sLOX-1 is present as a dimer. Monomers are detectable only when cells were grown in the absence of ox-LDL. Notably, sLOX-1 may undergo partial degradation, which results, in some experiments, in the presence of 2 close immunoreactive bands (Fig. 8C).
Discussion
LOX-1 receptors are distributed within lipid rafts in the plasma membranes and chronic exposure of cells to statins leads to a spatial disorganization of LOX-1 and a marked loss of LOX-1 function.23 Here we show, for the first time, that statins, besides their indirect effect on LOX-1 activity derived from lowering intracellular cholesterol, inhibit LOX-1 by direct interaction with the CTLD recognition domain, indicating a previously unrecognized pleiotropic effect of this class of drugs. Acute exposure of cells with different statins (lovastatin, atorvastatin, fluvastatin and pravastatin), at 4°C and for a time length not sufficient to induce any cholesterol synthesis reduction, results in a very marked decrease of ox-LDL bound to LOX-1 (Fig. 3). Direct inhibition is confirmed by molecular docking of statins over the CTLD recognition domain (Fig. 4), indicating that these drugs completely fill the hydrophobic tunnel crossing the entire LOX-1 dimer using a common strategy characterized by small differences (Fig. 4 and Fig. S2, supplementary data). Thus, all tested statins show favorable binding energies, quantifiable by the docking simulations in less than −10.0 kcal/mol (Table 1), independently on the chemical structure and lipophilicity.
The ability of LOX-1 to adapt to various molecules is expected, being LOX-1 a scavenger receptor, participating in the removal of many foreign substances and waste materials by extensive ligand specificity. The hydrophobic tunnel plays a crucial role in the recognition of ox-LDL and other ligands, as also demonstrated by site-directed mutagenesis. Point mutations on Ile149, which points to the empty space in the center of the tunnel, markedly reduces the binding of ox-LDL and a series of oxidized phospholipids.14 Accordingly, we have recently identified a modified oxidized phospholipid, named PLAzPC, with favorable binding energy and high LOX-1 inhibitory activity, that interacts with residues in the tunnel and significantly prevents the binding ability of LOX-1 receptor to ox-LDL.15 Similarly to this compound, our docking simulations confirm that statins completely fill the hydrophobic tunnel using a common mechanism and that residue Ile149 is always involved in stabilizing contacts with these drugs (Fig. 4).
Interestingly, the MD simulations of the entire receptor indicate that the presence of the NECK domain alters the dynamic behavior of CTLD that has a crucial role in the mechanisms of substrate recognition, binding and internalization. The different dynamical coupling of the monomers in the LOX-1.apo and LOX-1.lig simulations allow us to propose that oscillations of LOX-1 receptors, reorientation of CTLDs and the observed “clamp motion” may be involved in the substrate recognition improving the interactions with the substrate. The “clamp motion” mechanism, which we hypothesize as a mechanism for ox-LDL catching, is only observed in the absence of ligands and blocked when the CTLD hydrophobic tunnel is filled (Figs. 5, 6, 7).
A direct interaction of statins with the CTLD recognition domain highlights 2 distinct mechanisms of LOX-1 inhibition: I) by lowering membrane cholesterol and a consequent mislocalization of LOX-1 in plasma membranes23 and II) by a direct interaction with the CTLD recognition domain. When cells are pre-incubated with statins for 24 hours before starting the binding experiment, to allow the cholesterol lowering, only a very moderate improvement (about 8%) of the inhibitory effect was observed (Fig. 2C), indicating that binding to LOX-1 is the primary effect of statins and the most efficient mechanism of inhibition in LOX-1 expressing cells. Thus, although cholesterol reduction may have impaired lipid rafts integrity and LOX-1 receptors distribution, LOX-1 receptors are not active anymore because bound to statins and no free CTLD sites are available. Other alternative indirect mechanisms on membrane physical properties, which may affect also other molecules involved in LOX-1 mediated ox-LDL binding and internalization, cannot however be excluded.
In our experimental conditions, we do not reach a complete inhibitory effect of LOX-1 activity even in the presence of higher concentration of statins. A residual 20–30% of bound fluorescent DiI-ox-LDL is always present. A possible explanation of this finding is that, while the hydrophobic component of the recognition, mainly due to the LOX-1 tunnel, is completely disabled by statins, the basic spine electrostatic attraction to ApoB100 protein located on the ox-LDL surface is still active.7,8 Moreover, in statin-treated cells, the fluorescence intensity does not uniformly decreases. On the contrary, there are cells with high fluorescence intensity and cells without fluorescence (Fig. 3A). Transient transfection in COS cells results in a very high LOX-1 expression variability. We have evidence that only cells with lower expression level are “switched off,” while those with higher LOX-1 level are still able to bind enough DiI-ox-LDL molecules to appear fluorescent. It is important to underline that multimerization and cluster organization of LOX-1 in plasma membrane are important requisites for its activity and largely influence the ox-LDL binding affinity.9,11,12 Clusters develop regardless of the presence of ox-LDL, their formation is LOX-1 concentration-dependent34 and may interfere with the binding of the inhibitors. The in vitro biochemical characterization of ox-LDL/LOX-1 binding with purified or recombinant receptors is, for all these considerations, challenging.
