

Abstract
NFκB signaling plays a significant role in human disease, including breast and ovarian carcinoma, insulin resistance, embryonic lethality and liver degeneration, rheumatoid arthritis, aging and Multiple Myeloma (MM). Inhibitor of κB (IκB) kinase β (IKKβ) regulates canonical Nuclear Factor κB (NFκB) signaling in response to inflammation and cellular stresses. NFκB activation requires Lys63-linked (K63-linked) ubiquitination of upstream proteins such as NEMO or TAK1, forming molecular complexes with membrane-bound receptors. We demonstrate that IKKβ itself undergoes K63-linked ubiquitination. Mutations in IKKβ at Lys171, identified in Multiple Myeloma and other cancers, lead to a dramatic increase in kinase activation and K63-linked ubiquitination. These mutations also result in persistent activation of STAT3 signaling. Liquid chromatography (LC)-high mass accuracy tandem mass spectrometry (MS/MS) analysis identified Lys147, Lys418, Lys555 and Lys703 as predominant ubiquitination sites in IKKβ. Specific inhibition of the UBC13-UEV1A complex responsible for K63-linked ubiquitination establishes Lys147 as the predominant site of K63-ubiquitin conjugation and responsible for STAT3 activation. Thus, IKKβ activation leads to ubiquitination within the kinase domain and assemblage of a K63-ubiquitin conjugated signaling platform. These results are discussed with respect to the importance of upregulated NFκB signaling known to occur frequently in MM and other cancers.
Keywords: cell proliferation, inflammatory signaling, IKKβ, K-63 ubiquitination, mass spectrometry, multiple myeloma, NFκB, oncogenesis, polyubiquitination, STAT3
NFκB signaling has been implicated in breast and ovarian carcinoma,1,2 insulin resistance,3,4 embryonic lethality and liver degeneration,5 rheumatoid arthritis,6,7 aging,8 and Multiple Myeloma.9,10 The initiation of the inflammatory response is dependent upon the ubiquitination cascade of proteins upstream of the Inhibitor of κB (IκB) kinase β (IKKβ), which plays a direct role in phosphorylating and targeting IκB proteins for degradation, releasing NFκB for nuclear translocation. Upon stimulation of cells with TNFα and LPS, K63-linked polyubiquitin modifications occur in proteins such as TRAFs (tumor necrosis factor receptor-associated factors), RIP1 (receptor interacting protein-1), IRAKs (IL-1 receptor-associated kinases), and TAK1 (TGFβ-activated kinase 1).11-14 Moreover, the NFκB Essential Modulator (NEMO, or IKKγ) is ubiquitinated recruiting TAB1/2 and TAK1 proteins assembled on this platform, which leads to activation of the IKK complex, composed of IKKα, IKKβ, and NEMO.15,16
Ubiquitin, a highly conserved 76-amino acid protein, is post-translationally conjugated to a wide variety of substrates. Ubiquitination is essential in myriad biological functions, including DNA damage repair, cell growth, apoptosis and immune responses.17-19 Ubiquitin has 7 lysine residues that can be conjugated to the C-terminal Gly of a subsequent ubiquitin to form polyubiquitin chains with specific linkages and topologies. The most studied polyubiquitinations are Lys48-linked (K48-linked), which serves as a signal for proteasome-mediated degradation of substrates, and K63-linked, which serves as a stabilizing signal for protein function and scaffolding interactions.11,18 K63-linked ubiquitination is a proteasomal-independent modification that plays an extensive role in NFκB activation by contributing to the stability and scaffolding functions of proteins such as IRAK-1, RIP1, NEMO, and TAK1.13,20-24 Polyubiquitination of NEMO recruits TAK1 for activation of the IKK complex whereas the recruitment of the A20 zinc finger protein onto the ubiquitin platform limits IKK activation.16
Mutations in the IKBKB gene resulting in Lys171Glu (K171E) and Lys171Arg (K171R) have been identified in Multiple Myeloma (MM), Spleen Marginal Zone Lymphoma (SMZL) and Mantle Cell Lymphoma (MCL).25-27 We used these mutations as a starting point to investigate the mechanism of IKKβ activation and downstream effects on cellular signaling. This work reveals that IKKβ is conjugated to both K48- and K63-linked ubiquitin that regulate the wild-type (WT) protein. However, mutation of Lys171 to either Glu or Arg leads to constitutive phosphorylation activity and also increased polyubiquitination of IKKβ, both K48- and K63-linked. Surprisingly, mutations at Lys171 also resulted in STAT3 activation. Thus, mutations at Lys171 of IKKβ differ from typical activation mediated by the S177/S181 phosphorylation sites, resulting in hyperactivation of kinase activity as well as increased ubiquitination and, specifically, K63-linked ubiquitin scaffolding that induces STAT3 signaling.
Results
Mutations at Lys171 activate IKKβ
Lys171 is located within a highly conserved region in the activation loop of the kinase domain of IKKβ, close to the major activating phosphorylation sites Ser177 and Ser181 (Fig. 1A).28 Lys171 also lies near Tyr169, recently shown to undergo tyrosine phosphorylation in the presence of FGFR2, resulting in IKKβ activation.29 In addition, Lys171 contributes to the stability of the activated protein via ionic interactions with phosphorylated Ser181.30 Hence, this residue plays a critical regulatory role in the activated structure.
Figure 1.

Phosphorylation of IKKβ Induced by Mutations at Lys171. (A) Schematic of IKKβ with the activation loop within the N-terminal kinase domain expanded to show amino acids critical for phosphorylation and signaling. The ubiqutin-like domain (ULD), the scaffold/dimerization domain (SDD) which contains the leucine zipper (LZ) and helix-looop-helix (HLH) regions, and NEMO binding domain (NBD) are indicated. (B) IKKβ mutant proteins were expressed in HEK293 cells and the IKK complex was immunoprecipitated with IKKγ antisera and assayed for in vitro phosphorylation. Samples were separated by SDS-PAGE and detected by autoradiography. IKKβ expression is shown by immunoblotting for IKKβ (middle panel). Assays were quantitated relative to 32P incorporation of the IKKβ S177E/S181E mutant, +/− sem. (bottom) (C) HEK293T cells expressing IKKβ mutants were analyzed for activation loop serine phosphorylation using Phospho-IKKα/β antisera (top). The membrane was reprobed for IKKβ (middle panel). Serine 177/181 phosphorylation was quantitated relative to the K171T mutant, +/− sem. (bottom panel). (D) HEK293T cells expressing IKKβ WT or K171E were treated with 50 μg/ml cycloheximide (CHX) for 2, 5, 7, 12 and 24 h. Lysates were examined for IKKβ serine phosphorylation and total IKKβ as in (C) (top 2 panels). The membrane was reprobed for β-tubulin (third panel). Serine 177/181 phosphorylation was quantitated relative to K171E at time zero, +/− sem. (bottom).
Using site-directed mutagenesis, we constructed the mutant K171E observed in the human cancers MM and SMZL.25,26 We also constructed all other mutants at this position resulting from single base changes, thus creating the mutants K171R, M, N, Q and T, recognizing that some of these mutations might be identified in future patient cancers. Indeed, while this work was in progress, the mutation K171R was identified in a case of Mantle Cell Lymphoma.27 IKKβ constructs were expressed in HEK293 cells: WT, K171-mutated, the positive control S177E/S181E (constitutively activated “EE”), and the negative control S177A/S181A (non-activatable “AA”). IKK complexes were recovered using antisera against NEMO and assayed by in vitro kinase assay using [32P]-ATP. The results show that all the mutations at Lys171 exhibited increased phosphorylation of IKKβ relative to the activated “EE” mutant, with the K171E exhibiting approximately 5-fold greater activity (Fig. 1B).
The increase in phosphorylation was confirmed to be occurring on the activation loop residues S177/S181 by phospho-specific immunoblotting (Fig. 1C). Whereas the activated “EE” mutant is unrecognizable by the antiserum, each of the mutants at Lys171 showed strong activation in the order: R > T > Q > M > E > N. We also examined the stability of S177/S181 phosphorylation in the K171E mutant versus WT in a cycloheximide (CHX) time course over 24 h, with K171E exhibiting higher initial phosphorylation and slower decay (t1/2 ∼20 h for K171E, t1/2 ∼7 h for WT) (Fig. 1D). Together, the data presented in Fig. 1 suggest that the important feature for mutational activation at Lys171 is not the introduction of a phosphomimic (K171E), nor retention of a basic charge (K171R), but rather the specific loss of the Lys residue.
