

Abstract
Apoptosis- and proliferation-effector genes are substantially regulated by the same transactivators, with E2F-1 and Oct-1 being notable examples. The larger proliferation-effector genes have more binding sites for the transactivators that regulate both sets of genes, and proliferation-effector genes have more regions of active chromatin, i.e, DNase I hypersensitive and histone 3, lysine-4 trimethylation sites. Thus, the size differences between the 2 classes of genes suggest a transcriptional regulation paradigm whereby the accumulation of transcription factors that regulate both sets of genes, merely as an aspect of stochastic behavior, accumulate first on the larger proliferation-effector gene “traps,” and then accumulate on the apoptosis effector genes, thereby effecting sequential activation of the 2 different gene sets. As IRF-1 and p53 levels increase, tumor suppressor proteins are first activated, followed by the activation of apoptosis-effector genes, for example during S-phase pausing for DNA repair. Tumor suppressor genes are larger than apoptosis-effector genes and have more IRF-1 and p53 binding sites, thereby likewise suggesting a paradigm for transcription sequencing based on stochastic interactions of transcription factors with different gene classes. In this report, using the ENCODE database, we determined that tumor suppressor genes have a greater number of open chromatin regions and histone 3 lysine-4 trimethylation sites, consistent with the idea that a larger gene size can facilitate earlier transcriptional activation via the inclusion of more transactivator binding sites.
Keywords: apoptosis, bioinformatics, chromatin, cell cycle arrest, gene size, genomics, transcription factors
Introduction
Several studies have noted that apoptosis-effector genes are relatively small, with early work suggesting that small size represented a small target for DNA damage.1 Thus, apoptosis genes would be less likely to be damaged under circumstances that would require apoptosis. Apoptosis-effector genes are, on average, smaller than proliferation-effector genes.2,3 This contrast in gene size led to the hypothesis that the extra gene “space” could have a functional relevance and be populated by more transcription factor binding sites, which is indeed the case.2,3 This knowledge provided a key insight into a novel paradigm for explaining a long-time conundrum, how a molecular-logic dictates that both proliferation genes and apoptosis-effector genes could be regulated by the same set of transcription factors.
The dichotomous effects of shared transcription factors have been dramatic in several experimental models. For example, E2F-1 knockout mice, as observed decades ago, have tumors,4-6 despite the fact that E2F-1 clearly stimulates the transcription of many genes required for progression through S-phase, such as the dehydrofolate reductase and histone genes.7,8 The E2F-1 knock out mouse results are consistent with retinoblastoma protein (Rb) mediated inhibition of apoptosis,9 as Rb also inhibits E2F-1 function as a transcriptional activator.10 NF-kappaB is important for S-phase in the Hodgkin's disease, Reed-Sternbeg cells11,12 but leads to apoptosis in the closely related anaplastic lymphoma blast cells.13,14 Recently, we reported the pro-proliferative effects of low levels of IFN-γ, and the pro-apoptotic effects of high levels of IFN-γ, with both phenomena demonstrating an Oct-1 dependence,15 consistent with a feed-forward mechanism of apoptosis, which would be dictated by the distinct sizes of the proliferation- and apoptosis-effector genes coupled with a stochastic aspect to the loading of Oct-1 on different classes of genes.
Thus, in previous reports, we have determined that proliferation-effector genes have more open chromatin regions, and more regions with histone 3 (H3) lysine-4 (K04) pro-transcription, trimethylation sites, than do apoptosis-effector genes,2,3 again consistent with the idea that a larger gene size provides for more functionality, and in particular, represents a larger trap for transcriptional activators.
We have noted that both IRF-1 and p53 activate both tumor suppressor and apoptosis-effector genes; and have noted that tumor suppressor genes are larger and contain more of these transcriptional activator binding sites than do apoptosis-effector genes.2 The average size of the tumor suppressor genes used in ref.2 for that determination is 114,360 base pairs, while the average size of the apoptosis-effector genes used in that analysis is 31,256 base pairs. These data are also consistent with a feed forward mechanism, whereby increases in IRF-1 and p53 that accompany the halting of S-phase, for DNA repair, can eventually lead to apoptosis via “over-accumulation” and eventual population of apoptosis-effector genes.
