

Abstract
Serine-threonine kinase receptor-associated protein (STRAP) is a TGF-β receptor-interacting protein that participates in the regulation of cell proliferation and cell death in response to various stresses. Here, we demonstrate that STRAP phosphorylation plays an important role in determining the pro- or anti-apoptotic function of STRAP. Murine protein serine/threonine kinase 38 (MPK38) phosphorylates STRAP at Ser188 via direct interaction. Complex formation between STRAP and MPK38 is mediated by Cys152 and Cys270 of STRAP and Cys339 and Cys377 of MPK38, suggesting the redox dependency of this interaction. MPK38-mediated STRAP Ser188 phosphorylation contributes to the pro-apoptotic function of STRAP by modulating key steps in STRAP-dependent ASK1, TGF-β, p53, and PI3K/PDK1 signaling pathways. Moreover, knockdown of endogenous MPK38 using an inducible MPK38 shRNA system and in vivo activation of MPK38 by treatment of HEK293 and STRAP-null MEF cells with 1-chloro-2,4-dinitrobenzene (DNCB), a specific inhibitor of Trx reductase, provide evidence that STRAP Ser188 phosphorylation plays a key role in STRAP-dependent cell death. Adenoviral delivery of MPK38 in mice also demonstrates that STRAP Ser188 phosphorylation in the liver is tightly associated with cell death and proliferation through ASK1, TGF-β, p53, and PI3K/PDK1 pathways, resulting in apoptotic cell death.
Keywords: ASK1, MPK38/MELK, p53, PI3K/PDK1, STRAP, TGF-β
Abbreviations
- STRAP
serine-threonine kinase receptor-associated protein
- MPK38
murine protein serine/threonine kinase 38
- ASK1
apoptosis signal-regulating kinase 1
- PDK1
3-phosphoinositide-dependent protein kinase-1
- PI3K
phosphoinositide 3-kinase
- ZPR9
zinc finger-like protein 9
Introduction
Serine-threonine kinase receptor-associated protein (STRAP) was originally identified as a transforming growth factor-β (TGF-β) receptor-interacting protein that inhibits TGF-β signaling by stabilizing the association between TGF-β receptors and Smad7, and was shown to localize to both the cytoplasm and nucleus.1 STRAP has been reported to positively regulate 3-phosphoinositide-dependent protein kinase-1 (PDK1) by dissociating a 14-3-3 protein that acts as a negative regulator from the PDK1-14-3-3 complex.2 Similarly, STRAP has been shown to contribute to tumor progression by blocking TGF-β-mediated signaling, especially in colon and lung carcinomas, indicating that it has an anti-apoptotic function.3,4,5 Recently, STRAP was shown to associate with ASK1 and subsequently inhibit ASK1 activity in a phosphorylation-dependent manner.6 STRAP also participates in the regulation of GSK3β function and Notch3 stabilization through direct interaction with GSK3β and Notch3.7 These observations strongly support the oncogenic functions of STRAP. By contrast, STRAP has been shown to be involved in inducing cell death through direct interaction with p53,8 suggesting that STRAP also possesses a pro-apoptotic function. However, the mechanism by which the pro- or anti-apoptotic functions of STRAP are determined remains unknown.
Murine protein serine/threonine kinase 38 (MPK38)/maternal embryonic leucine zipper kinase (Melk) is a member of the AMP-activated protein kinase (AMPK)-related kinase family and controls a variety of biological processes, including cell cycle, spliceosome assembly, gene expression, cell proliferation, carcinogenesis, and apoptosis.9,10 An understanding of the post-translational modifications, such as phosphorylation,11 that play an important role in the regulation of protein stability and activity will help to identify the mechanisms by which MPK38 affects its substrates. MPK38 has been shown to phosphorylate Bcl-GL, a pro-apoptotic member of the Bcl-2 family, resulting in the suppression of BCL-GL-induced apoptosis.12 MPK38-mediated phosphorylation of ZPR9, a zinc finger protein, was shown to stimulate its nuclear localization, leading to enhanced B-myb transactivation.13 MPK38 physically interacts with, and phosphorylates, PDK1 at Thr354, thereby inhibiting its activity and function.14 MPK38 also functions as a novel positive regulator for promoting p53 activity through phosphorylation of p53 Ser15.15 MPK38-mediated phosphorylation of Smad proteins (Ser245 of Smad2, Ser204 of Smad3, Ser343 of Smad4, and Thr96 of Smad7) affect Smad(s)-mediated signaling, leading to the stimulation of TGF-β signaling.16 Recently, we provided evidence of the importance of MPK38-mediated phosphorylation of ASK1 at Thr838 in the enhancement of ASK1 activity and function.10 We also found that Thr76 phosphorylation of Trx by MPK38 plays a critical role in the negative regulation of MPK38-induced ASK1, TGF-β, and p53 signaling;17 thus the elucidation of MPK38-mediated phosphorylation of target proteins could lead to a better understanding of the biological functions of MPK38 and its target proteins.
Here, we have analyzed the role of phosphorylation of STRAP Ser188 in the regulation of STRAP-dependent ASK1, TGF-β, p53, and PI3K/PDK1 signaling. Our findings show that MPK38 directly interacts with, and phosphorylates, STRAP at Ser188. STRAP phosphorylation subsequently affects complex formation between ASK1 and MKK3, the TGF-β receptor and Smad3, p53 and Mdm2, or PDK1 and AKT1, which are critical for the activation of ASK1, TGF-β, p53, and PI3K/PDK1 signaling pathways, respectively, eventually leading to apoptotic cell death. Moreover, our present results, together with those of a recent study showing the inhibitory role of STRAP phosphorylation at Thr175 and Ser179 in ASK1-mediated cell death,6 suggest a possible mechanism by which the pro- or anti-apoptotic function of STRAP is determined.