It is worth noting that, during inflammation processes, LOX-1 expression is upregulated in endothelial cells, smooth muscle cells, monocyte/macrophages, advanced atherosclerotic plaque and atherosclerotic aorta from hyperlipidemic animals and humans.18,35 Whether sequestering statin molecules by LOX-1 receptors in these districts results in a measurable decrease in the availability of circulating statins, is an interesting issue that requires additional studies.
A number of clinical investigations strongly support that statins have a marked beneficial activity in preventing myocardial damage after coronary angioplasty. The trial NAPLES II demonstrated that even a single dose (80 mg) of atorvastatin, administered within 24 hours before stenting, is effective.36,37 This benefit is clearly independent of the cholesterol-lowering effect of atorvastatin. In patients with acute myocardial infarction, the ox-LDL plasma level is markedly elevated and LOX-1 receptors are up-regulated and appear to be associated with apoptosis, necrosis and left ventricular functional deterioration. In addition, LOX-1 deficient mice showed decreased infarct size after ischemia/reperfusion.38 Blocking LOX-1 activation with specific anti-LOX-1 therapeutic molecules represents an important achievement for cardio-protection.
Based on our experimental and simulative data we suggest that the beneficial effect of statin loading for acute coronary syndromes, may be due to their inhibitory activity on LOX-1 receptors, opening a new field of investigation about possible different uses of these drugs.
Materials and Methods
DNA constructs
For the expression in mammalian cells, human LOX-1 was subcloned into pEF/V5-His vectors (Invitrogen, Inchinnan, Paisley, UK), as previously described.9
Antibodies and reagents
Mab α-V5 (Invitrogen), mouse anti-caveolin 1 (Santa Cruz Biotechnology), rabbit anti-LOX-1 (Aviva Systems Biology) were used as primary antibodies and goat anti-mouse IgG HRP and Rhodamine Red X-conjugated AffiniPure donkey anti-mouse IgG (Jackson Immunoresearch Laboratories Inc., West Grove, PA) as secondary. Lovastatin (Alexis Biochemicals, San Diego, CA) was activated by NaOH addiction with a lovastatin/NaOH ratio (v/v) of 2:3, at 50°C and neutralized with HCl to pH 7. Atorvastatin, fluvastatin, pravastatin and Hoechst 33342 were purchased from Sigma-Aldrich, St.Louis, MO.
Cell cultures and transfection
COS, HEK-293 and human umbilical vein endothelial EA.hy92639 cells were grown in DMEM (Dulbecco's modified Eagle's medium) (Biowest, Miami, FL, USA) supplemented with 10% fetal bovine serum (Gibco, Inchinnan, Paisley, UK) and 100U/ml penicillin/streptomycin (Euroclone, Devon, UK). COS cells were transiently transfected with JetPEI (Polyplus Transfection, Illkirch, France).
Purification of caveolae-enriched membrane fractions and detection of soluble LOX-1
Caveolae-enriched membrane fractions were prepared by a detergent-free purification.23,24 Protein concentration was measured in each fraction by Bradford assay (Sigma-Aldrich, St.Louis, MO). Proteins were precipitated with 10% trichloroacetic acid (TCA) and solubilized in SDS-PAGE sample buffer, run in 10% acrylamide gels and transferred to polyvinylidene difluoride (PVDF) membranes (GE Healthcare, Chalfont St. Giles, UK). Immunoreactive bands were visualized by enhanced chemiluminescence (ECL, Sigma-Aldrich, St.Louis, MO). For detection of sLOX-1 in the medium, conditioned media from transiently transfected COS cells cultured in Opti-MEM (GIBCO) were centrifuged at 10.000xg, precipitated with TCA and solubilized in SDS-PAGE sample buffer for western blot analysis as described.17
Immunofluorescence analysis
Cell membrane immunofluorescence was carried out as described.40 Samples were examined with a DMRA Leica fluorescence microscope, equipped with CCD camera.