IKKβ is conjugated to K63-linked ubiquitin polymers
To examine IKKβ for potential K63-linked ubiquitination, IKKβ immune complexes were collected from cells expressing IKKβ (WT, K171-mutated, EE, AA), HA-Ub and HA-NEMO. Immunoprecipitation and immunoblotting revealed the presence of K63-linked ubiquitin conjugates which were significantly increased by each of the mutations examined at Lys171 (Fig. 2A). As the K171E mutation was initially identified in human lymphomas,25,26 we examined a time course of TNFα stimulation from 0–24 h (Fig. 2B). The time course reveals that although WT never exhibited a significant increase, the activated control “EE” mutant displayed a significant K63-linked signal visible out to 12 h, but absent in the 24 h sample. In contrast, the mutant K171E exhibits a very robust K63-linked signal at intermediate time points (2, 5, 8, 12 h) and, significantly, this signal persists even at the 24 h time point. A similar pattern holds true for the total ubiquitination signal, demonstrating the persistence of the signal in the K171E mutant even at 24 h (Fig. 2B, 3rd Panel). A similar time course from 0–24 h carried out in the presence of CHX also revealed that under these conditions the K171E mutation leads to a significant increase in the strength and persistence of the K63-linked signal in comparison with the WT protein (Fig. 2C). Clearly, the K171E mutation potentiates and stabilizes the total as well as the specific K63-linked ubiquitination of IKKβ during prolonged stimulation of cells with TNFα.
Figure 2.

IKKβ Ubiquitination Regulated by Lys171. (A) HEK293T cells expressing IKKβ WT or Lys171 mutants, HA-Ub and HA-NEMO, were lysed and immunoprecipitated with IKKβ antisera. Samples were immunoblotted for K63-linkage specific polyubiquitin (top panel). The membrane was reprobed for IKKβ (bottom panel). (B) Cells as in Panel A were treated with 10 ng/ml of TNFα for 2, 5, 8, 12 and 24 h. Samples were immunoprecipitated and blotted as decribed in (A) (top 2 panels). Duplicate samples were immunoprecipitated with HA-probe antisera and immunoblotted for IKKβ (third panel). The membrane was reprobed for K63-linkage specific polyubiquitin (bottom). (C) Cells as in Panel A were treated with 50 μg/ml CHX for 2, 5, 7, 12 and 24 h. IKKβ immunoprecipitates were blotted as in (A). (D) Cells as in Panel A were treated with +/− 10ng/ml TNFα and +/− 10 μM MG132 for 12 h. IKKβ immunoprecipitates were blotted for K63-linkage specific polyubiquitin (top panel) or K48-linkage specific polyubiquitin (third panel). Membranes were reprobed for IKKβ (2nd and 4th panels).
IKKβ was previously shown to be degraded by the 26S proteasome pathway via KEAP1-mediated ubiquitination.31 Therefore, we looked into the effects of simultaneous treatment of cells with TNFα to activate inflammatory signaling plus MG132 to inhibit proteasomal degradation (Fig. 2D). For the WT protein, for both K48-linked and K63-linked ubiquitination, treatment with MG132 significantly increased ubiquitination (Lanes 5 vs 8). In contrast, the K48-linked and K63-linked ubiquitination of the “EE” and K171E proteins was largely unaffected by TNFα treatment alone or in the presence of MG132 (Lanes 6, 7, 9, 10). Perhaps the most striking observation from Fig. 2D is the strength of the K171E signal which, under all conditions, exceeds the maximal signal from either the WT or “EE” protein. These results show that the K171E mutant, and to a lesser extent the activated “EE” control, exhibit constitutive K48-linked and K63-linked ubiquitination of IKKβ.
IKKβ K171E constitutively activates STAT3 signaling independently of NFκB
Interleukin-6 (IL-6) is deregulated in various carcinomas resulting in abnormal cellular proliferation, drug-resistance, cell cycle progression and migration.32 IL-6 signaling activates STAT proteins which have been implicated in cellular transformation and oncogenesis in MM, endometrial, lung, colorectal and breast carcinomas, and other human diseases.33,34 In addition, STAT3 activation has been linked to inflammation-induced tumorigenesis initiated by malignant cells,35-38 including persistent activation of NFκB signaling, establishing a positive tumorigenic feedback loop.39-41
We investigated the effects of IL-6 stimulation on K171E-expressing cells (Fig. 3A). When HEK293T cells expressing WT, K171E, “EE” and “AA” IKKβ proteins were stimulated with IL-6, activation of endogenous STAT3 was observed by immunoblotting for phospho-Y705 (Lanes 11–15). In contrast, TNFα treatment showed little or no observable STAT3 activation in all samples except for K171E (Lanes 6–10). Surprisingly, IKKβ K171E expression leads to activation of STAT3 in the absence of IL-6 (Lanes 3, 8).
Figure 3.

IKKβ Lys171 Activation of STAT3. (A) HEK293T cells expressing IKKβ derivatives were treated with 10 ng/ml TNFα or 10 ng/ml IL-6 for 10 min. Lysates were immunoblotted for Phospho-STAT3 (Tyr705) (top), STAT3 (middle panel) and IKKβ (bottom). (B) HEK293T cells expressing IKKβ mutants were treated with 10 ng/ml of TNFα for 5, 15, 45 and 60 min. Lysates were immunoblotted for Phospho-STAT3 (Tyr705) (top), STAT3 (2nd panel), Phospho-IκBα (Ser32/36) (3rd panel), IκBα (4th panel), and IKKβ (bottom). Phosphorylation of STAT3 was quantitated relative to the K171E mutant at time zero, +/− sem. (bottom panel). (C) HEK293T cells expressing IKKβ WT, K171E and S177E/S181E were treated with +/− 10 ng/ml TNFα and +/− 10 μM MG132 for 12 h. Lysates were immunoblotted as in (B). (D) Lysates from HEK293T cells expressing IKKβ derivatives were immunoblotted as in (A). STAT3 phosphorylation was quantitated relative to the Lys171Thr mutant, +/− sem. (bottom).
We further examined the effects of TNFα stimulation over a 1 h time course, comparing cells expressing WT, K171E and “EE” mutants (Fig. 3B). The K171E mutant induced robust constitutive STAT3 activation that increased following TNFα and peaked at 150% at 15 min. Although the WT and “EE” mutant proteins exhibited TNFα-responsiveness, the magnitude of STAT3 phosphorylation was always significantly less than for K171E. The phosphorylation of S32/S36 of IκBα provides another downstream effect of NFκB signaling stimulated by TNFα. In contrast to the P-STAT3 signal, the appearance of IκBα P-S32/S36 was rapid, peaking at 5 min and then decaying, similar to WT, K171E or “EE” mutants (Fig. 3B, 3rd Panel, Lanes 5–7).
When treating cells with TNFα and the proteasome inhibitor MG132 for a period of 12 h, the K171E mutant activation of P-STAT3 was largely TNFα-independent, although it did increase modestly in response to MG132 (Fig. 3C, Lanes 3, 6, 9). TNFα-stimulation resulted in detectable phosphorylation of IκBα S32/S36 for both the WT and “EE” mutant proteins, which increased massively when combined with MG132 treatment, demonstrating that these proteins are typically subject to proteasomal degradation (Fig. 3C, 3rd Panel, Lanes 8, 10). In addition, the various mutations at Lys171 induced significant activation of STAT3 (Fig. 3D). Thus, although K171E significantly activated P-STAT3, the other substitutions (M, N, Q, R, T) at this position were more robust.
The observations presented in Figure 3 demonstrate that mutational activation of IKKβ at Lys171 is significantly different than the phospho-mimic S177E/S181E “EE” mutant. Mutations at Lys171 lead to constitutive activation of STAT3 that is independent of TNFα or IL-6 stimulation, and only marginally regulated by proteasomal degradation.