Results
MPK38 is a STRAP-interacting protein. We recently demonstrated that MPK38 physically interacts with Smad proteins and stimulates TGF-β-induced apoptosis and cell-cycle arrest.16 STRAP has been previously reported to interact with Smad2, Smad3, Smad7, and ASK1, which are also associated with MPK38.1,3,6 Based on these findings, we addressed the question of whether MPK38 influences STRAP-mediated function through direct interaction and phosphorylation. To this end, we first performed in vivo binding assays to determine whether MPK38 physically binds to STRAP. Immunoblot analysis revealed that MPK38 interacted with STRAP (Fig. 1A, left). To verify the binding between MPK38 and STRAP, we performed co-immunoprecipitation experiments using cell lysates from HEK293 and MCF7 cells. STRAP was clearly detectable in the MPK38 immunoprecipitates, whereas no complex formation was observed in the immunoprecipitates of pre-immune serum used as a nonspecific control (Fig. 1A, middle). This observation was verified by reciprocal immunoprecipitation experiments in which an anti-STRAP antibody, instead of an anti-MPK38 antibody, was used for immunoprecipitation (data not shown). Furthermore, we performed nondenaturing PAGE analysis using purified recombinant MPK38 and STRAP proteins to examine whether this interaction is direct. A shift in the mobility of 32P-labeled autophosphorylated MPK38 was detectable when incubated with unlabeled, recombinant STRAP proteins, whereas no shift was observed in the presence of recombinant glutathione S-transferase (GST) alone (Fig. 1A, right), indicating that MPK38 and STRAP directly interact.
Figure 1.

Redox dependency of the interaction between MPK38 and STRAP. (A) Interaction between MPK38 and STRAP in cells. FLAG-tagged STRAP was co-transfected with glutathione S-transferase (GST)-MPK38 or vector alone (GST) into HEK293 cells. GST fusion proteins were purified on glutathione-Sepharose beads (GST Purification), and complex formation was determined by immunoblot analysis using an anti-FLAG antibody (left). To examine the endogenous interaction between MPK38 and STRAP, cell lysates from HEK293 and MCF7 cells were subjected to immunoprecipitation using either rabbit pre-immune serum (Preimm.) or anti-MPK38 antibody (α-MPK38) followed by immunoblot analysis using an anti-STRAP antibody (middle). MPK38-STRAP complex formation was also analyzed by nondenaturing PAGE (8%). Autophosphorylated recombinant MPK38 (32P-MPK38) was incubated with unlabeled recombinant GST alone or GST-STRAP, as described in “Materials and methods” (right). (B) Mapping of the binding domains involved in the MPK38-STRAP interaction. The schematic structures of wild-type (WT) and deletion constructs of MPK38 (left, upper) and STRAP (right, upper) are shown. The schematic structures of STRAP are somewhat modified compared to those reported previously.6 Numbers indicate the amino acid residues corresponding to the domain boundaries. For the mapping of MPK38 domains involved in STRAP binding, HEK293 cells were co-transfected with GST alone or GST-MPK38 constructs (WT, MCAT, and MPKC) together with FLAG-STRAP, and purified with glutathione-Sepharose beads (GST purification). The amount of STRAP proteins bound to the MPK38 constructs was determined by immunoblot analysis using an anti-FLAG antibody (lower left, top panel). The same stripped blot was re-probed with an anti-GST antibody to determine the expression of GST fusion proteins in the co-precipitates (lower left, middle panel). For the mapping of STRAP domains involved in MPK38 binding, HEK293 cells were transfected with vector alone (GST) or GST-MPK38 in combination with the indicated FLAG-STRAP deletion constructs [WD1–3, WD4–7, WD4, WD4(C152S), WD6, WD7, WD7(C270S), and CT], and cell lysates were purified using glutathione-Sepharose beads (GST purification). Complex formation was determined by immunoblot analysis using an anti-FLAG antibody (lower middle and right, top panels). (C) Involvement of cysteine residues in the MPK38-STRAP interaction. HEK293 cells transfected with the indicated expression vectors were lysed and GST precipitates were analyzed for MPK38-STRAP complex formation by immunoblot analysis using an anti-FLAG antibody (upper, top panels). To determine the redox dependency of the interaction between MPK38 and STRAP, HEK293 cell lysates were treated with the indicated concentrations of H2O2, dithiothreitol (DTT), and β-mercaptoethanol (β-ME) on ice for 0.5–1 h and then subjected to immunoprecipitation with an anti-MPK38 antibody (IP:α-MPK38). Immune complexes were analyzed for the presence of STRAP by immunoblot analysis using an anti-STRAP antibody (lower, top panel). The amount of immunoprecipitated MPK38 (lower, middle panel) and the expression levels of STRAP (lower, bottom panel) in the cell lysates were analyzed in parallel using anti-MPK38 and anti-STRAP antibodies, respectively. (D) Modulation of MPK38-STRAP complex formation by ASK1/TGF-β/p53/PDK1 signaling. HEK293 cell lysates were treated with or without the following stimuli: H2O2 (2 mM, 30 min), TNF-α (500 ng/ml, 30 min), thapsigargin (Tg: 20 μM, 30 min), ionomycin (IONO: 1 μM, 24 h), TGF-β1 (100 pM, 20 h), 5-fluorouracil (5FU: 0.38 mM, 30 h), doxorubicin (Dox: 100 ng/ml, 24 h), or insulin (100 nM, 20 min). They were then immunoprecipitated with an anti-MPK38 antibody (IP:α-MPK38), followed by immunoblotting with an anti-STRAP antibody to determine the endogenous association between MPK38 and STRAP proteins (top panels). The expression level of MPK38 in the immunoprecipitates was determined using an anti-MPK38 antibody (3rd panels). WT, wild-type; IP, immunoprecipitation; re., recombinant.
