

Abstract
Cancer therapeutics that target a signaling pathway to which the cancer cells are addicted can deliver dramatic initial responses, but resistance is nearly always inevitable. A variety of mechanisms that cancer cells employ to escape from targeted cancer drugs have been described. We review here the role of Hepatocyte Growth Factor (HGF) and its receptor MET in drug resistance. We present data demonstrating that HGF can confer resistance to a number of kinase inhibitors in a variety of cancer cell lines and discuss our results in relation to the findings of others. Together, these data point at a major role for HGF/MET signaling in resistance to a variety of targeted cancer drugs.
Keywords: drug resistance, EMT, HGF, MET, targeted therapies
Introduction
In the last decade, cancer treatment has entered a new era with the introduction of therapeutics that specifically target oncogenic driver. Well-known examples include BCR-ABL (imatinib), EGFR (erlotinib, gefitinib), HER2 (trastuzumab) and BRAF (vemurafenib). These targeted therapies can lead to considerable inhibition of tumor growth by blocking important signaling pathways required for proliferation and/or survival. However, very often the tumor quickly becomes resistant to the therapy. It poses a formidable challenge to prolong the progression free survival of such patients. The molecular mechanisms of drug resistance have been studied extensively and have been reviewed equally extensively in the recent past.1-3 Understanding the underlying mechanism of resistance has already contributed to development of second-generation drug inhibitors that overcome resistance mechanisms that involve secondary mutations of the drug target. Here we focus on the role of Hepatocyte Growth Factor (HGF) and its receptor MET in resistance to targeted cancer drugs as this growth factor is emerging as an important factor in mediating resistance to these drugs.
A Short History of Hepatocyte Growth Factor
Hepatocyte growth factor, also known as Scatter Factor (SF), is a growth factor that was discovered as a mitogen for hepatocytes and other tissues.4,5 Roughly at the same time it was also found to be a protein secreted by fibroblasts that increased the motility of epithelial cells.6-8 In addition, HGF was recognized as the ligand for the MET tyrosine kinase receptor.9,10 Activation of the MET receptor by HGF can activate the MAPK and PI3-kinase/AKT pathways.
HGF/MET signaling plays an important role in many processes during embryogenesis. Proper development of the liver,11 placenta12, neurons13 and skeletal muscles of the limbs14 are just a few examples. In adult tissues, it is among others involved in wound healing15 and organ regeneration, including liver regeneration.16,17 In part, the importance of HGF in these processes lies in its ability to induce an Epithelial-to-Mesenchymal-Transition (EMT).18 This transformation of epithelial cells into cells with mesenchymal characteristics explains in part the increased capacity to migrate (hence the name ‘Scatter Factor’). In normal cells, EMT is vital for certain embryonic cells to enable them to migrate to other parts of the embryo where they subsequently can develop into new structures.19 HGF can induce EMT by inducing expression of SNAIL1 or SNAIL2 transcription factors via the MAPK pathway.20,21 The SNAIL transcription factors play a vital role in the execution of the EMT program.22
HGF Signaling in Drug Resistance
EMT also appears to play an important role in cancer progression. Cancer cells are thought to undergo EMT to acquire the migratory abilities necessary to invade and metastasize. Carcinoma cells that become more mesenchymal through EMT can invade surrounding tissue and eventually metastasize distantly. Also, EMT makes cancer cells less prone to apoptosis and senescence, which is associated with resistance to cancer therapies.19 Indeed, EMT has been implicated in resistance to several targeted therapies.23-26 Since HGF can induce an EMT as well as activate pro-survival pathways via its receptor MET, a link between HGF and resistance to targeted therapies can be readily made. For instance, activation of HGF or MET as well as amplification of MET have been shown to mediate resistance to EGFR Tyrosine Kinase Inhibitors (TKIs) in NSCLC.27-29 Recently, it was shown that HGF/MET signaling also plays a role in resistance to EGFR inhibition in colorectal cancer in vitro and in vivo.30,31 This was also described in a colorectal cancer stem cell-like population derived from primary human colon cancers transplanted in mice (referred to as xenopatients).32
In melanoma, stromal secretion of HGF has been associated with resistance to BRAF inhibitors.33 Interestingly, immunohistochemistry experiments showed that stromal cell expression of HGF in patients with BRAF mutant melanoma was correlated with innate resistance to BRAF inhibitor treatment. Wilson et al. found that increased pre-treatment plasma HGF levels were associated with worse outcome as measured by progression-free survival in the BRIM2 study of metastatic melanoma treated with BRAF inhibitor.34 Similarly, high pre-treatment HGF levels were associated with resistance to EGFR antibody therapy in metastatic colorectal cancer patients.35
These latter 3 studies highlight the potential of HGF to limit the effect of kinase inhibitors in patients and highlight the role of paracrine HGF/MET signaling in mediating resistance to multiple targeted therapies. More generally, these studies indicate that interactions between tumors and their microenvironment can play a pivotal role in drug resistance.
A gain-of-Function Screen Confirms Autocrine HGF Production as a Resistance Mechanism to Gefitinib in NSCLC
To study mechanisms of resistance to targeted cancer drugs, we performed an unbiased genetic screen to identify genes whose increased expression confers resistance to the EGFR inhibitor gefitinib in the NSCLC cell line PC9. This cell line carries a mutated EGFR gene and is consequently very sensitive to gefitinib. We used the lentiviral VBIM (Validation-Based Insertional Mutagenesis) vector system. This vector upon integration inserts the strong cytomegalovirus (CMV) promoter into the genome of mammalian cells. This method allows the identification of any protein or RNA whose overexpression confers a selectable phenotype through the use of 3 different reading frame vectors (SD1, SD2 and SD3).36
Three pools of 3×106 PC9 cells were infected with the 3 different VBIM reading frame vectors. After infection each pool was plated in 15 dishes at low density (150,000 cells per dish) in medium containing 400 nM gefitinib. The cells were cultured for 3 weeks to allow drug-resistant colonies to form. Uninfected PC9 cells were cultured in 400 nM gefitinib containing medium as a negative control (Fig. 1A). Drug-resistant clones were picked for further analysis. This yielded 8 clones for SD1, 0 for SD2 and 2 for SD3. PC9 parental cells did not yield any gefitinib resistant colonies (Fig. 1B).