Ox-LDL preparation, labeling and binding assays
Human LDL was prepared from fresh healthy normolipidemic plasma of volunteers by ultracentrifugation41 and in vitro oxidized as described.23 Ox-LDL was labeled with 1,1′-dioctadecyl-3,3,3′,3′-tetramethyllindocarbocyanine perchlorate (DiI, Invitrogen, Inchinnan, Paisley, UK).9,26 Binding assays were performed as previously described.17 COS cells were plated in 48 multiwells at 70% confluence, transfected with LOX-1-V5 plasmid and, 24 hours after transfection, used for binding assay. Cells were incubated 30 min at 4°C with statins diluted in DMEM with 10% FCS. Cells were then incubated with 10μg/ml (DiI)-labeled ox-LDL in the presence of statins on ice for 1 hour. Quantitation of bound Dil-ox-LDL was assayed by DiI extraction in isopropanol26 and fluorescence determined in a Perkin Elmer spectrofluorometer with excitation and emission wavelengths set at 520 and 578 nm, respectively. Dil-ox-LDL binding was also performed in transfected COS cells plated in glass coverslips. After 1 hour incubation with (DiI)-labeled ox-LDL, with or without statins, cells were washed twice in PBS, fixed 10 min in paraformaldehyde, incubated 5 min with NaBrH and 5 min with Hoechst 33342 0.3 μg/ml before mounting. Dil-ox-LDL bound cells were counted and the percentage of positive cells with respect of all Hoechst-positive nuclei was calculated. The results shown in the figures of this paper are the average from 3 different experiments for each statin used. For statistical analysis, at least 150 cells were counted and checked for red fluorescence Dil-ox-LDL positivity for each coverslip.
Statistical data analysis
All data were inserted into an Excel database (Microsoft, Redmond, Washington, USA) and analyzed with the Statistical Package for the Social Sciences Windows version 15.0 (SPSS, Chicago, Illinois, USA). Descriptive statistics used for continuous variables was the average ± standard deviation. Normality assumptions were demonstrated with histograms and the Kolgomorov-Smirnov test. Comparison among groups was performed using one-way ANOVA with multiple comparisons by Bonferroni test. A p value < 0.05 was considered statistically significant.
Molecular docking procedure
The protein-ligand docking has been executed with the AutoDock Vina 1.1.2 program,42 using the AutoDock/Vina PyMOL plugin.43,44 The statins SDF files, downloaded from the PubChem compound database, have been converted into PDB files and filled with hydrogens using the Open Babel program.45 The program SYBYL 6.0 (Tripos Inc. 1699, South Hanley Road St. Louis, Mo, USA) has been used to minimize the structures of the compounds. The docking runs have been carried out using as a receptor the high resolution X-ray structure of the wild type human LOX-1 receptor CTLD deposited in the Protein Data Bank (PDB, http://www.rcsb.org/pdb), with PDB code 1YPQ.7 The dioxane molecule, that in the X-ray structure obstructs the hydrophobic tunnel, has been removed to permit a full interaction of the compounds within the tunnel.7,10 The docking simulation has been performed using the genetic algorithm with local gradient optimization.42 For each docking simulation a box of 65 × 65 × 65 (grid points in X, Y, Z axes, corresponding to a 24.375 Å3 grid) has been centered in the CTLD tunnel empty cavity. The residues side chains within 9.0 Å from the LOX-1 tunnel center have been considered flexible in the calculations to increase the search space of solutions. The affinity of the docked compounds has been expressed in terms of binding energy (kcal/mol). Each docking simulation run takes about 8 hours (elapsed real time) on a dedicated Intel i7 – 950 – 3.07 GHz CPU workstation.
LOX-1 dimer model
The LOX-1 model, used to simulate the almost entire receptor (i.e., without the cytoplasmic domain) (Fig. 1), has been assembled as follows. The high resolution X-ray structure of the wild type human LOX-1 receptor CTLD deposited in the Protein Data Bank (PDB, http://www.rcsb.org/pdb), with PDB code 1YPQ,7 was chosen to model the recognition domain. In the 1YPQ CTLD structure the terminal ends, not uniformly determined by X-ray diffraction, have been regularized adding 3 residues (Arg271, Ala272, Gln273) extracted from the 1YPO structure,7 to the C-terminus of both 1YPQ subunits and 4 residues (Arg136, Val137, Ala138 and Asn139) at the N-terminus of subunit A. The dioxane molecule, bound within the tunnel7, has been removed.10 The inter-subunits disulfide bridge, occurring between the Cys140 of the 2 subunits, has been restored. The NECK domain model, obtained as previously described,17 has been connected to the CTLD dimer and 2 hydrophobic α-helices, representing the transmembrane domain (residues from Trp35 to Leu60), that were built using the Chimera program46 and joined to the NECK domain.
The LOX-1 cytoplasmic domain, for which no template is known, was not included in the model to reduce the MD system size, hypothesizing that its presence, in the approximation of the simulation and with the membrane constraint, should not influence the motions of the rest of the protein. The double layer phospholipid membrane was built using the VMD program.47 The phospholipid POPE (1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine) has been used to model the bilayer, because it well reproduces the experimental behavior of a phospholipid membrane.48 Phospholipids within a distance lower than 3.0 Å from the protein have been removed from the system.