LC-MS/MS analysis identifies ubiquitination sites in IKKβ
Samples from HEK293T cells expressing either IKKβ WT or K171E proteins were analyzed by LC-MS/MS to identify significant posttranslational modifications. Proteins to be analyzed were recovered in immune complexes with antisera against IKKβ or the HA-tag of HA-Ub and HA-NEMO. Lys147, Lys418, Lys555 and Lys703 were identified as predominant ubiquitination sites on tryptic peptides based on the additional mass of 114 Da arising from the C-terminal di-Gly residues of Ub bonded by an isopeptide linkage to the modified Lys residue in IKKβ (Table 1). Lys301 was also identified as a minor site. A representative spectrum is presented identifying each of the diGly-tagged residues Lys147, Lys418, Lys555 and Lys703 (Fig. 4A). The frequency which different sites are detected with, termed spectral counting, is a widely accepted method of label-free quantification.29,42,43 While spectral counts of different peptides are not strictly comparable, large differences correlate with relative abundance for the same peptide despite limitations arising from differing ionization efficiencies comparing different peptides. For each of the predominant sites of ubiquitination, Lys147, Lys418, Lys555 and Lys703, spectral counts in Table 1 suggest that ubiquitination at each of these sites was increased modestly in cells expressing K171E vs. WT IKKβ, except for Lys147 which exhibited a ∼5-fold increase in K171E samples.
Table 1.
Ubiquitination IKKβ peptides identified by LC-MS/MS. In addition to the peptide sequences identified, also shown here are TPP adjusted minimum probabilities, charge states, Xcorr values, initial and combined spectral count of each identified ubiquitinated peptides
| Residue | IKKβ | Prep # | Precursor Ion Charge | Peptide Sequence | Adjusted Probability | Instances | Xcorr | Partial Spectral Count | Total Spectral Count |
|---|---|---|---|---|---|---|---|---|---|
| K147 | WT | #6628 | 3 | DLK[242]PENIVLQQGEQR | 0.9998 | 2 | 3.55-3.71 | 2 | 13 |
| K171E | #6629 | 2 | DLK[242]PENIVLQQGEQR | 0.9998 | 5 | 3.28-4.50 | 11 | ||
| #6629 | 3 | DLK[242]PENIVLQQGEQR | 0.9998 | 4 | 2.29-4.54 | ||||
| #6645 | 3 | DLK[242]PENIVLQQGEQR | 0.9984 | 1 | 2.92 | ||||
| |
|
#6629 |
2 |
LK[242]PENIVLQQGEQR |
0.8414 |
1 |
2.84 |
|
|
| K301 |
K171E |
#6629 |
3 |
GTDPTYGPNGCFK[242]ALDDILNLK |
0.2656 |
2 |
3.30-3.75 |
2 |
2 |
| K418 | WT | #6628 | 3 | PQPESVSCILQEPK[242]R | 0.237 | 3 | 3.37-4.15 | 15 | 32 |
| #6644 | 3 | PQPESVSCILQEPK[242]R | 0.2112 | 12 | 3.34-3.88 | ||||
| K171E | #6629 | 2 | PQPESVSCILQEPK[242]R | 0.3313 | 4 | 3.75-4.33 | 17 | ||
| #6629 | 3 | PQPESVSCILQEPK[242]R | 0.3117 | 7 | 3.50-4.53 | ||||
| |
|
#6645 |
3 |
PQPESVSCILQEPK[242]R |
0.304 |
6 |
3.28-4.38 |
|
|
| K555 | WT | #6628 | 3 | K[242]QGGTLDDLEEQAR | 0.9998 | 7 | 3.53-4.46 | 9 | 22 |
| #6644 | 3 | K[242]QGGTLDDLEEQAR | 0.9997 | 2 | 2.83-3.94 | ||||
| K171E | #6629 | 2 | K[242]QGGTLDDLEEQAR | 0.9998 | 7 | 3.63-4.64 | 13 | ||
| #6629 | 3 | K[242]QGGTLDDLEEQAR | 0.9998 | 5 | 3.63-4.49 | ||||
| |
|
#6645 |
3 |
K[242]QGGTLDDLEEQAR |
0.9835 |
1 |
3.58 |
|
|
| K703 | WT | #6628 | 3 | LSQPGQLMSQPSTASNSLPEPAK[242]K | 0.9815 | 4 | 2.68-3.42 | 4 | 10 |
| K171E | #6629 | 3 | LSQPGQLMSQPSTASNSLPEPAK[242]K | 0.9613 | 6 | 2.84-3.63 | 6 |
Figure 4.

Identification and Biological Function of IKKβ ubiquitination Sites. (A) Representative spectrum of peptides showing the relative intensity of selected b and y ions for the major sites of IKKβ ubiquitination. Note that not all identified ions are labeled due to space constraints. (B) HEK293T cells expressing IKKβ Lys to Arg mutants were immunoprecipitated and immunoblotted as in Figure 2A. (C) Lysates from HEK293T cells expressing IKKβ Lys to Arg mutants were immunoblotted as in Figure 1(C).
To assess the importance of individual IKKβ ubiquitination sites, each was mutated to Arg, either alone or in combination with the activating mutation K171E. When assayed for K63-linked ubiquitination, each single mutant resembled the WT protein except for K147R, which exhibited little or no detectable K63-linked ubiquitin (Fig. 4B, Lane 3). When combined with the activating K171E mutation, all but one double mutant protein exhibited K63-linked ubiquitination similar to the K171E mutant alone (Lanes 8, 10–13). The notable exception was the double mutant K171E/K147R (Lane 9), which exhibited little or no detectable K63-linked ubiquitination. These results are consistent with a preliminary identification of Lys147 as the principal site of K63-linked ubiquitination in IKKβ.
Next, we examined phosphorylation of the single and double mutants of IKKβ (Fig. 4C). Each of the single mutations resulted in significantly increased phosphorylation (Lanes 4–7), with the exception of K147R (Lane 3). The single ubiquitination site mutations in combination with K171E did not result in any further increase in phosphorylation (Lanes 10–13). The unique exception was again provided by the double mutant K171E/K147R which exhibited only slight phosphorylation, as best seen in the long exposure (2nd Panel, Lane 9). Thus, each Lys identified in our MS/MS analysis contributes to the regulatory function of IKKβ, including those distant from the kinase domain.
K63-linked ubiquitination is required for STAT3 activation
The identification of residues Lys147, Lys418, Lys555 and Lys703 as predominant ubiquitination sites does not reveal whether each site is K48-linked versus K63-linked nor its role in STAT3 activation. Because K63-linked ubiquitination is preferentially involved in cellular signaling rather than proteasomal degradation, we hypothesized that STAT3 activation by IKKβ K171E may be dependent upon its increased K63-linked ubiquitination. Therefore, we used the small molecule inhibitor NSC697923 to inhibit the E2 ubiquitin-conjugating enzyme UBC13-UEV1A complex, selectively inhibiting K63-linked ubiquitination.44
HEK293T cells expressing WT or K171E were treated with the inhibitor NSC697923 and examined for IKKβ S177/S181 phosphorylation. Although some increased phosphorylation was observed for WT IKKβ, the activation of IKKβ kinase in the K171E mutant protein was largely independent of K63-linked ubiquitination (Fig. 5A). We also examined the effect of the K63-linked inhibitor NSC697923 on STAT3 activation in IKKβ WT- and K171E-expressing cells (Fig. 5B). The activation of STAT3 by the IKKβ mutant K171E (Lane 5) was blocked by treatment with NSC697923 (Lanes 6, 7).
Figure 5.