Figure 1.

A gain-of-function screen to identify genes that mediate gefitinib resistance in NSCLC cell line PC9. (A) Setup of the screen. PC9 cells were infected with VBIM lentivirus, plated at low density in 400 nM gefitinib and gefitinib resistant colonies were picked after 3 weeks selection. Uninfected PC9 cells were used as negative control. (B) Gefitinib resistant colony SD1.1 after 3 weeks selection. Parental PC9 cells are sensitive to gefitinib and do not form colonies. (C) Conformation of the gefitinib resistant phenotype of clone SD1.1. PC9 parental cells are sensitive to 200, 400 and 800 nM gefitinib, whereas SD1.1 cells can grow in gefitinib containing medium. (D) Growth curves of PC9 parental cells that were either untreated (blue) or treated with 400 nM gefitinib (red). (E) Growth curves of PC9 SD1.1 cells that were either untreated or treated with 400 nM gefitinib. (F) Western blotting on lysates of PC9 parental and PC9 SD1.1 cells that were untreated or treated for 6 hours with 400 nM gefitinib. In PC9 parental cells MAPK and AKT signaling is abrogated upon gefitinib treatment, inhibiting proliferation. In SD1.1 cells, MAPK and AKT signaling is sustained after gefitinib treatment, resulting in continued proliferating. This is explained by overexpression of the HGF gene, which re-activates the MAPK and PI3K/AKT pathways by activating MET.
To identify the integration sites of the virus in drug-resistant cells, an inverse PCR was performed on the genomic DNA isolated from these resistant clones. We could only recover a PCR product for clone SD1.1. In this clone, the integration site was situated approximately ∼30 kb upstream of the HGF gene. To validate the drug resistant phenotype, clone SD1.1 was once more subjected to gefitinib treatment. Indeed, these cells were able to grow in gefitinib concentrations up to 800 nM (Fig. 1C). In addition, a growth curve shows the differential growth between PC9 parental and SD1.1 cells upon gefitinib treatment. Untreated PC9 parental cells grew to confluency, but upon treatment with 400 nM gefitinib their growth was largely inhibited (Fig. 1D). In contrast, SD1.1 cells were growing as fast in 400 nM gefitinib as the untreated cells (Fig. 1E). To evaluate pathway activity downstream of the EGF receptor and to confirm overexpression of HGF in clone SD1.1, western blotting was performed on cell lysates of PC9 parental and SD1.1 cells, either untreated or treated with gefitinib for 6 hours. Indeed, HGF was found to be overexpressed in clone SD1.1. MET receptor activity was evaluated through measurement of the phosphorylation levels of its tyrosines 1234 and 1235, which are critical for the kinase activation.37 In SD1.1 cells, the MET receptor showed higher phosphorylation than in the parental PC9 cells. Total MET levels seem slightly lower in SD1.1. This can be explained by increased internalization of the activated receptor. Assessment of signaling downstream of EGFR upon gefitinib treatment indicated phosphorylation of both ERK and AKT are inhibited in PC9 parental cells. However, in SD1.1 cells both ERK and AKT phosphorylation levels are hardly decreased after treatment, providing a biochemical explanation for the drug-resistant phenotype (Fig. 1F).
As described in the introduction, MET receptor gene amplification or activation of its ligand HGF have previously been described as mechanisms of resistance to gefitinib.27-29 With this unbiased genetic screen we confirm that endogenous upregulation of HGF activates MET and can indeed confer resistance to gefitinib. However, the pathways activated by HGF-MET signaling are potentially relevant in resistance to other targeted therapies as well. We therefore extended our analysis to the potential role of HGF signaling in resistance to other kinase inhibitors.
HGF Can Confer Resistance to Multiple Kinase Inhibitors
We set out to investigate the possible role of HGF in mediating resistance to targeted therapies beyond gefitinib as well as in cancer cells originating in other tissues than the lung. We selected 6 kinase inhibitors, each acting against different oncogenic driver proteins that interfere with signaling at different levels in signaling cascades. We used the second generation EGFR inhibitor afatinib, the monoclonal antibody against EGFR cetuximab, the ALK inhibitor TAE684, the MEK inhibitor selumetinib (AZD6244), the PDGFR/VEGFR inhibitor sunitinib and the BRAF inhibitor PLX4032. To assess the potential of HGF to confer resistance to these drugs we used a cell line panel with sensitivity to the respective drugs. NSCLC cell lines PC9, H1975 and H3122 are sensitive to gefitinib, afatinib and TAE684 respectively. Proliferation of colon cancer cell lines Difi and SKCO-1 is inhibited by cetuximab and AZD6244. 768O is a renal cancer cell line that responds to sunitinib and Mel888 is sensitive to BRAF inhibition by PLX4032. In addition, we evaluated the capacity of crizotinib, a MET inhibitor, to reverse drug resistance mediated by HGF.
We first asked whether autocrine HGF signaling could cause drug resistance in each of the 6 situations described above as it is known that autocrine production of growth factors and the subsequent activation of their receptors can lead to insensitivity to EGFR inhibitors. One such example of autocrine growth factor resistance is production of Fibroblast Growth Factors in NSCLC cells, rendering these cells resistant to gefitinib.38 Indeed, also autocrine HGF signaling can lead to resistance to gefitinib in NSCLC.39 To assess if autocrine HGF production can cause resistance to the drugs we selected, the cell line panel described above was infected with either the empty pBabePuro vector as a control or the same vector carrying the HGF cDNA. This mimics the situation in our insertional mutagenesis screen and reflects a process in which cancer cells create an autocrine loop by upregulating the endogenous expression of HGF.
