

Heart injuries such as those induced by acute ischemia can lead to heart failure, the most common cardiac ailment and a serious health problem worldwide. This occurs mainly due to the inability of the mammalian heart to regenerate after injury. Developing strategies to boost cardiac regeneration processes in humans is therefore clinically imperative.
During embryonic development, cardiac muscle cells (cardiomyocytes) actively proliferate to make a functional heart. The growth factor Neuregulin-1 (NRG1) and its tyrosine kinase receptors, ERBB4 and ERBB2, are essential for heart development. In the first days after birth, mouse neonates are able to regenerate their hearts following injury, through proliferation of pre-existing cardiomyocytes.1 However, this regenerative window is closed after one week,1 corresponding to the time at which most cardiomyocytes exit the cell cycle.2 After that, cardiomyocytes continue to grow mainly by hypertrophy, i.e. increase in cell size, and, in response to injury, fibrotic scar formation predominates over tissue replacement. The very low proliferative rate of adult cardiomyocytes in addition to the absence of well-defined cardiac stem cells in the adult heart account for the poor cardiac regenerative response seen in adult mammals.
Recent studies demonstrate that NRG1 administration improves cardiac function in injured mice and in heart failure patients. The beneficial effects of NRG1 can be due to increased cardiomyocyte survival, stress resistance, improved contractility, enhanced angiogenesis, and cardiomyocyte proliferation,3 although the latter remains controversial.4 Whether NRG1 could boost cardiomyocyte proliferation and heart regeneration is a critical issue with implications for clinical therapeutic treatments of heart diseases.
Recently, we characterized the impact of NRG1-ERBB2 signaling in cardiomyocyte proliferation and heart regeneration(Fig. 1).5 We first observed that NRG1 can stimulate cardiomyocyte proliferation at birth, but that this activity declines after the first week of post-natal life in mice.5 This observation is consistent with another study showing that the ability of NRG1 to induce heart regeneration following cryoinjury in mice and the proliferation of human cardiomyocytes (derived from biopsies of congenital heart disease babies) rapidly declines in early post-natal life (the first week in the mouse and within 6 months in humans, respectively).6 Importantly, we revealed that declining ERBB2 (but not ERBB4) levels shortly after birth are the limiting factor, the bottleneck, for NRG1-induced proliferative response of cardiomyocytes (Fig. 1).5 Our findings establish that ERBB2 is necessary for NRG1-induced proliferation in neonatal cardiomyocytes.5 Likewise in zebrafish, Nrg1 activation promotes cardiomyocyte proliferation and cardiomegaly demonstrating that heart regeneration in fish is dependent on Erbb2 activity.7
Post-natal induction of constitutively active ERBB2 resulted in cardiomegaly, a pronounced increase in heart size.5 Analysis of the underlying mechanisms demonstrated that ERBB2 signaling promoted cardiomyocyte dedifferentiation, proliferation, and hypertrophy. Dedifferentiation was characterized by disassembly of the sarcomeric architecture, replacement with immature components (α-SMA) and increased expression of stem/progenitor cell markers Runx1, DAB2 and cKIT.5 Therefore, ERBB2 signaling reverts cardiomyocytes to a lessdifferentiated state, allowing them to proliferate again. In line with the observed dedifferentiation phenotype, ERBB2 activation promoted proliferation of mono and bi-nucleated cardiomyocytes,5 the latter thought to be incapable of cell division.
We next demonstrated that transient activation of ERBB2 for 10–20 days after ischemic injury, either in juvenile or adult hearts, triggered a series of events starting with cardiomyocyte dedifferentiation, proliferation, neovascularization and, after ERBB2-signaling termination, proceeding to cardiomyocyte re-differentiation that together lead to anatomical and functional heart regeneration (Fig. 1).5 Taken together, our findings in mice suggest that low ERBB2 levels in the adult heart decrease NRG1-induced cardiomyocyte proliferation, therefore limiting postnatal cardiac regenerative plasticity. Thus, enhancing ERBB2 activity in adult cardiomyocytes represents a promising therapeutic approach for heart regeneration during heart failure. Future studies should focus on developing strategies to fine tune ERBB2 levels in cardiomyocytes in vivo for a short time to repair the damaged heart. Modulating this pathway within the infarcted heart could provide an optimal route for maximizing endogenous regenerative capacity by inducing cardiomyocyte proliferation and tissue replacement, reducing scarring, and improving cardiac function.