In this report, we show that the larger tumor suppressor genes have more regions of active chromatin than the smaller apoptosis genes, as indicated by regions of open chromatin and H3 K04 trimethylation.
Results
We obtained the number and intensity of the DNase I HS sites from the ENCODE database, for multiple cell lines, for a set of tumor suppressor and apoptosis-effector genes (Garcia et al SOM gene list). These data were plotted as the average number regions per gene as a function of a minimum, average signal intensity for each of the sets, respectively (Methods, SOM). That is, as the average signal intensities for the gene-sets, respectively, increase in value along the X-axis, the average number of sites per gene decreases until genes have zero sites at the higher signal intensities and therefore drop out of the plot. These data and the plots are present in the SOM for all cell lines from ENCODE for which the analysis could be performed. An example result is shown in Figure 1A, i.e., for the T47D cell line, with a difference plot shown in Figure 1B.
Figure 1.
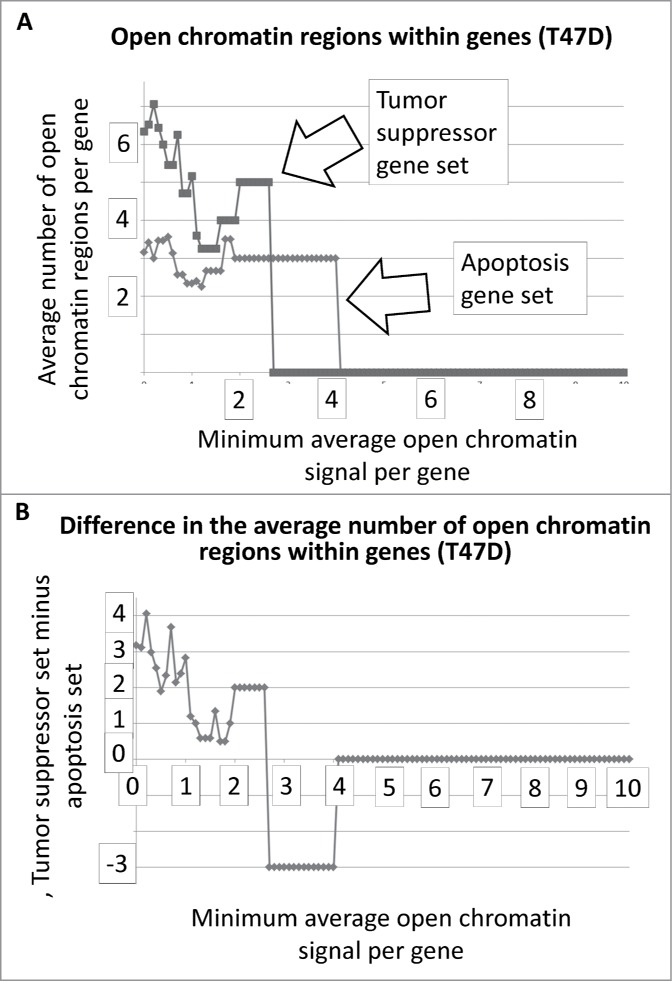
Average number of open chromatin regions within tumor suppressor and apoptosis-effector genes as a function of the minimum average signal intensity for each gene set. The T47D cell line is shown as an example. As the minimum average signal intensity increases, genes are removed from the calculation of the average number of open chromatin regions. The Perl (version 5) code for this analysis is in the SOM of refs2,3. The outputted Excel files for all ENCODE cell lines available for this analysis are in the SOM.
The results for Fig. 1A and B represent DNAse I HS sites within the genes represented by the 2 gene sets. In the case of T47D, the tumor suppressor genes have, on average, more DNase I HS sites within the genes at the lower signal intensities. The same result holds true when the DNase I HS sites within 5000 base pairs on either side of the each of the genes, in both sets, are included in the analyses (Fig. 2A, B). The Student's t-test p-values for the low-level signals, for Figs. 1 and 2 and for Figs. 3 and 4, are in Table 1.
Figure 2.