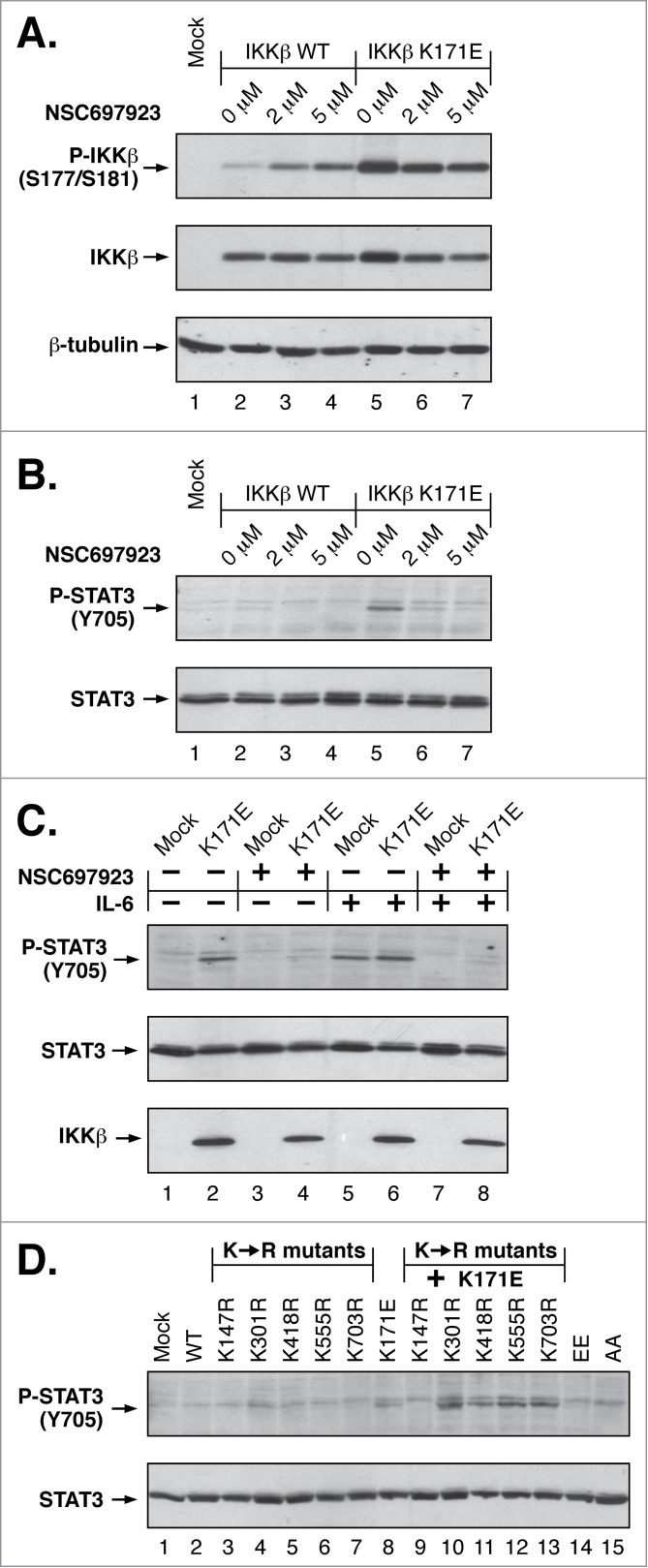
Inhibition of K63-linked ubiquitination Blocks IKKβ Lys171 Signaling. (A) Lysates from HEK293T cells expressing IKKβ WT and K171E were treated with 2 μM and 5 μM NSC697923 for 2 h and immunoblotted for Phospho-IKKα/β (top), IKKβ (middle panel) and β-tubulin (bottom). (B) Duplicate samples from (A) were immunoblotted for Phospho-STAT3 (Tyr705) (top) and STAT3 (bottom). (C) Lysates from HEK293T cells expressing IKKβ K171E treated with +/− 5 μM NSC697923 for 2 h and +/− 10 ng/ml IL-6 for 10 min were immunoblotted as in (B). (D) Lysates from HEK293T cells expressing IKKβ Lys to Arg mutants were immunoblotted as in (B).
Following IL-6 treatment, both mock and K171E-expressing cells exhibited STAT3 activation (Fig. 5C, Lanes 5, 6). When cells were pretreated with the inhibitor NSC697923, the P-STAT3 signal was largely ablated (Lanes 7, 8). These results show that STAT3 activation, whether in response to IL-6 stimulation or IKKβ K171E expression, is dependent upon K63-linked ubiquitination. Lastly, among the IKKβ ubiquitination sites identified, the activation of STAT3 was dependent upon Lys147 as shown by the ability of K147R to block the K171E-induced STAT3 phosphorylation (Fig. 5D, Lane 9).
Taken together, the results presented in Figure 5 demonstrate that STAT3 activation in response to the IKKβ K171E mutation is dependent on the presence of the ubiquitination site Lys147, and that ubiquitination at this site is K63-linked as shown by its sensitivity to the selective inhibitor NSC697923.
Discussion
This is the first report to show that IKKβ is modified by the addition of K63-linked ubiquitination. We demonstrate that the mutation K171E leads to constitutive activation of the intrinsic kinase activity of IKKβ, suggesting a role of the normal Lys171 side chain in stabilizing an inactive kinase in addition to its documented importance in the activated structure.30 Other mutations at this position also result in constitutive activation of IKKβ, demonstrated by in vitro kinase assay and phosphorylation of the activation loop residues S177/S181. Such mutations also result in significant activation of STAT3 signaling, which is independent of TNFα or IL-6 stimulation.
Other members of the NFκB pathway also undergo K63-linked regulatory ubiquitination. The K63-linked ubiquitination of NEMO has been reported as critical for the activation of T-cell receptor signaling; specifically, overexpression of Bcl10 recruits MALT-1 which induces K63-linked ubiquitination of Lys399 in NEMO that activates NFκB in lymphocytes.45 In addition, misregulation of K63-ubiquitination at Lys285 in NEMO occurs in Crohn's Disease via Nod2.46 In breast cancer cells, IKKε was shown to undergo K63-linked ubiquitination at Lys30 and Lys401. K63-linked ubiquitination is essential for the oncogenic activity of IKKε that contributes to the hyperactivation of NFκB.47
Previous reports, examining IKKβ ubiquitination, showed by site-directed mutagenesis and immunoblotting that IKKβ is mono-ubiquitinated at Lys163 in Tax-transfected HEK293T cells.48 The mono-ubiquitination of IKKβ was attributed to the E3 ligase RING-finger Ro52, which then leads to autophagy-mediated degradation of the protein and subsequent downregulation of NFκB signaling.31,49 Furthermore, the K171R mutation was shown to cause constitutive phosphorylation of IKKβ during a screen of kinase domain Lys residues.48 Another report illustrated that IKKβ is polyubiquitinated at Lys555 by the E3 ligase KEAP1 in TNFα-stimulated HEK293T cells, resulting in a downregulation of NFκB signaling due to 26S-proteasomal degradation of the protein.50
Using LC-MS/MS, we identified 4 predominant ubiquitination sites in IKKβ, Lys147, Lys418, Lys555 and Lys703. Spectral counts suggest the extent of ubiquitination at each of these sites was increased modestly in cells expressing K171E vs. WT IKKβ. Lys147 was the only major site to show a significant increase in K171E samples, exhibiting a 5-fold increase. This is significant because Lys147 was also the only ubiquitination site required for STAT3 activation and the primary site of K63-linked ubiquitination. Ubiquitination at a different Lys residue, Lys163, suggested by site-directed mutagenesis and immunoblotting in an earlier study, was not observed in our work, possibly reflecting the inclusion of Tax expression in this prior work.48 Although the ubiquitination sites identified here were all initially identified in a study of the human cellular ubiquitinome,51 we have determined here that each site contributes to the regulation of IKKβ kinase function.