Using protein gel blotting, we confirmed that HGF was overexpressed in the cells infected with the pBp-HGF vector, but not the cells infected with the empty vector. (Fig. 2A) Subsequently, we tested if the HGF overexpressing cell lines were resistant to the selected inhibitors. In long-term colony formation assays, HGF overexpression could indeed confer resistance to all drugs tested (Fig. 2B, second column of each colony formation panel). The MET inhibitor crizotinib was tested for its potential to reverse the HGF induced resistance. Where crizotinib alone had no effect on proliferation, it potently reversed the resistance in the HGF overexpressing cells in combination with the kinase inhibitors (Fig. 2B, third and fourth columns of each panel). An exception here was the EML4-ALK driven H3122 cell line, where crizotinib treatment alone inhibits proliferation, since crizotinib also inhibits the oncogenic driver in these cells: EML4-ALK. These experiments show that autocrine HGF expression can lead to resistance to 7 kinase inhibitors in different cell types and that blocking of the MET receptor reverts this drug resistance.
Figure 2.

Autocrine production of HGF induces resistance to multiple kinase inhibitors in a cancer cell line panel. (A) Western blot showing overexpression of HGF in pBp-HGF infected cells. pBp empty vector serves as control. (B) Overexpression of HGF confers resistance to 7 kinase inhibitors in a drug sensitive cell line panel. Control pBp empty vector infected cells remain drug sensitive. MET inhibitor crizotinib has no effect as a single drug (except in EML4-ALK driven H3122 cells, which crizotinib also inhibits), but reverts HGF induced drug resistance.
Next, we repeated the colony formations with the cell line panel, now using recombinant HGF to mimic HGF paracrine MET receptor signaling. This would reflect a situation in which cancer cells use HGF secreted by stroma cells or that is otherwise available in the tumor microenvironment. Wilson et al. tested a variety of recombinant growth factors, among which HGF, and found that they can induce resistance to a range of inhibitors.34 This shows that activation of several individual RTKs can be a potent mechanism to reactivate or bypass inhibited driver pathways and hence acquire drug resistance. We confirm their findings for HGF and extend these findings to include the kinase inhibitors afatinib, cetuximab and AZD6244. Our data indicate that recombinant HGF confers resistance to all kinase inhibitors tested in a concentration dependent fashion (Fig. S1, upper rows). Higher concentrations of HGF almost completely nullify the effect of the inhibitors. However, as expected, combination with crizotinib reverses the resistance effectively (Fig. S1, lower rows).
Tumor cells interact actively and intimately with their surrounding stroma cells. This can potentially affect the response of tumor cells to drugs.40 For instance, stromal cells can produce growth factors and cytokines that could be exploited in a paracrine fashion by the tumor cells. HGF produced and secreted by the stroma cells is such a growth factor.
As a second model to study the effects of paracrine HGF signaling, we created HGF-overexpressing mouse NIH3T3 fibroblasts through retroviral infection with the pBp-HGF vector. This created NIH3T3 cells that produced HGF (Fig. S2B). These NIH3T3 cells were then co-cultured with the cells from the panel, following treatment of the NIH3T3 cells with mitomycin C to prevent their proliferation. Clearly, HGF secreted by the NIH3T3 cells was able to make the cancer cell lines resistant to all kinase inhibitor treatments, whereas combination treatment with crizotinib counteracted the HGF and resensitized the cells to the drugs (Fig. S2A). This shows that resistance to multiple kinase inhibitors can also be mediated by HGF secreted by cells in proximity of cancer cells or possibly by HGF that is made available by cells elsewhere in the body via the circulation.
All these data point toward HGF/MET signaling being a very powerful and general mechanism for cancer cells to evade kinase inhibitor treatment. Our data show that the source of the HGF appears to be relatively unimportant as, at least in vitro, both autocrine and paracrine signaling results in potent drug resistance.
HGF Induces Resistance to Kinase Inhibitors by (re)Activation of the MAPK and AKT Pathways
To evaluate the effects of HGF at a biochemical level, we performed western blotting on protein lysates taken from the cell line panel treated with one of the selected inhibitors, crizotinib, HGF or a combination thereof. All the inhibitors tested block MAPK and/or AKT signaling (in addition, sunitinib also inhibits STAT3 signaling in 786O cells), which was verified by decreased pERK and pAKT levels (and pSTAT3 in 786O cells) compared to the untreated control condition (Fig. 3, first and second columns in each protein gel blot panel). However, activation of the MET receptor with HGF in cells that were treated with drug resulted in (re)activation of the MAPK and AKT pathways, as pERK and pAKT levels increased (Fig. 3, sixth column). Treatment with crizotinib alone had hardly effect on either pERK or pAKT, except in H3122 as crizotinib potently inhibits the EML4-ALK oncoprotein present in these cells. However, crizotinib was able to block the increased phosphorylation of ERK and AKT induced by HGF, explaining the reversal of drug resistance.
Figure 3.