Molecular Dynamics simulations
The protein topologies and the coordinates files of the entire LOX-1 models (Fig. 1), used as input for the MD program NAMD 2.7,49 have been obtained using VMD.47 The protein structure has been modeled using the CHARMM22/CMAP force field50,51 and the membrane using the CHARMM36 force field48 (Table S1, supplementary data). The full receptor has been simulated also in the presence of the atorvastatin ligand that, as indicated by the docking analysis, fits with the lowest binding energy in the LOX-1 tunnel (Table 1). The protein structure has been immersed in a parallelepiped water box, using TIP3P water molecules,52 neutralizing the protein charges with Na+ counterions (Table S1, supplementary data), placed in electrostatically preferred positions, imposing a minimal distance between the solute and the box walls of 15.0 Å. Before running the simulation, the water has been minimized for 2000 steps, setting restraints of 500 kcal/mol on protein atoms, then a second minimization run of 2000 steps, without restraints, has been carried out. The minimized structure has been thermalized at 0 K for 50 ps, using the NVT ensemble, with a time-step of 1.0 fs, setting restraint of 500 kcal/mol on the backbone only. The temperature has been increased of 50 K, every 400 steps, until a temperature of 300 K has been reached. At this point an additional 200 ps MD run, with a 1.0 fs time step, has been carried out. The optimized and relaxed system has been simulated for a total of 100 ns using the NPT ensemble with a 2.0 fs time-step. Simulation has been carried out using periodic boundary conditions, with a cut-off radius of 12.0 Å for the non-bonded interactions, which has been smoothed after 8.0 Å. The neighbor pair lists have been updated every 10 steps with an inclusion distance of 13.5 Å. The electrostatic interactions have been calculated every 2 steps (4.0 fs) using the particle-mesh Ewald method,53 with 1.0 Å grid spacing. The simulation has been carried out at a constant temperature of 300 K, using Langevin dynamics parameters,54 with a damping coefficient of 5/ps, as coupling coefficient to be applied to all atoms. For the pressure control the Langevin piston Nose-Hoover method, a combination of the Nose-Hoover constant pressure method55 with piston fluctuation control implemented using Langevin dynamics,56 has been used specifying the desired pressure at 1.01325 bar (1 atm), the oscillation period at 100 fs, the decay times of the piston with the barostat damping time scale at 50 fs and the temperature of the piston at 300 K. The SHAKE algorithm57 was applied to restrain the protein hydrogen atoms and the SETTLE algorithm58 was applied to restrain the water atoms positions. Atomic coordinates were saved every 250 steps (0.5 ps) and the first 25 ns were not used for the analysis. The atorvastatin structure has been optimized using Molden 5.159 and the resulting structure has been used as input for the SwissParam server (www.swissparam.ch),60 to obtain the CHARMM compatible force field parameters of the molecule.
Calculators
Both the systems have been simulated using 72 cores on the MATRIX cluster at CASPUR calculation center composed by 320 nodes, each with 2 AMD Opteron quadcore CPU, for a total of 2560 cores.
MD simulations analyses
The root mean square deviation (RMSD), principal component analysis (PCA)30,31 and time evolution of the secondary structures,61 have been carried out using the GROMACS MD package version 4.6.5 program,62,63 while the distance distribution between the monomers mass centers has been evaluated using the VMD program.47 The elements of the correlation analysis (Cij) were computed as:
where Δri is the displacement from the mean position of the i-th atom and the 〈 〉 represents the time average over the whole trajectory. Positive Cij values represent a correlated motion between residues i and j (i.e. the residues move in the same direction). Negative values of Cij represent an anti-correlated motion between residues i and j (i.e., they move in opposite directions).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to Giuseppe Novelli for helpful discussions, Roberto Sorge for assistance in statistical analysis and Graziano Bonelli for his expert graphical support. EA.hy926 cell line was a generous gift from Dr. Giovanni Fasciglione.
Funding
We thank FILAS for the fellowship granted to F.O. and G.V. and TECNO.TIB.E.R.I.S. under the project “MOLBLOX.”
SUPPLEMENTAL MATERIAL
Supplemental data for this article can be accessed on the publisher's website.