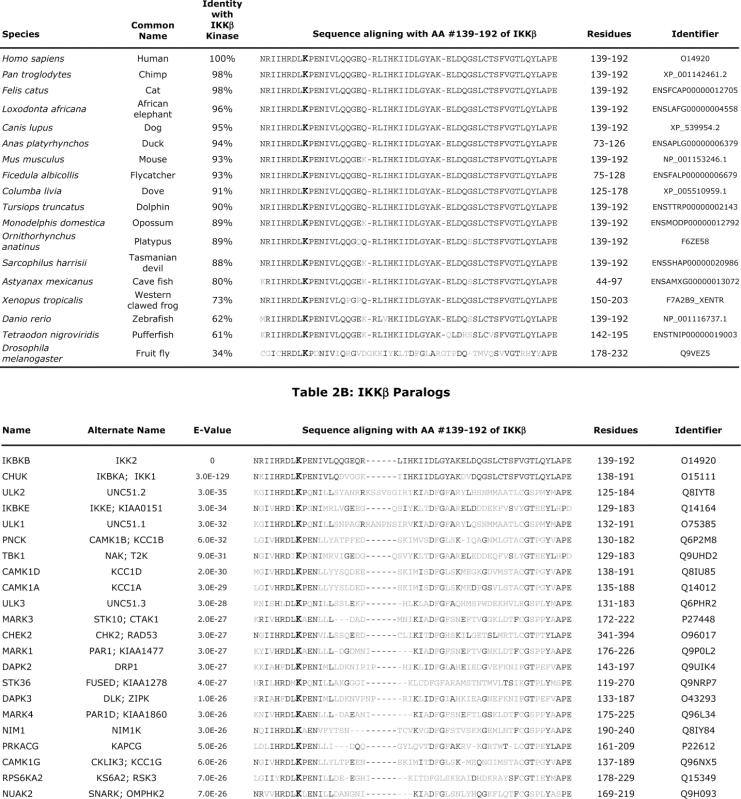
Lys147 in IKKβ lies within the highly conserved sequence DLKPEN between the beta6 and beta7 regions of the kinase.30 A comparison of other species shows this region to be highly conserved among IKKβ orthologs (Table 2A). Even more surprising is the very strict conservation of this region, including the Lys corresponding to Lys147 of IKKβ, in a very large number of human kinase paralogs (Table 2B). This raises the possibility that many kinases exhibiting a Lys at the corresponding position may undergo K63-linked ubiquitination as a regulatory modification. This has already been demonstrated for the corresponding Lys158 of TAK1, required for the subsequent phosphorylation of MKK6 which activates the JNK-p38 kinase pathway.52 Lys158 residue in TAK1 has also been suggested as a site of K63-linked ubiquitination involved in the activation of TAK1 in Helicobacter pilori infections,53 and in genotoxic stress-induced NFκB activation.54 Notably, the kinase activity of TAK1 is abrogated when Lys158 is mutated to Arg, as is the case for the K147R mutation in IKKβ. Another clinically important example is provided by the K63-linked ubiquitination at the corresponding Lys578 residue of BRAF, required for BRAF-mediated signaling.55 The unequivocal mass spectrometry evidence presented here that Lys147 of IKKβ undergoes ubiquitination, together with the biochemical evidence that this is the major site of K63-linked ubiquitination in IKKβ, will likely be relevant to TAK1 and other related protein kinases.
Table 2.
Conservation of IKKβ Lys171 in orthologous and paralogous proteins. Amino acids shown are the residue corresponding to Lys171 (bold), identical (black), and non-identical residues (gray).
 |
As previously mentioned, the K171E mutation was initially identified via genetic sequencing of MM and SMZL.25,26 As this work was in progress, the K171R mutation was identified in MCL27 and shown in the data above to be even more strongly activating than K171E. A survey of mutations in IKKβ that occur in human cancer is presented in Table 3, some of which may be without effect whereas others, such as the Lys171 mutations we have characterized, have significant biochemical ramifications. Previously, in a receptor tyrosine kinase of importance both in development and in human cancer, we demonstrated profound FGFR3 kinase activation due to the K650E mutation as responsible for the lethal developmental disorder Thanatophoric Dysplasia type II (TDII).56 A comparison of other amino acid substitutions at Lys650 of FGFR3 also identified K650M as strongly activating, which was identified several years later in individuals with the rare developmental syndrome Severe Achondroplasia with Delayed Development and Acanthosis Nigricans (SADDAN).57 In the case of IKKβ, as genetic sequencing of additional cancers becomes widely available, our results predict that other substitutions at this position which are accessible by single nucleotide changes in addition to K and R will be observed, such as M, N, Q and T.
Table 3.
Genetic changes identified in IKKβ in human cancer
| Mutation | Cancer | Effect Reported | Biological Effect | Ref |
|---|---|---|---|---|
| E81Q |
Breast Cancer |
Disease causing |
(−) |
61 |
| K171E | Multiple Myeloma | Activating | IKKβ activation; STAT3 activation; Lys63-Ub | 25 |
| K171E | Spleen Marginal Zone Lymphoma | Damaging | 26 | |
| K171R |
Mantle Cell Lymphoma |
(−) |
|
27 |
| A360S |
Breast Cancer |
Disease causing |
(−) |
61-64 |
| Amplification |
Ovarian Cancer |
(−) |
(−) |
65 |
| R144Q | Colorectal Cancer | Benign | (−) | 66 |
| R446W | Probably damaging | |||
| I598S | Probably damaging | |||
| E707* |
|
(−) |
|
|
| G667C |
Lung Adenocarcinoma |
(−) |
(−) |
67 |
| D484Y |
Esophageal Adenocarcinoma |
(−) |
(−) |
68 |
| R53W |
Spinal Cord Ependynoma |
(−) |
(−) |
69 |
| R47R | Transitional Cell Carcinoma (Bladder Cancer) | (−) | (−) | 70 |
| Q175* | ||||
| I314M |
|
|
|
|
| L679_fs |
Clear-Cell Renal Carcinoma |
(−) |
(−) |
71 |
| R288S |
Chondrosarcoma |
(−) |
(−) |
72 |
| A212T | Colorectal Cancer | (−) | (−) | 73 |
| A357S | ||||
| S474Y | ||||
| M454I |
|
|
|
|
| K603R |
Small-Cell Lung Carcinoma |
(−) |
(−) |
74 |
| N667D | Gastric Cancer | Benign | (−) | 75 |
Future work is necessary to identify the interacting proteins that associate with a K63-ubiquitination scaffold assembled by IKKβ, which we show is responsible for IL-6-dependent STAT3 activation and for the unusual STAT3 signaling demonstrated by the K171E mutation. The activation of additional signaling pathways such as STAT3 may be expected to confer significant proliferative or antiapoptotic properties to human cancer cells expressing IKKβ mutant proteins. In addition, the identification of K63-linked ubiquitination potentially provides a novel selective chemotherapeutic target.
Methods
Cell culture
HEK293 and HEK293T cells were grown in DMEM with 10% FBS and 1% Pen/Strep and maintained in 10% CO2 at 37°C. Cells were transfected with plasmid DNA using calcium phosphate precipitation at 3% CO2, as previously described.29
Plasmid constructs
IKKβ wild-type (WT) has been described previously,58 which was not epitope-tagged. Mutations in IKKβ were created by Quikchange site-directed mutagenesis and confirmed by DNA sequencing. The haemagglutinin (HA)-tagged ubiquitin (HA-Ub3) expression plasmid was a gift from Dr. Tony Hunter and Dr. Andrea Carrano (Salk Institute, La Jolla CA).59 The expression plasmid containing murine NEMO (HA-NEMO) was a gift from Dr. Mark Hannink (University of Missouri, Columbia MO).
Antibodies, immunoprecipitation and immunoblot
Antibodies were obtained from the following sources: IKKγ (FL-419), IKKβ (H-4), IKKβ (G-8), β-tubulin (H-235), HA-probe (F-7), STAT3 (C-20), IκBα (C-21) from Santa Cruz Biotechnology; Phospho-IKKα/β (Ser176/180) (16A6), K63-linkage Specific Polyubiquitin (D7A11), K48-linkage Specific Polyubiquitin (D9D5), Phospho-STAT3 (Tyr705) (D3A7), Phospho-IκBα (Ser32/36) (5A5) from Cell Signaling Technology; horseradish peroxidase (HRP) anti-mouse, HPR anti-rabbit from GE Healthcare. Enhanced chemiluminence (ECL) reagents were from GE Healthcare. TNFα and MG132 were obtained from Bio-techne; recombinant human Interleukin-6 (IL-6) from Life Technologies; and NSC697923 from Santa Cruz Biotechnology.
HEK293 and HEK293T cells were transfected, starved and collected in RIPA Buffer.29 Immunoprecipitation and immunoblotting experiments were performed as described.29,60 5 mM N-Ethylmaleimide (NEM) was added to RIPA buffer when lysates were examined for ubiquitination by immunoblotting.
Mass spectrometry analysis
HEK293T cells were plated one day prior to transfection at 3.0 × 106 cells per 15 cm tissue culture plate. 10 plates per sample were transfected with IKKβ WT or IKKβ K171E, HA-Ub3 and HA-NEMO. Cells were treated with 10 μM MG132 for approximately 5 h prior to lysis. Lysates were collected and immunoprecipitated as described.29 Analysis of tryptic peptides by the Sanford-Burnham Medical Research Institute Proteomics Facility was carried out by high-resolution, high-accuracy 1D LC-MS/MS, consisting of a Michrom HPLC, a 15-cm Michrom Magic C18 column, a low-flow ADVANCED Michrom MS source, and LTQ-Orbitrap XL and Velos Pro with ETD (Thermo Fisher Scientific). MS/MS spectra were submitted to Sorcerer Enterprise v.3.5 release (Sage-N Research Inc..) with SEQUEST algorithm as the search program for peptide/protein identification. Search results were further analyzed using Peptide/Protein prophet v.4.5.2 (ISB). Spectra annotated with fragment ions were captured using the Javascript MS/MS spectrum viewer Lorikeet, and annotated using Adobe Illustrator to show selected b and y ions.