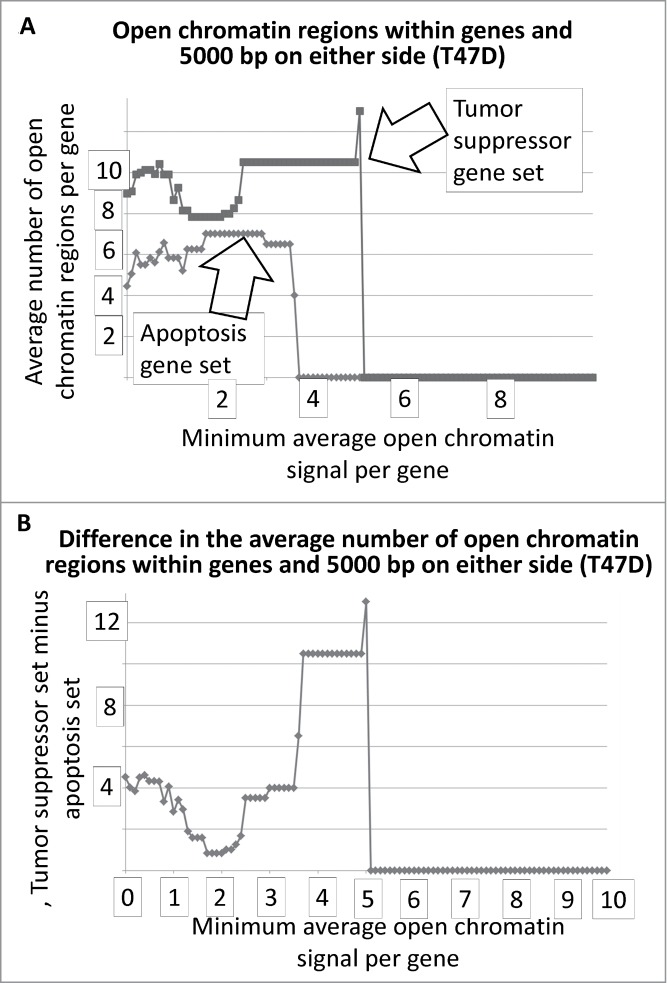
Average number of open chromatin regions within tumor suppressor and apoptosis-effector genes, and within 5000 bp on either side, as a function of the minimum, average signal intensity for each gene set. The T47D cell line is shown as an example.
Figure 3.

Average number of H3 K04me3 regions within tumor suppressor and apoptosis-effector genes as a function of the minimum average signal intensity for each gene set. The A549 cell line is shown as an example.
Figure 4.

Average number of H3 K04me3 regions within tumor suppressor and apoptosis-effector genes, and within 5000 bp on either side, as a function of the minimum average signal intensity for each gene set. The A549 cell line is shown as an example.
Table 1.
Student's t-test p-values for Figs. 1–4, for minimum signal intensity values below 1.0, in each plot. (NS = not significant; p-value calculations are present in the Excel files, represented by each figure, in the SOM)
| Figure | p-value |
|---|---|
| 1A | 9.30 E-08 |
| 2A | 2.43 E-12 |
| 3A | 0.026 |
| 3C | NS |
| 4A | 3.66 E-09 |
| 4C | 0.0002 |
To determine whether there was a similar indication of more active chromatin regions, per gene, among the tumor suppressor gene set, using a different approach, we determined the average number of H3 K04me3 regions, as a function of minimum average signal intensity, for the tumor suppressor and apoptosis-effector gene sets. Results indicated that indeed, a greater number of H3 K04me3 methylation regions were present in the tumor suppressor gene sets (Fig. 3A–D, as one example cell line; SOM). This result also held true when 5000 bp on either side of each gene was included in the analyses (Fig. 4A–D, as one example cell line; SOM).
To provide an indication of the overall trend for all ENCODE cell lines analyzed, the minimum signal intensity and moderate signal intensity results for the DNase I HS sites, for the entire collection of difference plots, representing the difference in signal intensities for the tumor suppressor gene and apoptosis-effector gene sets, were plotted as histograms (SOM: Garcia et al chromatin histograms, Figs. 5 and 6). For the data representing both the DNase I HS sites within genes, and the DNase I HS sites within genes and within 5000 bp on either side of the gene, there is an indication that the majority of cell lines available for this analysis have more DNase I HS sites per tumor suppressor gene that per apoptosis-effector gene, regardless of whether the DNase HS sites are tabulated solely within genes (Fig. 5A, B) or within genes plus 5000 bp on either side of the genes (Fig. 5C, D).