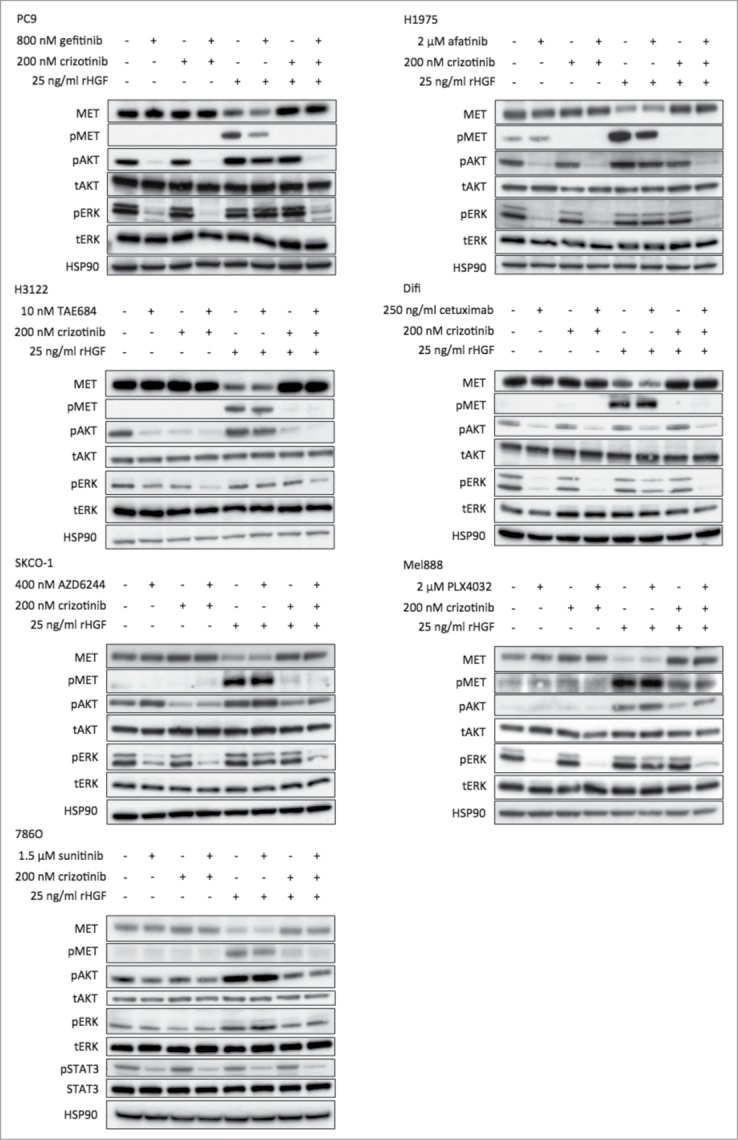
HGF (re-)activates MAPK and AKT signaling upon drug treatment. Western blotting on lysates of the cell line panel shows the effect on the MAPK and AKT signaling pathways after 6 hours of drug treatment with or without addition of HGF. Addition of HGF during drug treatment consistently results in (re-)activation of the MAPK and AKT pathways, evidenced by the increase in phosphorylated ERK and phosphorylated AKT. Crizotinib reversed this effect potently.
(Re-)activation of proliferation and pro-survival signaling pathways upon inhibition is a recurrent theme in drug resistance. Activation of RTK signaling plays an important role in such reactivation.41-43 We show that the HGF/MET axis can very potently reactivate initially inhibited ERK and AKT signaling upon treatment with a number of kinase inhibitors, which has also been shown by others for various kinase inhibitors.27,33,34 This makes it an attractive pathway for cancer cells to activate. Since many carcinoma cells express the MET receptor, its activation seems a common mechanism through which cancer cells can acquire drug resistance and could explain why this axis is involved in an increasing number of resistance mechanisms against a divergent series of drugs.
Conclusions and Future Perspectives
The findings reported here support and extend those described by others in showing that HGF/MET signaling can be a potent mediator of resistance to a diverse set of (tyrosine) kinase inhibitors in multiple tumor models via (re)activation of survival and proliferation pathways. We also show that simultaneous inhibition of MET and a kinase inhibitor reverses HGF driven resistance. Indeed, combination of the irreversible EGFR inhibitor afatinib with crizotinib has already been described to be effective against MET amplified and HGF overexpressing lung cancer cells both in vitro and in vivo.44 Therefore, in case of activation of the HGF/MET axis during kinase inhibitor treatment, simultaneous inhibition of MET could prove to be a viable strategy to overcome resistance and prolong therapeutic benefit for patients. Since HGF/MET signaling seems to be so potent in mediating resistance to many TKIs and is already found in a considerable proportion of EGFR TKI resistant tumors, it could be worthwhile to routinely check MET activation in patients with TKI resistant tumors. If so, exploring combination treatment with MET inhibitors could be a good option to treat such drug resistant tumors. Clinical trials exploring combination of crizotinib with another TKI have already started. One example is a phase I/II trial to combine erlotinib with crizotinib.45 Another phase 1b trial will try to enhance the effect of anti-angiogenic VEGF therapies, where HGF/MET signaling also seems to play a role in resistance, by co-administering crizotinib.46 However, these trials are currently only in safety and dosing assessment stages.
Apart from resistance to kinase inhibitors, there is evidence that EMT can also make cancer cells resistant to conventional chemotherapy. For instance, colon carcinoma cells made resistant to oxaliplatin exhibited features of EMT.47 Similarly, EMT was observed in ovarian carcinoma cells that were resistant to paclitaxel.48 In our own lab, TGFβ was shown to confer resistance to chemotherapy in colorectal carcinoma cells as well as to targeted therapies and this was accompanied by EMT.49,50 Because HGF has an ability to induce EMT, a role for HGF in chemoresistance is also plausible. Indeed, HGF has been implicated in chemoresistance.51-54 Targeting MET with inhibitors has recently been shown to revert chemoresistance in Small Cell Lung Cancer,55 showing the potential of MET inhibition beyond targeted therapies. Checking MET activity in chemoresistant tumors could therefore be an opening in attempting to resensitize such tumors to chemotherapy.
In conclusion, HGF/MET signaling has by now emerged as a factor with a seemingly extensive capability to make tumor cells resistant to many targeted and possibly conventional chemotherapies and should therefore be considered widely as an attractive drug target in combination strategies designed to overcome resistant tumors.
Materials and Methods
Cell culture and retroviral transduction
PC9, H1975, H3122, Difi, SKCO-1 and Mel888 cells were cultured in RPMI medium supplemented with 10% FBS, 1% glutamine and 1% penicillin/streptomycin. HEK293T, phoenix and 786O cells were cultured in high glucose Dulbecco's Modified Eagle Medium supplemented with 10% FBS, 1% glutamine and 1% penicillin/streptomycin. NIH3T3 cells were cultured in high glucose Dulbecco's modified Eagle medium supplemented with 10% NCS (newborn calf serum), 1% glutamine and 1% penicillin/streptomycin. All cell lines were cultured at 37°C and 5% CO2.