Presented in Table 1, Samples #6628 and #6629, from IKKβ WT and K171E expressing cells, respectively, were collected as IKKβ immune complexes. Samples #6644 and #6645, from IKKβ WT and K171E expressing cells, respectively, were collected as HA (hemagglutinin) immune complexes. Tryptic peptide coverage of IKKβ in the 4 samples was: #6628, 69%; #6629, 70%; #6644, 56%; #6645, 55%. Total IKKβ spectra analyzed in the 4 samples was: #6628, 2473 spectra; #6629, 2249 spectra; #6644, 2521 spectra; #6645, 1990 spectra.
In vitro kinase assays
HEK293 cells were transfected and starved overnight prior to lysing in Cytoplasmic Extract Buffer as described.29 Lysates were immunoprecipitated with IKKγ anitsera, collected on Protein A-Sepharose, washed with Cytoplasmic Extract Buffer and Wash Buffer and subjected to in vitro phosphorylation kinase assay. Kinase reactions containing 10 μCi of [γ-32P]- with 20 μM ATP were incubated at 30°C for 30 min, and separated by 12.5% SDS-PAGE. Gels were stained, destained, dried and exposed to film.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. Alexandre Rosa Campos of The Sanford-Burnham Medical Research Institute for technical assistance and Dr. Andrea C. Carrano of UC San Diego for valuable technical suggestions. We also thank Laura Castrejon for assistance with manuscript preparation and submission.
Funding
LHG received support from the Graduate Assistance in Areas of National Need (GAANN) Fellowship Program (L.H.G), and KM received support from grant P20 CA132386 from the National Institutes of Health which funds The Sanford-Burnham Medical Research Institute Proteomics Facility (K.M.).
References
- 1. Hernandez L, Hsu SC, Davidson B, Birrer MJ, Kohn EC, Annunziata CM. Activation of NF-kappaB signaling by inhibitor of NF-kappaB kinase beta increases aggressiveness of ovarian cancer. Cancer Res 2010; 70:4005-14; PMID:20424119; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-3912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hu MC, Lee DF, Xia W, Golfman LS, Ou-Yang F, Yang JY, Zou Y, Bao S, Hanada N, Saso H, et al. IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell 2004; 117:225-37; PMID:15084260; http://dx.doi.org/ 10.1016/S0092-8674(04)00302-2 [DOI] [PubMed] [Google Scholar]
- 3. Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, Wynshaw-Boris A, Poli G, Olefsky J, Karin M. IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med 2005; 11:191-8; PMID:15685170; http://dx.doi.org/ 10.1038/nm1185 [DOI] [PubMed] [Google Scholar]
- 4. Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, Shoelson SE. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat Med 2005; 11:183-90; PMID:15685173; http://dx.doi.org/ 10.1038/nm1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tanaka M, Fuentes ME, Yamaguchi K, Durnin MH, Dalrymple SA, Hardy KL, Goeddel DV. Embryonic lethality, liver degeneration, and impaired NF-kappa B activation in IKK-beta-deficient mice. Immunity 1999; 10:421-9; PMID:10229185; http://dx.doi.org/ 10.1016/S1074-7613(00)80042-4 [DOI] [PubMed] [Google Scholar]
- 6. Andreakos E, Smith C, Kiriakidis S, Monaco C, de Martin R, Brennan FM, Paleolog E, Feldmann M, Foxwell BM. Heterogeneous requirement of IkappaB kinase 2 for inflammatory cytokine and matrix metalloproteinase production in rheumatoid arthritis: implications for therapy. Arthritis Rheum 2003; 48:1901-12; PMID:12847684; http://dx.doi.org/ 10.1002/art.11044 [DOI] [PubMed] [Google Scholar]
- 7. Aupperle K, Bennett B, Han Z, Boyle D, Manning A, Firestein G. NF-kappa B regulation by I kappa B kinase-2 in rheumatoid arthritis synoviocytes. J Immunol 2001; 166:2705-11; PMID:11160335; http://dx.doi.org/ 10.4049/jimmunol.166.4.2705 [DOI] [PubMed] [Google Scholar]
- 8. Zhang G, Li J, Purkayastha S, Tang Y, Zhang H, Yin Y, Li B, Liu G, Cai D. Hypothalamic programming of systemic ageing involving IKK-beta, NF-kappaB and GnRH. Nature 2013; 497:211-6; PMID:23636330; http://dx.doi.org/ 10.1038/nature12143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, Lenz G, Hanamura I, Wright G, Xiao W, et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 2007; 12:115-30; PMID:17692804; http://dx.doi.org/ 10.1016/j.ccr.2007.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gilmore TD. Multiple myeloma: lusting for NF-kappaB. Cancer Cell 2007; 12:95-7; PMID:17692798; http://dx.doi.org/ 10.1016/j.ccr.2007.07.010 [DOI] [PubMed] [Google Scholar]
- 11. Wertz IE, Dixit VM. Signaling to NF-kappaB: regulation by ubiquitination. Cold Spring Harbor Perspect Biol 2010; 2:a003350; PMID:20300215; http://dx.doi.org/ 10.1101/cshperspect.a003350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen ZJ. Ubiquitination in signaling to and activation of IKK. Immunol Rev 2012; 246:95-106; PMID:22435549; http://dx.doi.org/ 10.1111/j.1600-065X.2012.01108.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell 2006; 22:245-57; PMID:16603398; http://dx.doi.org/ 10.1016/j.molcel.2006.03.026 [DOI] [PubMed] [Google Scholar]
- 14. Yamazaki K, Gohda J, Kanayama A, Miyamoto Y, Sakurai H, Yamamoto M, Akira S, Hayashi H, Su B, Inoue J. Two mechanistically and temporally distinct NF-kappaB activation pathways in IL-1 signaling. Sci Signal 2009; 2:ra66; PMID:19843958; http://dx.doi.org/ 10.1126/scisignal.2000387 [DOI] [PubMed] [Google Scholar]
- 15. Rahighi S, Ikeda F, Kawasaki M, Akutsu M, Suzuki N, Kato R, Kensche T, Uejima T, Bloor S, Komander D, et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell 2009; 136:1098-109; PMID:19303852; http://dx.doi.org/ 10.1016/j.cell.2009.03.007 [DOI] [PubMed] [Google Scholar]
- 16. Skaug B, Chen J, Du F, He J, Ma A, Chen ZJ. Direct, noncatalytic mechanism of IKK inhibition by A20. Mol Cell 2011; 44:559-71; PMID:22099304; http://dx.doi.org/ 10.1016/j.molcel.2011.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bhoj VG, Chen ZJ. Ubiquitylation in innate and adaptive immunity. Nature 2009; 458:430-7; PMID:19325622; http://dx.doi.org/ 10.1038/nature07959 [DOI] [PubMed] [Google Scholar]
- 18. Chen ZJ, Sun LJ. Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell 2009; 33:275-86; PMID:19217402; http://dx.doi.org/ 10.1016/j.molcel.2009.01.014 [DOI] [PubMed] [Google Scholar]
- 19. Yang WL, Zhang X, Lin HK. Emerging role of Lys-63 ubiquitination in protein kinase and phosphatase activation and cancer development. Oncogene 2010; 29:4493-503; PMID:20531303; http://dx.doi.org/ 10.1038/onc.2010.190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xia ZP, Sun L, Chen X, Pineda G, Jiang X, Adhikari A, Zeng W, Chen ZJ. Direct activation of protein kinases by unanchored polyubiquitin chains. Nature 2009; 461:114-9; PMID:19675569; http://dx.doi.org/ 10.1038/nature08247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lamothe B, Besse A, Campos AD, Webster WK, Wu H, Darnay BG. Site-specific Lys-63-linked tumor necrosis factor receptor-associated factor 6 auto-ubiquitination is a critical determinant of I kappa B kinase activation. J Biol Chem 2007; 282:4102-12; PMID:17135271; http://dx.doi.org/ 10.1074/jbc.M609503200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Windheim M, Stafford M, Peggie M, Cohen P. Interleukin-1 (IL-1) induces the Lys63-linked polyubiquitination of IL-1 receptor-associated kinase 1 to facilitate NEMO binding and the activation of IkappaBalpha kinase. Mol Cell Biol 2008; 28:1783-91; PMID:18180283; http://dx.doi.org/ 10.1128/MCB.02380-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Conze DB, Wu CJ, Thomas JA, Landstrom A, Ashwell JD. Lys63-linked polyubiquitination of IRAK-1 is required for interleukin-1 receptor- and toll-like receptor-mediated NF-kappaB activation. Mol Cell Biol 2008; 28:3538-47; PMID:18347055; http://dx.doi.org/ 10.1128/MCB.02098-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wu CJ, Conze DB, Li T, Srinivasula SM, Ashwell JD. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-kappaB activation [corrected]. Nat Cell Biol 2006; 8:398-406; PMID:16547522; http://dx.doi.org/ 10.1038/ncb1384 [DOI] [PubMed] [Google Scholar]