To identify the interaction domain of MPK38 responsible for STRAP binding, we carried out in vivo binding assays using 2 deletion mutants of wild-type (WT) MPK38,16 namely, MCAT (amino acids 7–269) and MPKC (amino acids 270–643). Both WT MPK38 and N-terminal-truncated MPKC clearly interacted with STRAP, whereas no binding of STRAP was observed in the presence of C-terminal-truncated MCAT (Fig. 1B, left), indicating that the C-terminal domain of MPK38 (MPKC) is responsible for STRAP binding. Next, we determined the interaction domains of STRAP required for MPK38 binding using 2 deletion mutants of FLAG-tagged WT STRAP,6 WD1–3 (amino acids 1–129) and WD4–7 (amino acids 129–350). MPK38 interacted with the N-terminal-truncated STRAP mutant WD4–7, but not with C-terminal-truncated WD1–3 (Fig. 1B, middle). This result indicates that the C-terminal domain of STRAP (WD4–7), which harbors 4 C-terminal WD40 repeats, is responsible for MPK38 binding. To more precisely define the specific MPK38 binding region of STRAP, we performed in vivo binding assays using 6 additional deletion mutants of WT STRAP,6 namely, WD4, WD4(C152S), WD6, WD7, WD7(C270S), and CT. MPK38 interacted with the WD4 and WD7 deletion mutants, but not with the other STRAP deletion mutants, WD4(C152S), WD6, WD7(C270S), and CT (Fig. 1B, right). These results indicate that the cysteine residues within the C-terminal WD40 repeats of STRAP, especially the fourth and seventh WD40 repeats, are responsible for MPK38 binding. We further analyzed these results using in vivo binding assays in HEK293 cells transfected with FLAG-tagged WT STRAP and the C152S, C270S, and C152S/C270S mutants together with GST-tagged MPK38. MPK38 still interacted with either the C152S or C270S single mutant, albeit at low levels compared to WT STRAP, whereas no binding was observed in the presence of the C152S/C270S double mutant (Fig. 1C, upper left), again supporting a critical role for STRAP Cys152 and Cys270 in MPK38 binding. To identify the corresponding cysteine residues of MPK38 required for STRAP binding, we performed in vivo binding assays with 4 MPK38 mutants:17 C286S, C339S, C377S, and C339S/C377S. WT MPK38 and the C286S mutant interacted with STRAP at similar levels, whereas the interaction was substantially decreased in the presence of the 2 MPK38 mutants C339S and C377S. Moreover, the C339S/C377S double mutant failed to bind STRAP (Fig. 1C, upper right). These results suggest that the interaction between MPK38 and STRAP is mediated by Cys339 and Cys377 of MPK38 and Cys152 and Cys270 of STRAP, and that this interaction may be redox-dependent. Based on this, we examined whether the cellular redox state modulated the interaction between MPK38 and STRAP. Treatment of HEK293 cells with the reductants dithiothreitol (DTT) and β-mercaptoethanol (β-ME) markedly decreased complex formation between MPK38 and STRAP in a dose-dependent manner, whereas H2O2, an oxidant, had no such effect (Fig. 1C, lower), confirming the redox dependency of the MPK38-STRAP interaction in cells. Together, these findings suggest that MPK38 and STRAP physically interact through their cysteine residues in a redox-dependent fashion.
Pro- and anti-apoptotic signals differently modulate the interaction between MPK38 and STRAP. Because STRAP has been implicated in ASK1-, TGF-β-, p53-, and PDK1-mediated function,1,2,6,18 we tested whether the pro- and anti-apoptotic stresses that trigger ASK1, TGF-β, p53, or PI3K/PDK1 signaling, such as H2O2, TNF-α, thapsigargin (Tg), ionomycin (IONO), TGF-β1, 5-fluorouracil (5FU), doxorubicin (Dox), and insulin, influenced the association between MPK38 and STRAP. All stimuli tested, except for insulin, considerably increased the MPK38-STRAP association compared to untreated control cells, whereas insulin treatment led to a decrease in MPK38-STRAP association (Fig. 1D). These results suggest that MPK38 plays a regulatory role in STRAP-mediated activities, which are functionally linked to ASK1, TGF-β, p53, and PI3K/PDK1 signaling pathways.
STRAP Ser188 phosphorylation by MPK38 enhances the kinase activity of MPK38. We next examined whether MPK38 phosphorylates STRAP through their interaction. HEK293 cells were transiently transfected with GST-tagged WT or kinase-dead (K40R) MPK38, and the GST precipitates were incubated with [γ-32P]ATP and recombinant STRAP as a substrate. WT MPK38, but not kinase-dead MPK38, induced STRAP phosphorylation, indicating that STRAP is a substrate of MPK38 (Fig. 2A, left). To identify potential MPK38 phosphorylation sites on STRAP, we performed an in vitro kinase assay using 3 recombinant STRAP mutants (S188A, S210A, and T326A) that were screened by alignment analysis with the MPK38/AMPK phosphorylation consensus sequence,15,18 together with recombinant MPK38 proteins. No phosphorylation of the S188A mutant by MPK38 was observed (Fig. 2A, right). However, the S210A and T326A STRAP mutants were phosphorylated by MPK38. These results indicate that Ser188 of STRAP is a potential phosphorylation site for MPK38. To evaluate whether STRAP can act as a regulator of MPK38, we examined whether phosphorylation of STRAP Ser188 has an effect on the kinase activity of MPK38 using an in vitro kinase assay. Cells expressing WT STRAP exhibited substantially increased MPK38 kinase activity compared to control cells, whereas the S188A mutant had no such effect (Fig. 2B, left). This finding suggests that STRAP may be a positive regulator of MPK38. A similar result was also obtained in an in vitro kinase assay using purified recombinant MPK38 and STRAP proteins (Fig. 2B, right) and thus it is likely that phosphorylation of STRAP Ser188 by MPK38 plays an important role in the regulation of MPK38-mediated ASK1, TGF-β, p53, and PDK1 function.
Figure 2.