To perform the gefitinib resistance screen, VBIM-SD1, -SD2 and –SD3 lentivirus was produced by transfection of HEK293T cells using polyethylenimine (PEI). Lentivirus containing supernatant was used to infect 3 pools of 3 × 106 PC9 cells (1 pool for each VBIM-SD) in 3 consecutive rounds. Infection efficiency was assessed by GFP expression in the PC9 cells. Subsequently, cells were plated at low density (150,000 per 10 cm dish) in 400 nM gefitinib containing medium. For each VBIM-SD, 15 dishes were plated. 10 dishes of uninfected PC9 parental cells were plated as control, also at 150,000 cells per dish. After 3 weeks of drug selection resistant colonies were picked and expanded in drug-free medium for further analysis.
pBp or pBp-HGF retrovirus was produced by transfection of phoenix cells using PEI. Retrovirus containing supernatant was used to infect target cells in 3 consecutive rounds and target cells were then selected for successful infection with 2 μg/ml puromycin.
Colony Formations
Cells were plated in 6-well plates. Recombinant HGF and drugs were added as indicated the next day and refreshed every 3 days. Cells were grown until the well with untreated control cells was confluent. Cells were fixed in 3.7% paraformaldehyde and stained with 0.1% crystal violet.
The plating densities for recombinant HGF and pBp-HGF colony formations were 20,000 cells per well for PC9, H1975, H3122, 786O and Mel888 and 50,000 cells per well for Difi and SKCO-1.
NIH3T3 co-culturing experiments: NIH3T3 cells were plated 1 day prior to start of co-culturing at 50,000 cells per well. On the next day, the NIH3T3 cells were treated with 10 μg/ml mitomycin C in serum-free medium for 3 hours. Subsequently, the NIH3T3 cells were washed twice with PBS and the cell lines to be co-cultured were plated on top. Cells were then co-cultured in presence or absence of drug as indicated for 5 days and subsequently fixed and stained. Plating densities for the NIH3T3 co-culture colony formations: 50,000 cells per well for PC9, H1975, H3122, 786O and Mel888. 100,000 cells per well for Difi and 200,000 cells per well for SKCO-1.
IncuCyte
PC9 parental and SD1.1 cells were plated in triplicates in 384-well plate at 1500 cells/well and either untreated or treated with 400 nM gefitinib. The plate was incubated in the IncuCyte (Essen BioScience) and cells were allowed to grow to confluency. The IncuCyte measured and recorded confluency every 4 hours. This data was subsequently converted into growth curves.
Plasmids
Plasmids pBp and pBp-HGF plasmid were purchased from AddGene.
Reagents
Gefitinib (S1025), TAE684 (S1108), AZD6244 (S1008), PLX4032 (S1267), sunitinib (S1042) and crizotinib (S1068) were purchased from Selleckchem. Afatinib (SM-101000) from Alpha Diagnostic International and cetuximab was obtained from the pharmacy at The Netherlands Cancer Institute.
Human recombinant HGF (H9661) and mitomycin C (M4287) were purchased from Sigma-Aldrich.
Protein Lysate Preparation and Western Blotting
Cells were lysed in RIPA buffer containing 150 mM NaCl, 50 mM Tris pH 8.0, 1% NP-40, 0.5% sodium deoxycholate and 0.1% SDS supplemented with protease inhibitors (Complete, Roche) and phosphatase inhibitor cocktails II and III (Sigma). 10x reducing agent and 4x sample preparation buffer (NuPage) was added. Subsequently, the samples were boiled for 5 minutes and centrifuged at 14,000 rpm for 5 minutes. Equal amounts of protein were subjected to SDS gel electrophoresis using NuPage precast gels and MOPS buffer. This was followed by western blotting.
Antibodies
MET (8198) pMET (3077), AKT (2920), pAKT (4060), STAT3 (4904) and pSTAT3 (9145) antibodies were purchased from Cell Signaling. HSP90 (7947), ERK1 (93), ERK2 (154), pERK1/2 (7383) were purchased from Santa Cruz Biotechnologies. HGF (MA5-14160) was purchased from Thermo Scientific.
Inverse PCR
Genomic DNA was prepared from cells using DNAzol. gDNA was used for subsequent nested PCR reaction. Primers for nested PCR reaction 1: forward 5′-GTAAGACCACCGCACAGC-3′, reverse 5′-CCAGAGTCACACAACAGACG-3′. For nested PCR reaction 2: forward 5′-GATCTTCAGACCTGGAGGAG-3′, reverse 5′-CCAGAGAGACCCAGTACAAGC-3′. PCR products were purified with Qiagen Gel Extraction Kit (28706). PCR products were Sanger sequenced using nested PCR reaction 2 forward primer. The resulting sequences were aligned to the human genome using BLAST.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank the George Stark laboratory for providing us with the plasmids and protocols of the VBIM system.