Author Contributions
Silvia Biocca and Mattia Falconi have drawn the research design; Sara Matarazzo, Giulia Vindigni and Silvia Biocca conducted the experiments; Federico Iacovelli, Francesco Oteri and Mattia Falconi performed the simulations; Silvia Biocca, Sara Matarazzo, Giulia Vindigni, Alessandro Desideri, Federico Iacovelli and Mattia Falconi performed data analysis; Silvia Biocca, Alessandro Desideri and Mattia Falconi wrote the manuscript
References
- 1.Istvan ES, Deisenhofer J. Structural mechanism for statin inhibition of HMG-CoA reductase. Science 2001; 292:1160–4; PMID:11349148; http://dx.doi.org/ 10.1126/science.1059344 [DOI] [PubMed] [Google Scholar]
- 2.Wang CY, Liu PY, Liao JK. Pleiotropic effects of statin therapy: molecular mechanisms and clinical results. Trends Mol Med 2008; 14:37–44; PMID:18068482; http://dx.doi.org/ 10.1016/j.molmed.2007.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Javanmoghadam-Kamrani S, Keyomarsi K. Synchronization of the cell cycle using lovastatin. Cell Cycle 2008; 7:2434–40; PMID:18677105; http://dx.doi.org/ 10.4161/cc.6364 [DOI] [PubMed] [Google Scholar]
- 4.Corsini A, Ferri N, Cortellaro M. Are pleiotropic effects of statins real? Vasc. Health Risk Manag 2007; 3:611–3; PMID:18078012 [PMC free article] [PubMed] [Google Scholar]
- 5.Patti G, Pasceri V, Colonna G, Miglionico M, Fischetti D, Sardella G, Montinaro A, Di Sciascio G. Atorvastatin pretreatment improves outcomes in patients with acute coronary syndromes undergoing early percutaneous coronary intervention: results of the ARMYDA-ACS randomized trial. J Am Coll Cardiol 2007; 49:1272–8; PMID:17394957; http://dx.doi.org/ 10.1016/j.jacc.2007.02.025 [DOI] [PubMed] [Google Scholar]
- 6.Sawamura T, Kume N, Aoyama T, Moriwaki H, Hoshikawa H, Aiba Y, Tanaka T, Miwa S, Katsura Y, Kita T, et al.. An endothelial receptor for oxidized low-density lipoprotein. Nature 1997; 386:73–7; PMID:9052782; http://dx.doi.org/ 10.1038/386073a0 [DOI] [PubMed] [Google Scholar]
- 7.Ohki I, Ishigaki T, Oyama T, Matsunaga S, Xie Q, Ohnishi-Kameyama M, Murata T, Tsuchiya D, Machida S, Morikawa K, et al.. Crystal structure of human lectin-like, oxidized low-density lipoprotein receptor 1 ligand binding domain and its ligand recognition mode to OxLDL. Structure 2005; 13:905–17; PMID:15939022; http://dx.doi.org/ 10.1016/j.str.2005.03.016 [DOI] [PubMed] [Google Scholar]
- 8.Park H, Adsit FG, Boyington JC. The 1.4 angstrom crystal structure of the human oxidized low density lipoprotein receptor lox-1. J Biol Chem 2005; 280:13593–9; PMID:15695803; http://dx.doi.org/ 10.1074/jbc.M500768200 [DOI] [PubMed] [Google Scholar]
- 9.Biocca S, Filesi I, Mango R, Maggiore L, Baldini F, Vecchione L, Viola A, Citro G, Federici G, Romeo F, et al.. The splice variant LOXIN inhibits LOX-1 receptor function through hetero-oligomerization. J Mol Cell Cardiol 2008; 44:561–70; PMID:18191942; http://dx.doi.org/ 10.1016/j.yjmcc.2007.11.017 [DOI] [PubMed] [Google Scholar]
- 10.Falconi M, Biocca S, Novelli G, Desideri A. Molecular dynamics simulation of human LOX-1 provides an explanation for the lack of OxLDL binding to the Trp150Ala mutant. BMC Struct Biol 2007; 7:73; PMID:17988382; http://dx.doi.org/ 10.1186/1472-6807-7-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cao W, Calabro V, Root A, Yan G, Lam K, Olland S, Sanford J, Robak A, Zollner R, Lu Z, et al.. Oligomerization is required for the activity of recombinant soluble LOX-1. FEBS J 2009; 276:4909–20; PMID:19664054; http://dx.doi.org/ 10.1111/j.1742-4658.2009.07190.x [DOI] [PubMed] [Google Scholar]
- 12.Ohki I, Amida H, Yamada R, Sugihara M, Ishigaki T, Tate S. Surface plasmon resonance study on functional significance of clustered organization of lectin-like oxidized LDL receptor (LOX-1). Biochim Biophys Acta 2011; 1814:345–54; PMID:21035571; http://dx.doi.org/ 10.1016/j.bbapap.2010.10.006 [DOI] [PubMed] [Google Scholar]