Phosphorylation of STRAP at Ser188 by MPK38. (A) MPK38 phosphorylates STRAP at Ser188. Approximately 2–3 μg of recombinant STRAP proteins were mixed with MPK38 proteins purified with glutathione-Sepharose beads from HEK293 cell lysates containing GST-tagged wild-type (WT) or kinase-dead (K40R) MPK38, and in vitro kinase assays were then performed (left). The in vitro kinase assay to identify MPK38 phosphorylation sites on STRAP was performed as follows: 2 μg of recombinant WT STRAP or one of its mutants were mixed with 10 μM ATP, 5 μCi of [γ-32P]-ATP, and 10 mM MgCl2 in 20 μl of kinase buffer and incubated with recombinant WT MPK38 (2 μg) for 15 min at 37°C (right, top panel). (B) STRAP enhances the kinase activity of MPK38. HEK293 cells were transfected with GST-MPK38 in the presence or absence of FLAG-tagged WT or mutant (S188A) STRAP. GST-MPK38 proteins were purified on glutathione-Sepharose beads and then subjected to an in vitro kinase assay using ZPR9 as a substrate, followed by SDS-PAGE and autoradiography (left, top panel). The same blot was stripped and re-probed with an anti-phospho-Ser/Thr antibody to determine the phosphorylation level of ZPR9 (left, 2nd panel), and the expression level of FLAG-STRAP in total cell lysates was examined by immunoblotting with an anti-FLAG antibody (left, 3rd panel). The presence of equivalent amounts of substrate was verified by immunoblotting with an anti-GST antibody (left, bottom panel). Recombinant MPK38 (re.MPK38) proteins, instead of GST-MPK38 proteins obtained from the transfected cell lysates, were used for an in vitro kinase assay in the presence or absence of recombinant WT or mutant (S188A) STRAP proteins (right). P-MPK38, P-STRAP, and P-ZPR9 indicate autophosphorylated MPK38 and phosphorylated STRAP and ZPR9, respectively.
Phosphorylation of STRAP Ser188 by MPK38 alleviates STRAP-mediated inhibition of ASK1 signaling. Given that STRAP interacts with ASK1 and inhibits ASK1 signaling,6 we investigated whether phosphorylation of STRAP Ser188 by MPK38 influences STRAP-mediated inhibition of ASK1 signaling using luciferase reporter assays with the transcription factor AP-1. MPK38 alleviated STRAP-mediated inhibition of AP-1 transcriptional activity in a dose-dependent manner in the presence of WT STRAP (Fig. 3A, upper). However, such an effect was not detected in the presence of the S188A STRAP mutant, suggesting that alleviation of STRAP-mediated inhibition of AP-1 transcriptional activity by MPK38 is dependent on the phosphorylation status of STRAP at Ser188. In addition, down-regulation of endogenous MPK38 using MPK38-specific siRNAs showed higher STRAP-mediated inhibition of AP-1 transactivation than that observed with control STRAP alone (Fig. 3A, lower). Because ASK1 activation by H2O2 induces apoptotic cell death,19,20 we further assessed the effect of MPK38 on H2O2-mediated cell death in the presence of WT STRAP or the S188A mutant. Indeed, MPK38 significantly alleviated STRAP-mediated suppression of H2O2-induced cell death in a dose-dependent manner in the presence of WT STRAP, whereas the S188A STRAP mutant had no such effect (Fig. 3B). We also employed an inducible MPK38 shRNA system to confirm the effect of MPK38-mediated STRAP Ser188 phosphorylation on ASK1 signaling in vivo. Knockdown of endogenous MPK38 considerably decreased the phosphorylation level of both ASK1 and its downstream targets, including MKK3/6, p38, and ATF2, compared to either parental HEK293 cells or cells expressing a control scrambled shRNA (Fig. 3C). These results suggest that STRAP Ser188 phosphorylation by MPK38 contributes to the alleviation of STRAP-mediated inhibition of ASK1 function.
Figure 3.

Alleviation of STRAP-mediated inhibition of ASK1 signaling by MPK38-mediated STRAP Ser188 phosphorylation. (A) The effect of STRAP Ser188 phosphorylation by MPK38 on STRAP-mediated inhibition of ASK1-mediated AP-1 transactivation. HEK293 cells were transfected with expression vectors for wild-type (WT) and mutant (S188A) STRAP (0.3 and 0.6 μg), WT MPK38 (0.5 and 1 μg), ASK1 (0.3 μg), 0.2 μg of AP-1 luciferase plasmid, MPK38-specific siRNAs (100 and 200 nM), and scrambled siRNA (100 and 200 nM), as indicated, in the presence or absence of c-fos (0.6 μg) followed by luciferase and β-galactosidase assays. Results were expressed as mean SEM (upper: *P < 0.05, **P < 0.01; lower: *P < 0.05, ***p < 0.001 versus STRAP (0.3 μg) in the presence of ASK1). (B) The effect of STRAP Ser188 phosphorylation by MPK38 on STRAP-mediated inhibition of H2O2-mediated apoptosis. 293T cells were transiently transfected with expression vectors for WT and mutant (S188A) STRAP (1 and 3 μg), WT MPK38 (1 and 2 μg), and ASK1 (1.7 μg), as indicated, in the presence or absence of H2O2 (1 mM). Cells exposed to 1 mM H2O2 for 9 h were used as a positive control (lane 6). Results were expressed as mean SEM (**P < 0.01 vs. STRAP (1 μg) + ASK1 (1.7 μg) in the presence of H2O2). (C) The effect of MPK38 knockdown on ASK1 downstream signaling. HEK293 cells expressing inducible MPK38 or scrambled shRNA treated with or without 1-chloro-2,4-dinitrobenzene (DNCB) and doxycycline (Dox) were lysed and subjected to immunoblot analysis using anti-phospho-specific antibodies for ASK1 Thr845 (corresponding to Thr838 in human), MKK3/6 Ser189/207, p38 Thr180/Tyr182, and ATF2 Thr71. The level of STRAP Ser188 phosphorylation in STRAP immunoprecipitates was determined by anti-phospho-STRAP Ser188 immunoblotting (top panel). β-actin was used as a loading control. Sc, scrambled.