Finally, analysis of ERBB2 downstream pathways revealed a surprising uncou-pling between cardiomyocyte proliferation, dedifferentiation and hypertrophy, differentially dominated by ERK, AKT and GSK3β/β-catenin pathways.5 Therefore NRG1 treatment for cardiac regenerative purposes could be improved either by increasing ERBB2 levels in cardiomyocytes or by targeting ERBB2 specific downstream signaling molecules. In conclusion our findings point to a central role of ERBB2 signaling in cardiomyocyte cell division and heart regeneration. Understanding how ERBB2 signaling pathway promotes heart regeneration may point to new molecular targets and therapies enabling us to develop novel regenerative approaches to heart disease.
Figure 1.
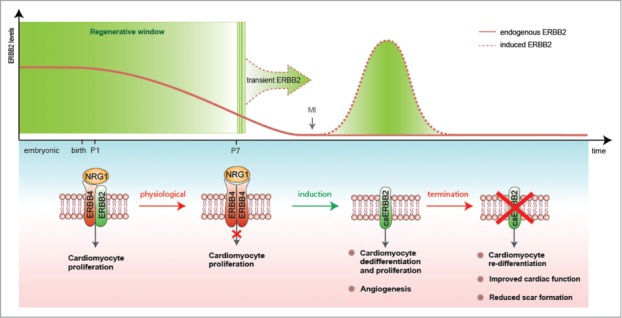
Transient activation of ERBB2 signaling as a strategy for heart regeneration. A schematic diagram of the role of ERBB2 in controlling post-natal cardiomyocyte proliferation and heart regeneration. ERBB2 levels diminish in cardiomyocytes during the first week after birth contributing to the cardiomyocyte cell cycle withdrawal. Transient induction of a constitutively active ERBB2 (caERBB2) in cardiomyocytes following myocardial infarction (MI) triggers cardiomyocyte dedifferentiation and proliferation as well as increased angiogenesis. Termination of the ERBB2 signaling facilitates cardiomyocyte re-differentiation leading to reduced scar formation and improved cardiac function.
References
- 1.Porrello ER, et al.. Science 2011; 331:1078-80; PMID:21350179; http://dx.doi.org/ 10.1126/science.1200708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Soonpaa MH, Field LJ. Circul Res 1998; 83:15-26; PMID:9670914; http://dx.doi.org/ 10.1161/01.RES.83.1.15 [DOI] [PubMed] [Google Scholar]
- 3.Bersell K, et al.. Cell 2009; 138:257-70; PMID:19632177; http://dx.doi.org/ 10.1016/j.cell.2009.04.060 [DOI] [PubMed] [Google Scholar]
- 4.Reuter S, et al.. PloS One 2014; 9:e115871; PMID:25545368; http://dx.doi.org/ 10.1371/journal.pone.0115871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.D'Uva G, et al.. Nat Cell Biol 2015; 17:627-38; PMID:25848746; http://dx.doi.org/ 10.1038/ncb3149 [DOI] [PubMed] [Google Scholar]
- 6.Polizzotti BD, et al.. Sci Transl Med 2015; 7:281ra45; PMID:25834111; http://dx.doi.org/ 10.1126/scitrans-lmed.aaa5171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gemberling M, et al.. Elife 2015; 4:e05871; PMID:25830562; http://dx.doi.org/ 10.7554/eLife.05871 [DOI] [PMC free article] [PubMed] [Google Scholar]