Funding
This work was supported by a grant from the European Research Council (grant #250043).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Chong CR, Janne PA. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat Med 2013; 19:1389-400; PMID:24202392; http://dx.doi.org/ 10.1038/nm.3388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ramos P, Bentires-Alj M. Mechanism-based cancer therapy: resistance to therapy, therapy for resistance. Oncogene 2014; PMID:25263438 [DOI] [PubMed] [Google Scholar]
- 3. Groenendijk FH, Bernards R. Drug resistance to targeted therapies: deja vu all over again. Mol Oncol 2014; 8:1067-83; PMID:24910388; http://dx.doi.org/ 10.1016/j.molonc.2014.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kan M, Zhang GH, Zarnegar R, Michalopoulos G, Myoken Y, McKeehan WL, Stevens JI. Hepatocyte growth factor/hepatopoietin A stimulates the growth of rat kidney proximal tubule epithelial cells (RPTE), rat nonparenchymal liver cells, human melanoma cells, mouse keratinocytes and stimulates anchorage-independent growth of SV-40 transformed RPTE. Biochem Biophys Res Commun 1991; 174:331-7; PMID:1846541; http://dx.doi.org/ 10.1016/0006-291X(91)90524-B [DOI] [PubMed] [Google Scholar]
- 5. Rubin JS, Chan AM, Bottaro DP, Burgess WH, Taylor WG, Cech AC, Hirschfield DW, Wong J, Miki T, Finch PW, et al. A broad-spectrum human lung fibroblast-derived mitogen is a variant of hepatocyte growth factor. Proc Natl Acad Sci U S A 1991; 88:415-9; PMID:1824873; http://dx.doi.org/ 10.1073/pnas.88.2.415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stoker M, Gherardi E, Perryman M, Gray J. Scatter factor is a fibroblast-derived modulator of epithelial cell mobility. Nature 1987; 327:239-42; PMID:2952888; http://dx.doi.org/ 10.1038/327239a0 [DOI] [PubMed] [Google Scholar]
- 7. Rosen EM, Meromsky L, Setter E, Vinter DW, Goldberg ID. Purified scatter factor stimulates epithelial and vascular endothelial cell migration. Proc Soc Exp Biol Med 1990; 195:34-43; PMID:2144630; http://dx.doi.org/ 10.3181/00379727-195-43115 [DOI] [PubMed] [Google Scholar]
- 8. Weidner KM, Behrens J, Vandekerckhove J, Birchmeier W. Scatter factor: molecular characteristics and effect on the invasiveness of epithelial cells. J Cell Biol 1990; 111:2097-108; PMID:2146276; http://dx.doi.org/ 10.1083/jcb.111.5.2097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bottaro DP, Rubin JS, Faletto DL, Chan AM, Kmiecik TE, Vande Woude GF, Aaronson SA. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 1991; 251:802-4; PMID:1846706; http://dx.doi.org/ 10.1126/science.1846706 [DOI] [PubMed] [Google Scholar]
- 10. Naldini L, Vigna E, Narsimhan RP, Gaudino G, Zarnegar R, Michalopoulos GK, Comoglio PM. Hepatocyte growth factor (HGF) stimulates the tyrosine kinase activity of the receptor encoded by the proto-oncogene c-MET. Oncogene 1991; 6:501-4; PMID:1827664 [PubMed] [Google Scholar]
- 11. Schmidt C, Bladt F, Goedecke S, Brinkmann V, Zschiesche W, Sharpe M, Gherardi E, Birchmeier C. Scatter factor/hepatocyte growth factor is essential for liver development. Nature 1995; 373:699-702; PMID:7854452; http://dx.doi.org/ 10.1038/373699a0 [DOI] [PubMed] [Google Scholar]
- 12. Uehara Y, Minowa O, Mori C, Shiota K, Kuno J, Noda T, Kitamura N. Placental defect and embryonic lethality in mice lacking hepatocyte growth factor/scatter factor. Nature 1995; 373:702-5; PMID:7854453; http://dx.doi.org/ 10.1038/373702a0 [DOI] [PubMed] [Google Scholar]
- 13. Maina F, Hilton MC, Ponzetto C, Davies AM, Klein R. Met receptor signaling is required for sensory nerve development and HGF promotes axonal growth and survival of sensory neurons. Genes Dev 1997; 11:3341-50; PMID:9407027; http://dx.doi.org/ 10.1101/gad.11.24.3341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bladt F, Riethmacher D, Isenmann S, Aguzzi A, Birchmeier C. Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature 1995; 376:768-71; PMID:7651534; http://dx.doi.org/ 10.1038/376768a0 [DOI] [PubMed] [Google Scholar]
- 15. Chmielowiec J, Borowiak M, Morkel M, Stradal T, Munz B, Werner S, Wehland J, Birchmeier C, Birchmeier W. c-Met is essential for wound healing in the skin. J Cell Biol 2007; 177:151-62; PMID:17403932; http://dx.doi.org/ 10.1083/jcb.200701086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Matsumoto K, Nakamura T. Hepatocyte growth factor: molecular structure and implications for a central role in liver regeneration. J Gastroenterol Hepatol 1991; 6:509-19; PMID:1834243; http://dx.doi.org/ 10.1111/j.1440-1746.1991.tb00897.x [DOI] [PubMed] [Google Scholar]
- 17. Huh CG, Factor VM, Sanchez A, Uchida K, Conner EA, Thorgeirsson SS. Hepatocyte growth factor/c-met signaling pathway is required for efficient liver regeneration and repair. Proc Natl Acad Sci U S A 2004; 101:4477-82; PMID:15070743; http://dx.doi.org/ 10.1073/pnas.0306068101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stern CD, Ireland GW, Herrick SE, Gherardi E, Gray J, Perryman M, Stoker M. Epithelial scatter factor and development of the chick embryonic axis. Development 1990; 110:1271-84; PMID:2151613 [DOI] [PubMed] [Google Scholar]
- 19. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell 2009; 139:871-90; PMID:19945376; http://dx.doi.org/ 10.1016/j.cell.2009.11.007 [DOI] [PubMed] [Google Scholar]