Phosphorylation of STRAP Ser188 by MPK38 alleviates STRAP-mediated inhibition of TGF-β signaling. Given that STRAP stabilizes the association between TGF-β receptors and Smad7 and inhibits TGF-β signaling,1 we examined whether phosphorylation of STRAP Ser188 by MPK38 affects STRAP-mediated inhibition of TGF-β-induced transcription. MPK38 significantly alleviated STRAP-mediated inhibition of TGF-β-induced transcription in a dose-dependent manner in the presence of WT STRAP, whereas this effect was not observed in the presence of the S188A STRAP mutant (Fig. 4A, upper). This result suggests that STRAP Ser188 phosphorylation by MPK38 is involved in the regulation of STRAP-mediated inhibition of TGF-β-induced transcription. Concordantly, knockdown of endogenous MPK38 using MPK38-specific siRNAs enhanced STRAP-mediated inhibition of TGF-β-induced transcription compared to STRAP alone, whereas such an effect was not observed in the presence of control scrambled siRNA (Fig. 4A, lower). We next examined whether MPK38-mediated STRAP Ser188 phosphorylation functionally affects STRAP-mediated inhibition of TGF-β-induced apoptosis. Indeed, MPK38 alleviated TGF-β-induced apoptosis suppressed by STRAP in a dose-dependent manner in the presence of WT STRAP, whereas this effect was not observed in the presence of the S188A STRAP mutant (Fig. 4B), consistent with the results obtained from luciferase reporter assays. We then confirmed these results in an MPK38 shRNA inducible HEK293 cell line. As indicated by the phosphorylation level of STRAP at Ser188, downregulation of endogenous MPK38 considerably modulated the up- or down-regulation of TGF-β targets,16 such as PAI-1, p21Cip1, Smad7, CDK4, and Cyclin D1, compared to either parental HEK293 cells or cells expressing a control scrambled shRNA (Fig. 4C). These findings suggest that phosphorylation of STRAP Ser188 by MPK38 also plays a key role in alleviating STRAP-mediated inhibition of TGF-β signaling.
Figure 4.

Alleviation of STRAP-mediated inhibition of TGF-β signaling by MPK38-mediated STRAP Ser188 phosphorylation. (A) The effect of STRAP Ser188 phosphorylation by MPK38 on STRAP-mediated inhibition of TGF-β-induced transcription. HepG2 cells were transfected with expression vectors for wild-type (WT) and mutant (S188A) STRAP (1 μg each), 0.3 μg p3TP-Lux, increasing amounts of WT and kinase-dead (K40R) MPK38 (upper, 0.5, 1.0, 1.5, and 2.0 μg; lower, 0.6 and 1.2 μg), MPK38-specific siRNAs (100 and 200 nM), and scrambled siRNA (100 and 200 nM), as indicated, in the presence or absence of 100 pM of TGF-β1, followed by luciferase and β-galactosidase assays. Luciferase activity was measured 48 h after transfection. Results were expressed as mean SEM (upper: ***P < 0.001; lower: *P < 0.05; ***P < 0.001 versus STRAP (1 μg) in the presence of TGF-β1). (B) The effect of STRAP Ser188 phosphorylation by MPK38 on STRAP-mediated inhibition of TGF-β-induced apoptosis. HaCaT cells were transfected with WT and mutant (S188A) STRAP (1 μg each), green fluorescent protein (2 μg), and increasing amounts of expression vectors for WT MPK38 (1 and 2 μg) in the presence or absence of TGF-β1 (100 pM). Apoptotic cell death was determined as described in Fig. 3B. Results were expressed as mean SEM (*P < 0.05; NS, not significant). (C) The effect of STRAP Ser188 phosphorylation by MPK38 on the expression of TGF-β target genes. HEK293 cells expressing inducible MPK38 or scrambled shRNA treated with or without 1-chloro-2,4-dinitrobenzene (DNCB) and doxycycline (Dox), as described in Fig. 3C, were lysed and subjected to immunoblot analysis using anti-PAI-1, anti-p21, anti-Smad7, anti-CDK4, anti-Cyclin D1, anti-MPK38, and anti-β-actin antibodies. Sc, scrambled.
Phosphorylation of STRAP Ser188 by MPK38 enhances STRAP-induced p53 signaling. To investigate whether STRAP Ser188 phosphorylation by MPK38 has an effect on STRAP-induced p53 signaling, we performed a luciferase reporter assay to measure the effect of STRAP Ser188 phosphorylation on STRAP-induced p53 transactivation. MPK38 dose-dependently increased p53 transcriptional activity in the presence of WT STRAP (Fig. 5A, upper), whereas no such effect was observed in the presence of the S188A STRAP mutant, which is defective in MPK38-mediated phosphorylation (see Fig. 2). This result suggests that MPK38-mediated STRAP Ser188 phosphorylation plays a critical role in the regulation of p53 function induced by STRAP. To confirm the positive role of MPK38 in STRAP-mediated p53 transactivation, we also performed a knockdown experiment using MPK38-specific siRNAs. Downregulation of endogenous MPK38 decreased STRAP-induced p53 transcriptional activity in a dose-dependent manner, whereas control scrambled siRNA had no such effect (Fig. 5A, lower). Consistent with these findings, MPK38 dose-dependently increased p53-mediated apoptosis in the presence of WT STRAP, and this effect was not observed in the presence of the S188A STRAP mutant (Fig. 5B, upper). We also examined the effect of STRAP Ser188 phosphorylation on p53-mediated cell-cycle arrest. Flow cytometry analysis of MCF7 cells using 5FU to induce apoptosis revealed that co-expression of MPK38 with STRAP enhanced G0/G1 arrest in a dose-dependent manner compared to expression of WT STRAP alone (Fig. 5B, lower). However, the MPK38-mediated stimulatory effect on p53-mediated cell-cycle arrest was not observed in the presence of the S188A STRAP mutant, indicating again a critical role for MPK38-mediated STRAP Ser188 phosphorylation in STRAP-induced p53 activity. To further verify the stimulatory effect of MPK38-mediated STRAP Ser188 phosphorylation on p53 signaling, we analyzed the expression of p53 target genes using an inducible MPK38 shRNA system in HEK293 cells. Knockdown of endogenous MPK38 remarkably decreased expression of the p53 target genes p53, p21, Mdm2, and Bax, in the presence or absence of 1-chloro-2,4-dinitrobenzene (DNCB), a specific inhibitor of Trx reductase, which is involved in MPK38 activation,17 compared to either parental HEK293 cells or cells expressing a control scrambled shRNA (Fig. 5C). A comparable result was observed in immunoblot analysis of STRAP Ser188 phosphorylation with an anti-phospho-STRAP(S188) antibody (Fig. 5C, IP:α-STRAP). Together, these findings indicate that the phosphorylation of STRAP at Ser188 by MPK38 plays an important role in enhancing STRAP-mediated p53 signaling.