Figure 5.

Histogram tally of the differences between the average number of open chromatin regions for tumor suppressor and apoptosis-effector genes. These values were taken from the J2 and J52 cells of the Excel files, present in the SOM, representing each available ENCODE cell line. For further details, see Methods.
Figure 6.

Histogram tally of the differences between the average number of H3 K04me3 regions for tumor suppressor and apoptosis-effector genes. These values were taken from the J2 and J52 cells of the Excel files, present in the SOM, representing each available ENCODE cell line.
We next performed an analogous tabulation of the H3 K04me3 data, with results indicating that the majority of ENCODE cell lines available for this analysis trended toward more H3 K04me3 sites per gene among the tumor suppressor gene set.
Discussion
Overall, the above data are consistent with the conclusion that, on average, the tumor suppressor genes in the indicated set have more active chromatin regions than do the apoptosis-effector genes, for the data available from the ENCODE dataset, particularly for the lower and moderate signal intensities for these 2 chromatin parameters. As noted previously, the average size of the largest, primary transcripts for the tumor suppressor genes used in the above analyses is about 3.7 times the average size of the largest primary transcripts for the apoptosis-effector genes. Also as noted previously, the tumor suppressor genes have more IRF-1 and p53 binding sites than do the apoptosis-effector genes. While the regions of active chromatin above are not aligned with any particular transcriptional activator binding sites, it is clear that, in the case of the gene sets indicated, a large gene size correlates with more regions of active chromatin, i.e., more functional regions. Because these extra regions, in the longer genes, are most obvious at the lower signal intensities, it is possible that these extra regions essentially function as “feeder regions” for more extensive, assembled, pro-transcription chromatin complexes. Alternatively, a similar result may hold for chromatin regions with greater intensity but the analyses presented may not be sufficient to make this determination.
The above data raise the question, does a long gene size represent a larger, potential trap for transcriptional activators, where the gene is accessible to transcriptional activators, i.e., not in a globally, heterochromatic state? And if so, does gene size dictate a process of sequential activation, whereby large transcriptional activator traps are transcribed before small traps, when genes are competing for the same transcription factors? As noted above, there are numerous examples of transcription factors that activate both proliferation- and apoptosis-effector genes whereby a feed forward mechanism of apoptosis, dependent on very high levels of these shared transcriptional activators has been indicated. Several examples, include over-activation of the T-cell receptor in deletion of self-reactive T-cells;16 high level IFN-γ treatments;9 and even high-dose estrogen therapy used at one time for breast cancer treatments.17 However, in the case of a transition from cell cycle arrest, the data regarding basic phenomena is sparse. The data in this report thus justify more specific questions regarding both the basic phenomena and a possible mechanistic feature of transitioning from cell cycle arrest to apoptosis, namely a temporal gradient of increase of transcription factors that activate cell cycle arrest and apoptosis genes.
Methods
The approaches used in this article have been extensively described in refs.2,3. In addition, the supporting online material (SOM) for refs.2,3 has original, Perl (version 5) script used for mining the ENCODE database. In refs.,2,3 the open chromatin region and H3 K04me3 assessments were done with proliferation- and apoptosis-effector genes. In this report, the tumor suppressor genes2 (SOM) were substituted for the proliferation-effector genes.