- 20. Savagner P, Yamada KM, Thiery JP. The zinc-finger protein slug causes desmosome dissociation, an initial and necessary step for growth factor-induced epithelial-mesenchymal transition. J Cell Biol 1997; 137:1403-19; PMID:9182671; http://dx.doi.org/ 10.1083/jcb.137.6.1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Grotegut S, von Schweinitz D, Christofori G, Lehembre F. Hepatocyte growth factor induces cell scattering through MAPK/Egr-1-mediated upregulation of Snail. EMBO J 2006; 25:3534-45; PMID:16858414; http://dx.doi.org/ 10.1038/sj.emboj.7601213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2014; 15:178-96; PMID:24556840; http://dx.doi.org/ 10.1038/nrm3758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yauch RL, Januario T, Eberhard DA, Cavet G, Zhu W, Fu L, Pham TQ, Soriano R, Stinson J, Seshagiri S, et al. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin Cancer Res 2005; 11:8686-98; PMID:16361555; http://dx.doi.org/ 10.1158/1078-0432.CCR-05-1492 [DOI] [PubMed] [Google Scholar]
- 24. Chung JH, Rho JK, Xu X, Lee JS, Yoon HI, Lee CT, Choi YJ, Kim HR, Kim CH, Lee JC. Clinical and molecular evidences of epithelial to mesenchymal transition in acquired resistance to EGFR-TKIs. Lung Cancer 2011; 73:176-82; PMID:21168239; http://dx.doi.org/ 10.1016/j.lungcan.2010.11.011 [DOI] [PubMed] [Google Scholar]
- 25. Lesniak D, Sabri S, Xu Y, Graham K, Bhatnagar P, Suresh M, Abdulkarim B. Spontaneous epithelial-mesenchymal transition and resistance to HER-2-targeted therapies in HER-2-positive luminal breast cancer. PLoS One 2013; 8:e71987; PMID:23991019; http://dx.doi.org/ 10.1371/journal.pone.0071987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Byers LA, Diao L, Wang J, Saintigny P, Girard L, Peyton M, Shen L, Fan Y, Giri U, Tumula PK, et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin Cancer Res 2013; 19:279-90; PMID:23091115; http://dx.doi.org/ 10.1158/1078-0432.CCR-12-1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen J, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007; 316:1039-43; PMID:17463250; http://dx.doi.org/ 10.1126/science.1141478 [DOI] [PubMed] [Google Scholar]
- 28. Yano S, Wang W, Li Q, Matsumoto K, Sakurama H, Nakamura T, Ogino H, Kakiuchi S, Hanibuchi M, Nishioka Y, et al. Hepatocyte growth factor induces gefitinib resistance of lung adenocarcinoma with epidermal growth factor receptor-activating mutations. Cancer Res 2008; 68:9479-87; PMID:19010923; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-1643 [DOI] [PubMed] [Google Scholar]
- 29. Turke AB, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D, Lifshits E, Toschi L, Rogers A, Mok T, Sequist L, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 2010; 17:77-88; PMID:20129249; http://dx.doi.org/ 10.1016/j.ccr.2009.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liska D, Chen CT, Bachleitner-Hofmann T, Christensen JG, Weiser MR. HGF rescues colorectal cancer cells from EGFR inhibition via MET activation. Clin Cancer Res 2011; 17:472-82; PMID:21098338; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-0568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bardelli A, Corso S, Bertotti A, Hobor S, Valtorta E, Siravegna G, Sartore-Bianchi A, Scala E, Cassingena A, Zecchin D, et al. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov 2013; 3:658-73; PMID:23729478; http://dx.doi.org/ 10.1158/2159-8290.CD-12-0558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Luraghi P, Reato G, Cipriano E, Sassi F, Orzan F, Bigatto V, De Bacco F, Menietti E, Han M, Rideout WM 3rd, et al. MET signaling in colon cancer stem-like cells blunts the therapeutic response to EGFR inhibitors. Cancer Res 2014; 74:1857-69; PMID:24448239; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-2340-T [DOI] [PubMed] [Google Scholar]
- 33. Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J, Frederick DT, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature 2012; 487:500-4; PMID:22763439; http://dx.doi.org/ 10.1038/nature11183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, Peng J, Lin E, Wang Y, Sosman J, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 2012; 487:505-9; PMID:22763448; http://dx.doi.org/ 10.1038/nature11249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Takahashi N, Yamada Y, Furuta K, Honma Y, Iwasa S, Takashima A, Kato K, Hamaguchi T, Shimada Y. Serum levels of hepatocyte growth factor and epiregulin are associated with the prognosis on anti-EGFR antibody treatment in KRAS wild-type metastatic colorectal cancer. Br J Cancer 2014; 110:2716-27; PMID:24800946; http://dx.doi.org/ 10.1038/bjc.2014.230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lu T, Jackson MW, Singhi AD, Kandel ES, Yang M, Zhang Y, Gudkov AV, Stark GR. Validation-based insertional mutagenesis identifies lysine demethylase FBXL11 as a negative regulator of NFkappaB. Proc Natl Acad Sci U S A 2009; 106:16339-44; PMID:19805303; http://dx.doi.org/ 10.1073/pnas.0908560106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Longati P, Bardelli A, Ponzetto C, Naldini L, Comoglio PM. Tyrosines1234-1235 are critical for activation of the tyrosine kinase encoded by the MET proto-oncogene (HGF receptor). Oncogene 1994; 9:49-57; PMID:8302603 [PubMed] [Google Scholar]