- 13.Biocca S, Falconi M, Filesi I, Baldini F, Vecchione L, Mango R, Romeo F, Federici G, Desideri A, Novelli G. Functional analysis and molecular dynamics simulation of LOX-1 K167N polymorphism reveal alteration of receptor activity. PLoS One 2009; 4:e4648; PMID:19247493; http://dx.doi.org/ 10.1371/journal.pone.0004648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Francone OL, Tu M, Royer LJ, Zhu J, Stevens K, Oleynek JJ, Lin Z, Shelley L, Sand T, Luo Y, et al.. The hydrophobic tunnel present in LOX-1 is essential for oxidized LDL recognition and binding. J Lipid Res 2009; 50:546–55; PMID:18845619; http://dx.doi.org/ 10.1194/jlr.M800474-JLR200 [DOI] [PubMed] [Google Scholar]
- 15.Falconi M, Ciccone S, D'Arrigo P, Viani F, Sorge R, Novelli G, Patrizi P, Desideri A, Biocca S. Design of a novel LOX-1 receptor antagonist mimicking the natural substrate. Biochem Biophys Res Commun 2013; 438:340–5; PMID:23892036; http://dx.doi.org/ 10.1016/j.bbrc.2013.07.073 [DOI] [PubMed] [Google Scholar]
- 16.Ishigaki T, Ohki I, Utsunomiya-Tate N, Tate S. Chimeric structural stabilities in the coiled–coil structure of the NECK domain in human lectin-like oxidized low-density lipoprotein receptor 1 (LOX-1). J Biochem 2007; 141:855–66; PMID:17416594; http://dx.doi.org/ 10.1093/jb/mvm093 [DOI] [PubMed] [Google Scholar]
- 17.Biocca S, Arcangeli T, Tagliaferri E, Testa B, Vindigni G, Oteri F, Giorgi A, Iacovelli F, Novelli G, Desideri A, et al.. Simulative and experimental investigation on the cleavage site that generates the soluble human LOX-1. Arch Biochem Biophys 2013; 540:9–18; PMID:24113299; http://dx.doi.org/ 10.1016/j.abb.2013.10.001 [DOI] [PubMed] [Google Scholar]
- 18.Mehta JL, Chen J, Hermonat PL, Romeo F, Novelli G. Lectin-like, oxidized low-density lipoprotein receptor-1 LOX-1: a critical player in the development of atherosclerosis and related disorders. Cardiovasc Res 2006; 69:36–45; PMID:16324688; http://dx.doi.org/ 10.1016/j.cardiores.2005.09.006 [DOI] [PubMed] [Google Scholar]
- 19.Vohra RS, Murphy JE, Walker JH, Ponnambalam S, Homer-Vanniasinkam S. Atherosclerosis and the Lectin-like Oxidized low-density lipoprotein scavenger receptor. Trends Cardiovasc Med 2006; 16:60–4; PMID:16473764; http://dx.doi.org/ 10.1016/j.tcm.2005.12.001 [DOI] [PubMed] [Google Scholar]
- 20.Li D, Williams V, Liu L, Chen H, Sawamura T, Romeo F, Mehta JL. Expression of lectin-like oxidized low-density lipoprotein receptors during ischemia-reperfusion and its role in determination of apoptosis and left ventricular dysfunction. J Am Coll Cardiol 2003; 41:1048–55; PMID:12651056; http://dx.doi.org/ 10.1016/S0735-1097(02)02966-2 [DOI] [PubMed] [Google Scholar]
- 21.Hu C, Chen J, Dandapat A, Fujita Y, Inoue N, Kawase Y, Jishage K, Suzuki H, Li D, Hermonat PL, et al.. LOX-1 abrogation reduces myocardial ischemia-reperfusion injury in mice. J Mol Cell Cardiol 2008; 44:76–83; PMID:18022184; http://dx.doi.org/ 10.1016/j.yjmcc.2007.10.009 [DOI] [PubMed] [Google Scholar]
- 22.Mehta JL, Sanada N, Hu CP, Chen J, Dandapat A, Sugawara F, Satoh H, Inoue K, Kawase Y, Jishage K, et al.. Deletion of LOX-1 reduces atherogenesis in LDLR knockout mice fed high cholesterol diet. Circ Res 2007; 100:1634–42; PMID:17478727; http://dx.doi.org/ 10.1161/CIRCRESAHA.107.149724 [DOI] [PubMed] [Google Scholar]
- 23.Matarazzo S, Quitadamo MC, Mango R, Ciccone S, Novelli G, Biocca S. Cholesterol-lowering drugs inhibit lectin-like oxidized low-density lipoprotein-1 receptor function by membrane raft disruption. Mol Pharmacol 2012; 82:246–54; PMID:22570368; http://dx.doi.org/ 10.1124/mol.112.078915 [DOI] [PubMed] [Google Scholar]
- 24.Song KS, Li S, Okamoto T, Quilliam LA, Sargiacomo M, Lisanti MP. Co-purification and direct interaction of Ras with caveolin, an integral membrane protein of caveolae microdomains. Detergent-free purification of caveolae microdomains. J Biol Chem 1996; 271:9690–7; PMID:8621645; http://dx.doi.org/ 10.1074/jbc.271.16.9690 [DOI] [PubMed] [Google Scholar]
- 25.Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 1990; 82:1107–12; PMID:2359136; http://dx.doi.org/ 10.1093/jnci/82.13.1107 [DOI] [PubMed] [Google Scholar]
- 26.Stephan ZF, Yurachek EC. Rapid fluorometric assay of LDL receptor activity by DiI-labeled LDL. J Lipid Res 1993; 34:325–30; PMID:8381454 [PubMed] [Google Scholar]