Figure 5.

Enhancement of STRAP-induced p53 signaling by MPK38-mediated STRAP Ser188 phosphorylation. (A) The effect of STRAP Ser188 phosphorylation by MPK38 on p53-mediated transcription induced by STRAP. MCF7 cells were transfected with expression vectors for wild-type (WT) and mutant (S188A) STRAP (0.5 μg each), WT and kinase-dead (K40R) MPK38 (upper, 0.5, 1.0, 1.5, and 2.0 μg; lower, 1 and 2 μg), MPK38-specific siRNAs (100 and 200 nM), scrambled siRNA (100 and 200 nM), and 0.2 μg of p53 luciferase plasmid, as indicated, in the presence or absence of p53 (0.3 μg). Results were expressed as mean SEM (upper: **P < 0.01, ***P < 0.001; lower: **P < 0.01, ***P < 0.001 vs. STRAP (1 μg) in the presence of p53). (B) The effect of STRAP Ser188 phosphorylation by MPK38 on p53-mediated apoptosis and cell-cycle arrest induced by STRAP. MCF7 cells were transiently transfected with increasing amounts of MPK38 (1 and 2 μg) as indicated, together with green fluorescent protein (GFP) (2 μg) and WT and mutant (S188A) STRAP (2 μg each), in the presence or absence of p53 (1 μg) or 5FU (0.38 mM). Apoptotic cell death was determined by a GFP-based cell-death assay, as described in “Materials and methods” (upper). MCF7 cells under the same conditions were treated with 100 ng/ml doxorubicin for 24 h, and G0/G1 populations were analyzed by FACScan. The indicated percentages represent G0/G1 (white bars) and G2/M (black bars) arrest. Results were expressed as mean SEM (*P < 0.05; NS, not significant). (C) The effect of STRAP Ser188 phosphorylation by MPK38 on the expression of p53 target genes. HEK293 cells expressing inducible MPK38 or scrambled shRNA treated with or without 1-chloro-2,4-dinitrobenzene (DNCB: 50 μM, 30 min) and doxycycline (Dox: 1 μg/ml, 72 h) were lysed and subjected to immunoblot analysis using anti-p53, anti-p21, anti-Mdm2, anti-Bax, anti-MPK38, and anti-β-actin antibodies. The level of STRAP Ser188 phosphorylation in STRAP immunoprecipitates was determined by immunoblotting with an anti-phospho-STRAP(S188) antibody (top panel). The endogenous expression level of MPK38 was determined by immunoblotting with an anti-MPK38 antibody (second from bottom). β-actin was used as a loading control. Sc, scrambled.
Phosphorylation of STRAP Ser188 by MPK38 inhibits STRAP-induced PI3K/PDK1 signaling. Given that STRAP interacts with PDK1 and enhances its activity,2 we analyzed whether STRAP Ser188 phosphorylation by MPK38 has an effect on STRAP-induced PDK1 activity. To this end, we performed a green fluorescent protein (GFP)-based apoptosis analysis using 293T cells, which are susceptible to TNF-α-induced apoptosis.2 Co-expression of WT MPK38 increased apoptotic cell death in a dose-dependent manner in the presence of WT STRAP compared to control cells expressing STRAP alone, whereas kinase-dead MPK38 had no such effect in the presence of WT STRAP (Fig. 6A). In addition, neither WT nor kinase-dead MPK38 had an effect in the presence of the S188A STRAP mutant (Fig. 6A). These results suggest that MPK38-mediated STRAP Ser188 phosphorylation contributes to the inhibition of STRAP-induced PI3K/PDK1 signaling. We then performed flow cytometry to investigate the effect of MPK38-mediated STRAP Ser188 phosphorylation on PDK1 activity with respect to serum-induced cell growth in HaCaT cells. Co-expression of MPK38 with STRAP decreased the percentage of cells in S phase compared to control cells expressing STRAP alone (51% vs. 45%), whereas this effect was not observed in the presence of the S188A STRAP mutant (52% vs. 52%) (Fig. 6B, upper). Because PDK1 has been shown to inhibit TGF-β signaling through physical interaction with Smad proteins,21 we also examined the effect of MPK38-mediated STRAP Ser188 phosphorylation on PDK1-mediated suppression of TGF-β-induced cell-cycle arrest. Co-expression of MPK38 with STRAP considerably decreased the accumulation of cells in S phase after 24 h of serum stimulation in the presence of TGF-β1 compared with control cells expressing STRAP alone (Fig. 6B, lower; 38% vs. 30%). To confirm the effect of MPK38-mediated STRAP Ser188 phosphorylation on downstream PI3K/PDK1 signaling in vivo, we also performed an endogenous knockdown experiment using an inducible MPK38 shRNA system. Down-regulation of endogenous MPK38 expression considerably increased the phosphorylation level of PDK1 and its downstream targets, AKT1 and Bad, compared to either parental HEK293 cells or cells expressing a control scrambled shRNA (Fig. 6C). Consistently, a decrease in the phosphorylation level of STRAP at Ser188 was observed upon knockdown of endogenous MPK38; thus these findings indicate that MPK38 inhibits STRAP-induced PI3K/PDK1 signaling by phosphorylating STRAP at Ser188.