- 25. Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, Harview CL, Brunet JP, Ahmann GJ, Adli M, et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011; 471:467-72; PMID:21430775; http://dx.doi.org/ 10.1038/nature09837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rossi D, Deaglio S, Dominguez-Sola D, Rasi S, Vaisitti T, Agostinelli C, Spina V, Bruscaggin A, Monti S, Cerri M, e al. Alteration of BIRC3 and multiple other NF-kappaB pathway genes in splenic marginal zone lymphoma. Blood 2011; 118:4930-4; PMID:21881048; http://dx.doi.org/ 10.1182/blood-2011-06-359166 [DOI] [PubMed] [Google Scholar]
- 27. Bea S, Valdes-Mas R, Navarro A, Salaverria I, Martin-Garcia D, Jares P, Gine E, Pinyol M, Royo C, Nadeu F, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci U S A 2013; 110:18250-5; PMID:24145436; http://dx.doi.org/ 10.1073/pnas.1314608110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Delhase M, Hayakawa M, Chen Y, Karin M. Positive and negative regulation of IkappaB kinase activity through IKKbeta subunit phosphorylation. Science 1999; 284:309-13; PMID:10195894; http://dx.doi.org/ 10.1126/science.284.5412.309 [DOI] [PubMed] [Google Scholar]
- 29. Meyer AN, Drafahl KA, McAndrew CW, Gilda JE, Gallo LH, Haas M, Brill LM, Donoghue DJ. Tyrosine phosphorylation allows integration of multiple signaling inputs by IKKbeta. PLoS One 2013; 8:e84497; PMID:24386391; http://dx.doi.org/ 10.1371/journal.pone.0084497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu S, Misquitta YR, Olland A, Johnson MA, Kelleher KS, Kriz R, Lin LL, Stahl M, Mosyak L. Crystal structure of a human IkappaB kinase beta asymmetric dimer. J Biol Chem 2013; 288:22758-67; PMID:23792959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wada K, Niida M, Tanaka M, Kamitani T. Ro52-mediated monoubiquitination of IKK{beta} down-regulates NF-kappaB signalling. J Biochem 2009; 146:821-32; PMID:19675099; http://dx.doi.org/ 10.1093/jb/mvp127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hideshima T, Anderson KC. Molecular mechanisms of novel therapeutic approaches for multiple myeloma. Nat Rev Cancer 2002; 2:927-37; PMID:12459731; http://dx.doi.org/ 10.1038/nrc952 [DOI] [PubMed] [Google Scholar]
- 33. Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE, Jr. Stat3 as an oncogene. Cell 1999; 98:295-303; PMID:10458605; http://dx.doi.org/ 10.1016/S0092-8674(00)81959-5 [DOI] [PubMed] [Google Scholar]
- 34. Hodge DR, Hurt EM, Farrar WL. The role of IL-6 and STAT3 in inflammation and cancer. Eur J Cancer 2005; 41:2502-12; PMID:16199153; http://dx.doi.org/ 10.1016/j.ejca.2005.08.016 [DOI] [PubMed] [Google Scholar]
- 35. Liang J, Nagahashi M, Kim EY, Harikumar KB, Yamada A, Huang WC, Hait NC, Allegood JC, Price MM, Avni D, et al. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell 2013; 23:107-20; PMID:23273921; http://dx.doi.org/ 10.1016/j.ccr.2012.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, Ciliberto G, Moscinski L, Fernandez-Luna JL, Nunez G, et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity 1999; 10:105-15; PMID:10023775; http://dx.doi.org/ 10.1016/S1074-7613(00)80011-4 [DOI] [PubMed] [Google Scholar]
- 37. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 2009; 9:798-809; PMID:19851315; http://dx.doi.org/ 10.1038/nrc2734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol 2007; 7:41-51; PMID:17186030; http://dx.doi.org/ 10.1038/nri1995 [DOI] [PubMed] [Google Scholar]
- 39. Chauhan D, Uchiyama H, Akbarali Y, Urashima M, Yamamoto K, Libermann TA, Anderson KC. Multiple myeloma cell adhesion-induced interleukin-6 expression in bone marrow stromal cells involves activation of NF-kappa B. Blood 1996; 87:1104-12; PMID:8562936 [PubMed] [Google Scholar]
- 40. Bharti AC, Shishodia S, Reuben JM, Weber D, Alexanian R, Raj-Vadhan S, Estrov Z, Talpaz M, Aggarwal BB. Nuclear factor-kappaB and STAT3 are constitutively active in CD138+ cells derived from multiple myeloma patients, and suppression of these transcription factors leads to apoptosis. Blood 2004; 103:3175-84; PMID:15070700; http://dx.doi.org/ 10.1182/blood-2003-06-2151 [DOI] [PubMed] [Google Scholar]
- 41. Ben-Neriah Y, Karin M. Inflammation meets cancer, with NF-kappaB as the matchmaker. Nat Immunol 2011; 12:715-23; PMID:21772280; http://dx.doi.org/ 10.1038/ni.2060 [DOI] [PubMed] [Google Scholar]
- 42. Liu H, Sadygov RG, Yates JR 3rd. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal Chem 2004; 76:4193-201; PMID:15253663; http://dx.doi.org/ 10.1021/ac0498563 [DOI] [PubMed] [Google Scholar]
- 43. Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, Villen J, Haas W, Sowa ME, Gygi SP. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 2010; 143:1174-89; PMID:21183079; http://dx.doi.org/ 10.1016/j.cell.2010.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pulvino M, Liang Y, Oleksyn D, DeRan M, Van Pelt E, Shapiro J, Sanz I, Chen L, Zhao J. Inhibition of proliferation and survival of diffuse large B-cell lymphoma cells by a small-molecule inhibitor of the ubiquitin-conjugating enzyme Ubc13-Uev1A. Blood 2012; 120:1668-77; PMID:22791293; http://dx.doi.org/ 10.1182/blood-2012-02-406074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhou H, Wertz I, O'Rourke K, Ultsch M, Seshagiri S, Eby M, Xiao W, Dixit VM. Bcl10 activates the NF-kappaB pathway through ubiquitination of NEMO. Nature 2004; 427:167-71; PMID:14695475; http://dx.doi.org/ 10.1038/nature02273 [DOI] [PubMed] [Google Scholar]
- 46. Abbott DW, Wilkins A, Asara JM, Cantley LC. The Crohn's disease protein, NOD2, requires RIP2 in order to induce ubiquitinylation of a novel site on NEMO. Curr Biol 2004; 14:2217-27; PMID:15620648; http://dx.doi.org/ 10.1016/j.cub.2004.12.032 [DOI] [PubMed] [Google Scholar]
- 47. Zhou AY, Shen RR, Kim E, Lock YJ, Xu M, Chen ZJ, Hahn WC. IKKepsilon-mediated tumorigenesis requires K63-linked polyubiquitination by a cIAP1/cIAP2/TRAF2 E3 ubiquitin ligase complex. Cell Rep 2013; 3:724-33; PMID:23453969; http://dx.doi.org/ 10.1016/j.celrep.2013.01.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Carter RS, Pennington KN, Arrate P, Oltz EM, Ballard DW. Site-specific monoubiquitination of IkappaB kinase IKKbeta regulates its phosphorylation and persistent activation. J Biol Chem 2005; 280:43272-9; PMID:16267042; http://dx.doi.org/ 10.1074/jbc.M508656200 [DOI] [PubMed] [Google Scholar]