- 27.Gaw A, Packard CJ. Comparative chemistry, pharmacology and mechanism of action of the statins Gaw A, Packard CJ, Shepherd J Eds, Statins. The HMG CoA reductase inhibitors in perspective. London: Taylor & Francis; 2000; 49–61. [Google Scholar]
- 28.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst 1993; 26:283–91. [Google Scholar]
- 29.Chen VB, Arendall WB III, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallographica D 2010; 66:12–21; PMID:20057044; http://dx.doi.org/ 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garcia AE. Large-amplitude nonlinear motions in proteins. Phys Rev Lett 1992; 68:2696–9; PMID:10045464; http://dx.doi.org/ 10.1103/PhysRevLett.68.2696 [DOI] [PubMed] [Google Scholar]
- 31.Amadei A, Linssen AB, Berendsen HJC. Essential dynamics of proteins. Proteins 1993; 17: :412–25; PMID:8108382; http://dx.doi.org/ 10.1002/prot.340170408 [DOI] [PubMed] [Google Scholar]
- 32.Chillemi G, Davidovich P, D'Abramo M, Mametnabiev T, Garabadzhiu AV, Desideri A, Melino G. Molecular dynamics of the full-length p53 monomer. Cell Cycle 2013; 12:3098–108; PMID:23974096; http://dx.doi.org/ 10.4161/cc.26162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Falconi M, Oteri F, Di Palma F, Pandey S, Battistoni A, Desideri A. Structural-dynamical investigation of the ZnuA histidine-rich loop: involvement in zinc management and transport. J Comput Aided Mol Des 2011; 25:181–94; PMID:21240623; http://dx.doi.org/ 10.1007/s10822-010-9409-6 [DOI] [PubMed] [Google Scholar]
- 34.Matsunaga S, Xie Q, Kumano M, Niimi S, Sekizawa K, Sakakibara Y, Komba S, Machida S. Lectin-like oxidized low-density lipoprotein receptor (LOX-1) functions as an oligomer and oligomerization is dependent on receptor density. Exp Cell Res 2007; 313:1203–14; PMID:17306253; http://dx.doi.org/ 10.1016/j.yexcr.2007.01.007 [DOI] [PubMed] [Google Scholar]
- 35.Kataoka H, Kume N, Miyamoto S, Minami M, Moriwaki H, Murase T, Sawamura T, Masaki T, Hashimoto N, Kita T. Expression of lectinlike oxidized low-density lipoprotein receptor-1 in human atherosclerotic lesions. Circulation 1999; 99:3110–7; PMID:10377073; http://dx.doi.org/ 10.1161/01.CIR.99.24.3110 [DOI] [PubMed] [Google Scholar]
- 36.Briguori C, Visconti G, Focaccio A, Golia B, Chieffo A, Castelli A, Mussardo M, Montorfano M, Ricciardelli B, Colombo A. Novel approaches for preventing or limiting events (Naples) II trial: impact of a single high loading dose of atorvastatin on periprocedural myocardial infarction. J Am Coll Cardiol 2009; 54:2157–63; PMID:19664895; http://dx.doi.org/ 10.1016/j.jacc.2009.07.005 [DOI] [PubMed] [Google Scholar]
- 37.Nusca A, Melfi R, Patti G, Di Sciascio G. Statin loading for acute coronary syndromes. Curr Opin Cardiol 2010; 25:373–8; PMID:20520540; http://dx.doi.org/ 10.1097/HCO.0b013e32833987ca [DOI] [PubMed] [Google Scholar]
- 38.Hu C, Dandapat A, Chen J, Fujita Y, Inoue N, Kawase Y, Jishage K, Suzuki H, Sawamura T, Mehta JL. LOX-1 deletion alters signals of myocardial remodelling immediately after ischemia-reperfusion. Cardiovasc Res 2007; 76:292–02; PMID:17707356; http://dx.doi.org/ 10.1016/j.cardiores.2007.07.003 [DOI] [PubMed] [Google Scholar]
- 39.Edgell CJ, McDonald CC, Graham JB. Permanent cell line expressing human factor VIII-related antigen established by hybridization. Proc Natl Acad Sci USA 1983; 80:3734–7; PMID:6407019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cardinale A, Filesi I, Vetrugno V, Pocchiari M, Sy MS, Biocca S. Trapping prion protein in the endoplasmic reticulum impairs PrPC maturation and prevents PrPSc accumulation. J Biol Chem 2005; 280:685–94; PMID:15513919; http://dx.doi.org/ 10.1074/jbc.M407360200 [DOI] [PubMed] [Google Scholar]
- 41.Sattler W, Bone P, Stocker R. Isolation of human VLDL, LDL, HDL and two subclasses in the TL-100 tabletop centrifuge using TLA-100.4 rotor. Fullerton, CA; Taylor & Francis; 1992. [Google Scholar]
- 42.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 2010; 31:455–61; PMID:19499576; http://dx.doi.org/ 10.1002/jcc.21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.The PyMOL Molecular Graphics System Version 1.5.0.4 Schrödinger, LLC. [Google Scholar]