The SOM for this article contains the output for the mining of the ENCODE databases, (using the above referenced computer script) in Excel files. To verify the output of the ENCODE data-mining for one case, the following steps can be performed: (i) Note the data of Fig. 3C (in this article), “H3 K04me3 modifications within genes (A549, Rep 2);” (ii) open the Excel file for Fig. 3, A549, Rep 2, attached to the PDF for “Garcia et al SOM for article Fig 3;” (iii) note column I, entitled “Tsupp Genes,” which represents the number of genes remaining in the plot as the minimum signal intensity increases along the X axis in Fig. 3C; (iv) on line 75 of the Excel file, one tumor suppressor gene remains in the plot, with a “Tsupp Avg Sig” (column F) of 8.3859629853369; (v) copy and paste this number into the “Find” function of the SOM Excel file, “A549 whole genome H3K04me3 Rep 2 output (example);” the Find function will thus locate line 6244 of the Excel file; (vi) copy and paste the transcript identifier of line 6244 (uc002itj.3) into the search field of the Genome Browser at genome.ucsd.edu, locating the NME1-NME2 primary transcript; (vii) Under “Regulation,” click on “ENC histone,” toggle the Genome Browser functions to display “Histone Modifications by ChIP-seq from ENCODE/University of Washington” and the A549 (Tier 2) cell line, for H3K04me3; (viii) note the display for “Sg 2” in the PDF labeled “Garcia et al SOM Fig S1,” which is a replica of the Genome Browser display; (ix) note the 8 gray boxes for this display, and verify the count of 8 “regions” under “Tsupp Avg Reg” (column G) in the SOM Excel file, “H3 K04me3 modifications within genes (A549, Rep 2)” (Note: column G represents the Y axis of the plot of Fig. 3C in the article; thus at the point in the X-axis of Fig. 3C where one gene remains, the Y-axis indicates a value of “8,” representing number of H3 K04me3 regions for that, single gene remaining in the plot); (x) click on each of the 8 gray boxes, in the live Genome Browser web page, representing 8 regions of H3K04me3 modification (the resulting web page will indicate the signal value and size in base pairs for each of the 8 regions); locate these signal values in the PDF, “Garcia et al SOM Table S1;” (xi) follow the column headings of Table S1 for the process of establishing an average signal value for each gene. This gene-specific, average value is used to determine whether a gene should be dropped from the plot as the X-axis value increases.
To generate Figs. 5 and 6, the values the representing the average number of open chromatin (Fig. 5) or H3 K04me3 (Fig. 6) regions per gene, in each set, at the lowest possible signal value level were collected for all ENCODE cell lines used in the indicated analyses, using a previously described macro.3 These values are located in cell J2 in every Excel file attached to the PDFs entitled “Garcia et al. SOM for article Figs. 1–4” (there are 4 separate PDFs, one for each figure, 1–4.) In addition, the analogous values were collected from cell J52 in the same set of Excel files. The J52 values represent the average number of open chromatin (Fig. 5) or H3 K04me3 (Fig. 6) regions per gene, in each set, at the moderate (middle) signal intensity value level. The values for all of the J2 and J52 cells from all of the indicated Excel files are collected in the Excel file entitled “Garcia et al chromatin histograms Figs. 5, 6” that is attached to a PDF of the same name. The final number of ENCODE cell lines available for each analyses (open chromatin and H3 K04me3; i.e., the Figs. 1–4 analyses) is due to various technical matters regarding the ENCODE database or the processing steps in the code.2,3 There was no pre-selection of cell lines or data in the course of generating Figs. 5 and 6.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.McKay BC, Stubbert LJ, Fowler CC, Smith JM, Cardamore RA, Spronck JC. Regulation of ultraviolet light-induced gene expression by gene size. Proc Natl Acad Sci U S A. 2004;101(17):6582-6; PMID:15087501; http://dx.doi.org/ 10.1073/pnas.0308181101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mauro JA, Blanck G. Functionally distinct gene classes as bigger or smaller transcription factor traps: a possible stochastic component to sequential gene expression programs in cancer. Gene 2014;536(2):398-406; PMID:24291030; http://dx.doi.org/ 10.1016/j.gene.2013.11.013. [DOI] [PubMed] [Google Scholar]