Figure 6.

Suppression of STRAP-induced PI3K/PDK1 signaling by MPK38-mediated STRAP Ser188 phosphorylation. (A) Suppression of STRAP-induced PDK1 activity toward TNF-α-induced cell death by MPK38-mediated STRAP Ser188 phosphorylation. 293T cells were transfected with expression vectors for wild-type (WT) and mutant (K40R) MPK38 (1 and 3 μg), WT and mutant (S188A) STRAP (4 μg each), and an expression vector encoding green fluorescent protein (3 μg), as indicated. Transfected cells were incubated for 24 h and then treated with TNF-α (20 ng/ml) and cycloheximide (10 μg/ml) for 14 h to induce apoptosis. Apoptotic cell death was determined as described in Fig. 3B. Results were expressed as mean SEM (*P < 0.05; NS, not significant). (B) The effect of MPK38-mediated STRAP Ser188 phosphorylation on serum-mediated cell growth induced by PDK1 and TGF-β-mediated cell-cycle arrest suppressed by PDK1. HaCaT cells (2 × 105/dish) transfected with the indicated combinations of plasmid vectors (vector alone, STRAP, S188A, and MPK38) were synchronized in G0/G1 by hydroxyurea treatment (2 mM) for 20 h. Cells were collected before (0 h starvation) or after 10% serum treatment for 24 h in the absence (24 h serum stimulation) or presence (24 h serum stimulation + TGF-β1) of TGF-β1 (2 ng/ml) to determine the number of cells in the G1, S, and G2/M phases by flow cytometry. Each experiment was repeated at least 3 times. (C) The effect of MPK38 knockdown on PDK1 downstream signaling. HEK293 cells expressing inducible MPK38 or scrambled shRNA treated with or without 1-chloro-2,4-dinitrobenzene (DNCB) and doxycycline (Dox), as described in Fig. 3C, were lysed and subjected to immunoblot analysis using anti-phospho-specific antibodies for PDK1 Ser241, AKT1 Thr308, AKT1 Ser473, and Bad Ser136. The Ser188 phosphorylation level of STRAP in the STRAP immunoprecipitates was determined by immunoblotting with an anti-phospho-STRAP(Ser188) antibody (top panel). β-actin was used as a loading control. Sc, scrambled.
MPK38-mediated phosphorylation of STRAP Ser188 enhances the stability of STRAP. To address how STRAP Ser188 phosphorylation by MPK38 contributes to the pro-apoptotic activity of STRAP, we examined whether MPK38 affects the stability of STRAP. HEK293 cells were transfected with expression vectors for WT MPK38 or its mutants, C286S, C339S, C377S, and C339S/C377S, and STRAP protein levels were assessed by immunoblot analysis with an anti-STRAP antibody. WT MPK38 and the 3 single MPK38 mutants (C286S, C339S, and C377S) increased the stability of STRAP compared to control cells expressing empty vector, whereas the MPK38 double mutant C339S/C377S, which is unable to bind to STRAP (see Fig. 1C), had no such effect (Fig. 7A, upper). Similar levels of STRAP Ser188 phosphorylation were also observed in this analysis (Fig. 7A, upper, middle panel), suggesting that phosphorylation of STRAP Ser188 by MPK38 is associated with enhanced stability of STRAP. We then performed an ubiquitination assay to analyze the role of MPK38-mediated STRAP Ser188 phosphorylation on STRAP ubiquitination. Consistently, expression of WT MPK38 and the 3 single MPK38 mutants (C286S, C339S, and C377S) resulted in a decrease in the degree of ubiquitination of STRAP, whereas no such effect was observed in the presence of the MPK38 double mutant C339S/C377S (Fig. 7A, lower). These results suggest that the direct interaction and phosphorylation of STRAP at Ser188 by MPK38 plays an important role in the ubiquitination and degradation of STRAP. To further verify whether MPK38-mediated STRAP Ser188 phosphorylation contributes to the stability of STRAP in vivo, we performed immunoblot analysis following treatment with DNCB, which stabilizes and activates MPK38,17 and performed a knockdown experiment using inducible HEK293 cells expressing a shRNA targeting MPK38. Treatment with DNCB increased the stability of STRAP as well as the phosphorylation level of STRAP at Ser188 compared to control untreated cells (Fig. 7B, upper). Consistent with this, knockdown of endogenous MPK38 considerably decreased the stability of STRAP compared to control cells expressing a scrambled shRNA (Fig. 7B, lower). Similar trends were also observed in the phosphorylation level of STRAP at Ser188 (Fig. 7B, lower, middle panel). These results suggest that STRAP Ser188 phosphorylation by MPK38 may contribute to the regulation of STRAP stability. Because Cys339 and Cys377 of MPK38 were previously shown to be involved in binding of Trx,17 a destabilizer of MPK38, we also examined the effect of MPK38-mediated STRAP Ser188 phosphorylation on MPK38-Trx complex formation in vitro and in vivo (Fig. 7C). In both cases, WT STRAP remarkably decreased the degree of complex formation between MPK38 and Trx, resulting in enhanced MPK38 stability. However, the S188A STRAP mutant had no such effect. This finding suggests a novel role for MPK38-mediated STRAP Ser188 phosphorylation in the positive regulation of MPK38 stability. Taken together, these data strongly support a role for MPK38-mediated STRAP Ser188 phosphorylation in enhancing the stability of STRAP, leading to pro-apoptotic STRAP activity through ASK1, TGF-β, p53, and PI3K/PDK1 signaling pathways.