- 44.Seeliger D, de Groot BL. Protein thermostability calculations using alchemical free energy simulations. Biophys J 2010; 98:2309–16; PMID:20483340; http://dx.doi.org/ 10.1016/j.bpj.2010.01.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O'Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR. Open Babel: an open chemical toolbox. J Cheminform 2011; 3:33; PMID:21982300; http://dx.doi.org/ 10.1186/1758-2946-3-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera – a visualization system for exploratory research and analysis. J Comput Chem 2004; 25:1605–12; PMID:15264254; http://dx.doi.org/ 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 47.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph 1996; 14:33–8; PMID:8744570 [DOI] [PubMed] [Google Scholar]
- 48.Klauda JB, Venable RM, Freites JA, O'Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD Jr, Pastor RW. Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J Phys Chem B 2010; 114:7830–43; PMID:20496934; http://dx.doi.org/ 10.1021/jp101759q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem 2005; 26:1781–802; PMID:16222654; http://dx.doi.org/ 10.1002/jcc.20289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, et al.. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B 1998; 102:3586–616; PMID:24889800; http://dx.doi.org/ 10.1021/jp973084f [DOI] [PubMed] [Google Scholar]
- 51.Mackerell AD Jr, Feig M, Brooks CL 3rd. Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem 2004; 25:1400–15; PMID:15185334; http://dx.doi.org/ 10.1002/jcc.20065 [DOI] [PubMed] [Google Scholar]
- 52.Jorgensen WL, Mahoney MW. A five-site model for liquid water and the reproduction of the density anomaly by rigid, non polarizable potential functions. J Chem Phys 2000; 112:8910–22; http://dx.doi.org/ 10.1063/1.481505 [DOI] [Google Scholar]
- 53.Darden T, York D, Pedersen L. Particle mesh Ewald – An N.log(N) method for Ewald sums in large systems. J Chem Phys 1993; 98:10089–92; http://dx.doi.org/ 10.1063/1.464397 [DOI] [Google Scholar]
- 54.Brünger A, Brooks CL, Karplus M. Stochastic boundary conditions for molecular dynamics simulations of st2 water. Chem Phys Lett 1984; 105:495–500; http://dx.doi.org/ 10.1016/0009-2614(84)80098-6 [DOI] [Google Scholar]
- 55.Martyna GJ, Tobias DJ, Klein ML. Constant pressure molecular dynamics algorithms. J Chem Phys 1994; 101:4177–89; http://dx.doi.org/ 10.1063/1.467468 [DOI] [Google Scholar]
- 56.Feller SE, Zhang Y, Pastor RW, Brooks BR. Constant pressure molecular dynamics simulation: the Langevin piston method. J Chem Phys 1995; 103:4613–21; http://dx.doi.org/ 10.1063/1.470648 [DOI] [Google Scholar]
- 57.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys 1977; 23:327–41; http://dx.doi.org/ 10.1016/0021-9991(77)90098-5 [DOI] [Google Scholar]
- 58.Miyamoto S, Kollman PA. Settle: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J Comp Chem 2004; 13:952–62; http://dx.doi.org/ 10.1002/jcc.540130805 [DOI] [Google Scholar]
- 59.Schaftenaar G, Noordik JH. Molden: a pre- and post-processing program for molecular and electronic structures. J Comput Aided Mol Des 2000; 14:123–34; PMID:10721501; 10.1023/A: http://dx.doi.org/1008193805436 [DOI] [PubMed] [Google Scholar]
- 60.Zoete V, Cuendet MA, Grosdidier A, Michielin O. SwissParam: a fast force field generation tool for small organic molecules. J Comput Chem 2011; 32:2359–68; PMID:21541964; http://dx.doi.org/ 10.1002/jcc.21816 [DOI] [PubMed] [Google Scholar]
- 61.Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983; 22:2577–637; PMID:6667333 [DOI] [PubMed] [Google Scholar]
- 62.Berendsen HJC, van der Spoel D, van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Comp Phys Commun 1995; 95:43–56; http://dx.doi.org/ 10.1016/0010-4655(95)00042-E [DOI] [Google Scholar]
- 63.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput 2008; 4:435–47; http://dx.doi.org/ 10.1021/ct700301q [DOI] [PubMed] [Google Scholar]
- 64.Stierand K, Maass PC, Rarey M. Molecular complexes at a glance: automated generation of two-dimensional complex diagrams. Bioinformatics 2006; 22:1710–6; PMID:16632493; http://dx.doi.org/ 10.1093/bioinformatics/btl150 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.