- 49. Niida M, Tanaka M, Kamitani T. Downregulation of active IKK beta by Ro52-mediated autophagy. Mol Immunol 2010; 47:2378-87; PMID:20627395; http://dx.doi.org/ 10.1016/j.molimm.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lee DF, Kuo HP, Liu M, Chou CK, Xia W, Du Y, Shen J, Chen CT, Huo L, Hsu MC, et al. KEAP1 E3 ligase-mediated downregulation of NF-kappaB signaling by targeting IKKbeta. Mol Cell 2009; 36:131-40; PMID:19818716; http://dx.doi.org/ 10.1016/j.molcel.2009.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Udeshi ND, Svinkina T, Mertins P, Kuhn E, Mani DR, Qiao JW, Carr SA. Refined preparation and use of anti-diglycine remnant (K-epsilon-GG) antibody enables routine quantification of 10000s of ubiquitination sites in single proteomics experiments. Mol Cell Proteomics 2013; 12:825-31; PMID:23266961; http://dx.doi.org/ 10.1074/mcp.O112.027094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang C, Deng L, Hong M, Akkaraju GR, Inoue J, Chen ZJ. TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001; 412:346-51; PMID:11460167; http://dx.doi.org/ 10.1038/35085597 [DOI] [PubMed] [Google Scholar]
- 53. Lamb A, Chen J, Blanke SR, Chen LF. Helicobacter pylori activates NF-kappaB by inducing Ubc13-mediated ubiquitination of lysine 158 of TAK1. J Cell Biochem 2013; 114:2284-92; PMID:23606331; http://dx.doi.org/ 10.1002/jcb.24573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liang L, Fan Y, Cheng J, Cheng D, Zhao Y, Cao B, Ma L, An L, Jia W, Su X, Yang J, Zhang H. TAK1 ubiquitination regulates doxorubicin-induced NF-kappaB activation. Cell Signal 2013; 25:247-54; PMID:22981905; http://dx.doi.org/ 10.1016/j.cellsig.2012.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. An L, Jia W, Yu Y, Zou N, Liang L, Zhao Y, Fan Y, Cheng J, Shi Z, Xu G, et al. Lys63-linked polyubiquitination of BRAF at lysine 578 is required for BRAF-mediated signaling. Sci Rep 2013; 3:2344; PMID:23907581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Webster MK, D'Avis PY, Robertson SC, Donoghue DJ. Profound ligand-independent kinase activation of fibroblast growth factor receptor 3 by the activation loop mutation responsible for a lethal skeletal dysplasia, thanatophoric dysplasia type II. Mol Cell Biol 1996; 16:4081-7; PMID:8754806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tavormina PL, Bellus GA, Webster MK, Bamshad MJ, Fraley AE, McIntosh I, Szabo J, Jiang W, Jabs EW, Wilcox WR, et al. A novel skeletal dysplasia with developmental delay and acanthosis nigricans is caused by a Lys650Met mutation in the fibroblast growth factor receptor 3 gene. Am J Hum Genet 1999; 64:722-31; PMID:10053006; http://dx.doi.org/ 10.1086/302275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Drafahl KA, McAndrew CW, Meyer AN, Haas M, Donoghue DJ. The receptor tyrosine kinase FGFR4 negatively regulates NFkappaB signaling. PLoS One 2010; 5:e14412; PMID:21203561; http://dx.doi.org/ 10.1371/journal.pone.0014412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Carrano AC, Dillin A, Hunter T. A Kruppel-like factor downstream of the E3 ligase WWP-1 mediates dietary-restriction-induced longevity in Caenorhabditis elegans. Nat Commun 2014; 5:3772; PMID:24805825; http://dx.doi.org/ 10.1038/ncomms4772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Meyer AN, McAndrew CW, Donoghue DJ. Nordihydroguaiaretic acid inhibits an activated fibroblast growth factor receptor 3 mutant and blocks downstream signaling in multiple myeloma cells. Cancer Res 2008; 68:7362-70; PMID:18794123; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-0575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jiao X, Wood LD, Lindman M, Jones S, Buckhaults P, Polyak K, Sukumar S, Carter H, Kim D, Karchin R, Sjoblom T. Somatic mutations in the Notch, NF-KB, PIK3CA, and Hedgehog pathways in human breast cancers. Genes Chromosomes Cancer 2012; 51:480-9; PMID:22302350; http://dx.doi.org/ 10.1002/gcc.21935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Stephens P, Edkins S, Davies H, Greenman C, Cox C, Hunter C, Bignell G, Teague J, Smith R, Stevens C, et al. A screen of the complete protein kinase gene family identifies diverse patterns of somatic mutations in human breast cancer. Nat Genet 2005; 37:590-2; PMID:15908952; http://dx.doi.org/ 10.1038/ng1571 [DOI] [PubMed] [Google Scholar]
- 63. Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006; 314:268-74; PMID:16959974; http://dx.doi.org/ 10.1126/science.1133427 [DOI] [PubMed] [Google Scholar]
- 64. Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, Shen D, Boca SM, Barber T, Ptak J, et al. The genomic landscapes of human breast and colorectal cancers. Science 2007; 318:1108-13; PMID:17932254; http://dx.doi.org/ 10.1126/science.1145720 [DOI] [PubMed] [Google Scholar]
- 65. Cancer Genome Atlas Research Network . Integrated genomic analyses of ovarian carcinoma. Nature 2011; 474:609-15; PMID:21720365; http://dx.doi.org/ 10.1038/nature10166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Seshagiri S, Stawiski EW, Durinck S, Modrusan Z, Storm EE, Conboy CB, Chaudhuri S, Guan Y, Janakiraman V, Jaiswal BS, et al. Recurrent R-spondin fusions in colon cancer. Nature 2012; 488:660-4; PMID:22895193; http://dx.doi.org/ 10.1038/nature11282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, Yue P, Haverty PM, Bourgon R, Zheng J, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature 2010; 466:869-73; PMID:20668451; http://dx.doi.org/ 10.1038/nature09208 [DOI] [PubMed] [Google Scholar]
- 68. Dulak AM, Stojanov P, Peng S, Lawrence MS, Fox C, Stewart C, Bandla S, Imamura Y, Schumacher SE, Shefler E, et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat Genet 2013; 45:478-86; PMID:23525077; http://dx.doi.org/ 10.1038/ng.2591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bettegowda C, Agrawal N, Jiao Y, Wang Y, Wood LD, Rodriguez FJ, Hruban RH, Gallia GL, Binder ZA, Riggins CJ, et al. Exomic sequencing of four rare central nervous system tumor types. Oncotarget 2013; 4:572-83; PMID:23592488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Guo G, Sun X, Chen C, Wu S, Huang P, Li Z, Dean M, Huang Y, Jia W, Zhou Q, Tang A, et al. Whole-genome and whole-exome sequencing of bladder cancer identifies frequent alterations in genes involved in sister chromatid cohesion and segregation. Nat Genet 2013; 45:1459-63; PMID:24121792; http://dx.doi.org/ 10.1038/ng.2798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sato Y, Yoshizato T, Shiraishi Y, Maekawa S, Okuno Y, Kamura T, Shimamura T, Sato-Otsubo A, Nagae G, Suzuki H, et al. Integrated molecular analysis of clear-cell renal cell carcinoma. Nat Genet 2013; 45:860-7; PMID:23797736; http://dx.doi.org/ 10.1038/ng.2699 [DOI] [PubMed] [Google Scholar]
- 72. Tarpey PS, Behjati S, Cooke SL, Van Loo P, Wedge DC, Pillay N, Marshall J, O'Meara S, Davies H, Nik-Zainal S, et al. Frequent mutation of the major cartilage collagen gene COL2A1 in chondrosarcoma. Nat Genet 2013; 45:923-6; PMID:23770606; http://dx.doi.org/ 10.1038/ng.2668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Cancer Genome Atlas Research Network . Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012; 487:330-7; PMID:22810696; http://dx.doi.org/ 10.1038/nature11252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, Bergbower EA, Guan Y, Shin J, Guillory J, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet 2012; 44:1111-6; PMID:22941189; http://dx.doi.org/ 10.1038/ng.2405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zang ZJ, Ong CK, Cutcutache I, Yu W, Zhang SL, Huang D, Ler LD, Dykema K, Gan A, Tao J, et al. Genetic and structural variation in the gastric cancer kinome revealed through targeted deep sequencing. Cancer Res 2011; 71:29-39; PMID:21097718; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-1749 [DOI] [PMC free article] [PubMed] [Google Scholar]