- 38. Marek L, Ware KE, Fritzsche A, Hercule P, Helton WR, Smith JE, McDermott LA, Coldren CD, Nemenoff RA, Merrick DT, et al. Fibroblast growth factor (FGF) and FGF receptor-mediated autocrine signaling in non-small-cell lung cancer cells. Mol Pharmacol 2009; 75:196-207; PMID:18849352; http://dx.doi.org/ 10.1124/mol.108.049544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Okamoto W, Okamoto I, Tanaka K, Hatashita E, Yamada Y, Kuwata K, Yamaguchi H, Arao T, Nishio K, Fukuoka M, et al. TAK-701, a humanized monoclonal antibody to hepatocyte growth factor, reverses gefitinib resistance induced by tumor-derived HGF in non-small cell lung cancer with an EGFR mutation. Mol Cancer Ther 2010; 9:2785-92; PMID:20716641; http://dx.doi.org/ 10.1158/1535-7163.MCT-10-0481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McMillin DW, Negri JM, Mitsiades CS. The role of tumour-stromal interactions in modifying drug response: challenges and opportunities. Nat Rev Drug Discov 2013; 12:217-28; PMID:23449307; http://dx.doi.org/ 10.1038/nrd3870 [DOI] [PubMed] [Google Scholar]
- 41. Duncan JS, Whittle MC, Nakamura K, Abell AN, Midland AA, Zawistowski JS, Johnson NL, Granger DA, Jordan NV, Darr DB, et al. Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 2012; 149:307-21; PMID:22500798; http://dx.doi.org/ 10.1016/j.cell.2012.02.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, Bernards R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012; 483:100-3; PMID:22281684; http://dx.doi.org/ 10.1038/nature10868 [DOI] [PubMed] [Google Scholar]
- 43. Sun C, Hobor S, Bertotti A, Zecchin D, Huang S, Galimi F, Cottino F, Prahallad A, Grernrum W, Tzani A, et al. Intrinsic resistance to MEK inhibition in KRAS mutant lung and colon cancer through transcriptional induction of ERBB3. Cell Rep 2014; 7:86-93; PMID:24685132; http://dx.doi.org/ 10.1016/j.celrep.2014.02.045 [DOI] [PubMed] [Google Scholar]
- 44. Nanjo S, Yamada T, Nishihara H, Takeuchi S, Sano T, Nakagawa T, Ishikawa D, Zhao L, Ebi H, Yasumoto K, et al. Ability of the Met kinase inhibitor crizotinib and new generation EGFR inhibitors to overcome resistance to EGFR inhibitors. PLoS One 2013; 8:e84700; PMID:24386407; http://dx.doi.org/ 10.1371/journal.pone.0084700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ou SI, Govindan R, Eaton KD, Otterson GA, Gutierrez M, Mita AC, Argiris A, Brega N, Usari T, Tan W, et al. Phase I/II dos-finding study of crizotinib (CRIZ) in combination with erlotinib (E) in patients (pts) with advanced non-small cell lung cancer (NSCLC). Poster session presented at: 2012. ASCO Annual Meeting; 2012 June 1-5; Chicago, IL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pfizer A Phase 1b Study Of Axitinib In Combination With Crizotinib In Patients With Advanced Solid Tumors. In: ClinicalTrials.gov ; Internet. Bethesda (MD): National Library of Medicine (US). 2013 cited 2014 Nov 05. Available from: http:(clinicaltrials.gov1487;show(NCT01999972 NLM identifier: NCT01999972. [Google Scholar]
- 47. Yang AD, Fan F, Camp ER, van Buren G, Liu W, Somcio R, Gray MJ, Cheng H, Hoff PM, Ellis LM. Chronic oxaliplatin resistance induces epithelial-to-mesenchymal transition in colorectal cancer cell lines. Clin Cancer Res 2006; 12:4147-53; PMID:16857785; http://dx.doi.org/ 10.1158/1078-0432.CCR-06-0038 [DOI] [PubMed] [Google Scholar]
- 48. Kajiyama H, Shibata K, Terauchi M, Yamashita M, Ino K, Nawa A, Kikkawa F. Chemoresistance to paclitaxel induces epithelial-mesenchymal transition and enhances metastatic potential for epithelial ovarian carcinoma cells. Int J Oncol 2007; 31:277-83; PMID:17611683 [PubMed] [Google Scholar]
- 49. Huang S, Holzel M, Knijnenburg T, Schlicker A, Roepman P, McDermott U, Garnett M, Grernrum W, Sun C, Prahallad A, et al. MED12 controls the response to multiple cancer drugs through regulation of TGF-beta receptor signaling. Cell 2012; 151:937-50; PMID:23178117; http://dx.doi.org/ 10.1016/j.cell.2012.10.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Brunen D, Willems SM, Kellner U, Midgley R, Simon I, Bernards R. TGF-β: an emerging player in drug resistance. Cell Cycle 2013; 12:2960-8; PMID:23974105; http://dx.doi.org/ 10.4161/cc.26034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fan S, Ma YX, Wang JA, Yuan RQ, Meng Q, Cao Y, Laterra JJ, Goldberg ID, Rosen EM. The cytokine hepatocyte growth factor/scatter factor inhibits apoptosis and enhances DNA repair by a common mechanism involving signaling through phosphatidyl inositol 3’ kinase. Oncogene 2000; 19:2212-23; PMID:10822371; http://dx.doi.org/ 10.1038/sj.onc.1203566 [DOI] [PubMed] [Google Scholar]
- 52. Bowers DC, Fan S, Walter KA, Abounader R, Williams JA, Rosen EM, Laterra J. Scatter factor/hepatocyte growth factor protects against cytotoxic death in human glioblastoma via phosphatidylinositol 3-kinase- and AKT-dependent pathways. Cancer Res 2000; 60:4277-83; PMID:10945642 [PubMed] [Google Scholar]
- 53. Chen JT, Huang CY, Chiang YY, Chen WH, Chiou SH, Chen CY, Chow KC. HGF increases cisplatin resistance via down-regulation of AIF in lung cancer cells. Am J Respir Cell Mol Biol 2008; 38:559-65; PMID:18096875; http://dx.doi.org/ 10.1165/rcmb.2007-0001OC [DOI] [PubMed] [Google Scholar]
- 54. Yu G, Jing Y, Kou X, Ye F, Gao L, Fan Q, Yang Y, Zhao Q, Li R, Wu M, et al. Hepatic stellate cells secreted hepatocyte growth factor contributes to the chemoresistance of hepatocellular carcinoma. PLoS One 2013; 8:e73312; PMID:24023859; http://dx.doi.org/ 10.1371/journal.pone.0073312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Canadas I, Rojo F, Taus A, Arpi O, Arumi-Uria M, Pijuan L, Menendez S, Zazo S, Domine M, Salido M, et al. Targeting epithelial-to-mesenchymal transition with Met inhibitors reverts chemoresistance in small cell lung cancer. Clin Cancer Res 2014; 20:938-50; PMID:24284055; http://dx.doi.org/ 10.1158/1078-0432.CCR-13-1330 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.