- 3.Mauro JA, Butler S, Ramsamooj M, Blanck G. Copy number loss or silencing of apoptosis-effector genes in cancer. Gene 2014; 554(1):50-57; PMID: 25307873; http://dx.doi.org/ 10.1016/j.gene.2014.10.021. [DOI] [PubMed] [Google Scholar]
- 4.Field SJ, Tsai FY, Kuo F, Zubiaga AM, Kaelin WG Jr., Livingston DM, Orkin SH, Greenberg ME.. E2F-1 functions in mice to promote apoptosis and suppress proliferation. Cell 1996;85(4):549-61; PMID:8653790; http://dx.doi.org/ 10.1016/S0092-8674(00)81255-6. [DOI] [PubMed] [Google Scholar]
- 5.Yamasaki L, Jacks T, Bronson R, Goillot E, Harlow E, Dyson NJ. Tumor induction and tissue atrophy in mice lacking E2F-1. Cell 1996; 85(4):537-48; PMID:8653789; http://dx.doi.org/ 10.1016/S0092-8674(00)81254-4. [DOI] [PubMed] [Google Scholar]
- 6.Wu X, Levine AJ. p53 and E2F-1 cooperate to mediate apoptosis. Proc Natl Acad Sci U S A. 1994; 91(9):3602-6; PMID:8170954; http://dx.doi.org/ 10.1073/pnas.91.9.3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oswald F, Dobner T, Lipp M. The E2F transcription factor activates a replication-dependent human H2A gene in early S phase of the cell cycle. Mol Cell Biol. 1996;16(5):1889-95; PMID:8628255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Slansky JE, Li Y, Kaelin WG, Farnham PJ. A protein synthesis-dependent increase in E2F1 mRNA correlates with growth regulation of the dihydrofolate reductase promoter. Mol Cell Biol. 1993;13(3):1610-8; PMID:8441401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berry DE, Lu Y, Schmidt B, Fallon PG, O'Connell C, Hu SX, Xu HJ, Blanck G. Retinoblastoma protein inhibits IFN-gamma induced apoptosis. Oncogene 1996;12(8):1809-19; PMID:8622902. [PubMed] [Google Scholar]
- 10.Kirch HC, Putzer B, Schwabe G, Gnauck HK, Schulte Holthausen H. Regulation of adenovirus 12 E1A transcription: E2F and ATF motifs in the E1A promoter bind nuclear protein complexes including E2F1, DP-1, cyclin A and/or RB and mediate transcriptional (auto)activation. Cell Mol Biol Res. 1993; 39(8):705-16; PMID:7951410. [PubMed] [Google Scholar]
- 11.Gruss HJ, Brach MA, Drexler HG, Bonifer R, Mertelsmann RH, Herrmann F. Expression of cytokine genes, cytokine receptor genes, and transcription factors in cultured Hodgkin and Reed-Sternberg cells. Cancer Res 1992;52(12):3353-60; PMID:1596893. [PubMed] [Google Scholar]
- 12.Bargou RC, Emmerich F, Krappmann D, Bommert K, Mapara MY, Arnold W, Royer HD, Grinstein E, Greiner A, Scheidereit C, Dorken B. Constitutive nuclear factor-kappaB-RelA activation is required for proliferation and survival of Hodgkin's disease tumor cells. J Clin Invest. 1997;100(12):2961-9; PMID:9399941; http://dx.doi.org/ 10.1172/JCI119849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bargou RC, Leng C, Krappmann D, Emmerich F, Mapara MY, Bommert K, Royer HD, Scheidereit C, Dorken B. High-level nuclear NF-kappa B and Oct-2 is a common feature of cultured Hodgkin/Reed-Sternberg cells. Blood 1996;87(10):4340-7; PMID:8639794. [PubMed] [Google Scholar]
- 14.Hirsch B, Hummel M, Bentink S, Fouladi F, Spang R, Zollinger R, Stein H, Durkop H. CD30-induced signaling is absent in Hodgkin's cells but present in anaplastic large cell lymphoma cells. Am J Pathol. 2008;172(2):510-20; PMID:18187570; http://dx.doi.org/ 10.2353/ajpath.2008.070858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szekeres K, Koul R, Mauro J, Lloyd M, Johnson J, Blanck G. An Oct-1-based, feed-forward mechanism of apoptosis inhibited by co-culture with Raji B-cells: towards a model of the cancer cell/B-cell microenvironment. Exp Mol Pathol. 2014;97(3):585-9; PMID:25236570; http://dx.doi.org/ 10.1016/j.yexmp.2014.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carreno LJ, Gonzalez PA, Kalergis AM. Modulation of T cell function by TCR/pMHC binding kinetics. Immunobiology 2006;211(1-2):47-64; PMID:16446170; http://dx.doi.org/ 10.1016/j.imbio.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 17.Stoll BA. Hypothesis: breast cancer regression under oestrogen therapy. Br Med J 1973;3(5877):446-50; PMID:4353621; http://dx.doi.org/ 10.1136/bmj.3.5877.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.