Figure 7.
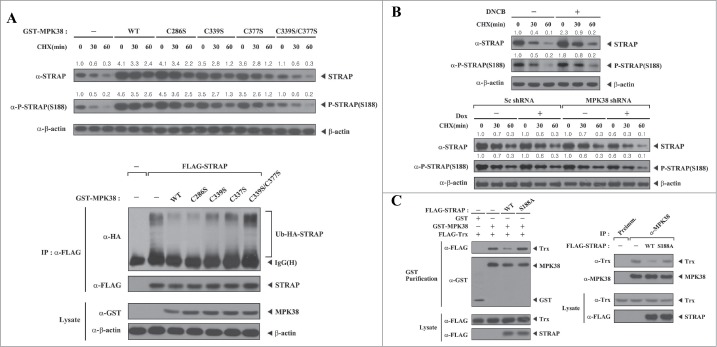
Enhanced STRAP stability induced by MPK38-mediated STRAP Ser188 phosphorylation. (A) The in vitro effect of MPK38-mediated STRAP Ser188 phosphorylation on the stability and ubiquitination of STRAP. HEK293 cells were transfected with pCMV2-FLAG empty vector (-) or expression vectors for wild-type (WT) or mutant forms (C286S, C339S, C377S, and C339S/C377S) of MPK38, and the stability and Ser188 phosphorylation level of STRAP were assessed by immunoblotting with anti-STRAP and anti-phospho-STRAP(S188) antibodies (upper). Time intervals indicate the number of minutes after cycloheximide (CHX) treatment (20 μg/ml). The relative level of STRAP stability and phosphorylation was quantified by densitometric analyses. The fold increase relative to untreated HEK293 cells expressing an empty vector was calculated. To determine the effect of MPK38-mediated STRAP Ser188 phosphorylation on STRAP ubiquitination (lower), HEK293 cells were transfected with expression vectors for HA-tagged ubiquitin (Ub), FLAG-STRAP, and glutathione S-transferase (GST)-tagged WT MPK38 or one of the cysteine mutants of MPK38 (C286S, C339S, C377S, and C339S/C377S; 2 μg each), as indicated. Cell lysates were subjected to immunoprecipitation using an anti-FLAG antibody (IP:α-FLAG) followed by immunoblotting with an anti-HA antibody to determine the level of STRAP ubiquitination (lower, top panel). The level of immunoprecipitated STRAP was verified by immunoblotting with an anti-FLAG antibody (lower, 2nd panel). (B) The in vivo effect of MPK38-mediated STRAP Ser188 phosphorylation on the stability of STRAP. HEK293 cells were treated with (+) or without (−) 1-chloro-2,4-dinitrobenzene (50 μM) for 30 min and further incubated with CHX (20 μg/ml) for the indicated time interval (min). Cell lysates were subjected to immunoblot analyses using anti-STRAP and anti-phospho-STRAP(S188) antibodies (upper). Inducible MPK38 or scrambled (Sc) shRNA HEK293 cells were treated with or without doxycycline (Dox: 1 μg/ml, 72 h) and then incubated with CHX (20 μg/ml) for the indicated time interval (min), followed by immunoblot analysis using anti-STRAP and anti-phospho-STRAP(S188) antibodies (lower). The relative level of STRAP stability and phosphorylation was quantified by densitometric analyses. The fold increase relative to untreated parental or scrambled shRNA HEK293 cells was calculated. (C) The effect of MPK38-mediated STRAP Ser188 phosphorylation on complex formation between MPK38 and Trx in vitro and in vivo. Exogenous MPK38-Trx complex formation in the presence of WT or mutant (S188A) STRAP was determined by immunoblot analysis using an anti-FLAG antibody (left). Endogenous MPK38-Trx complex formation in the presence of WT or mutant (S188A) STRAP was determined by immunoblot analysis using an anti-Trx antibody (right). β-actin was used as a loading control.
MPK38-mediated STRAP Ser188 phosphorylation is required for STRAP-dependent cell death in vivo. To investigate the importance of STRAP Ser188 phosphorylation by MPK38 for STRAP-dependent cell death in vivo, we first performed assays to determine binding and phosphorylation levels using DNCB. Exposure of HEK293 cells to DNCB increased MPK38-STRAP complex formation and resultant STRAP Ser188 phosphorylation in the presence or absence of apoptotic stimuli, including H2O2, TGF-β1, and 5FU (Fig. 8A, left top). On the other hand, compared to untreated control cells, DNCB treatment decreased the association of MPK38 with Trx and subsequent Trx Thr76 phosphorylation (Fig. 8A, left middle), consistent with the previous report.17 We also investigated whether MPK38 activity is proportional to the phosphorylation level of STRAP at Ser188 in in vitro kinase assays. Indeed, treatment with DNCB increased the kinase activity of MPK38 under the same conditions (Fig. 8A, left bottom), indicating that MPK38 activation is accompanied by STRAP Ser188 phosphorylation. We then confirmed these results using an inducible MPK38 shRNA system. The levels of Trx Thr76 phosphorylation and MPK38-Trx complex formation were considerably increased in MPK38-knockdown cells compared to control cells expressing a scrambled shRNA (Fig. 8B, left middle). By contrast, knockdown of endogenous MPK38 decreased MPK38-STRAP complex formation and resultant STRAP Ser188 phosphorylation (Fig. 8B, left top), suggesting that phosphorylation of STRAP Ser188 by MPK38 is tightly associated with the activation of MPK38. Furthermore, to demonstrate the correlation between MPK38-mediated STRAP Ser188 phosphorylation and STRAP-dependent cell death through ASK1, TGF-β, and p53 signaling pathways,10,15,16 we performed a GFP-based apoptosis assay under the same conditions. Indeed, exposure of HEK293 cells to DNCB markedly increased cell death in both the presence and absence of apoptotic stimuli (H2O2, TGF-β1, and 5FU) compared to untreated control cells (Fig. 8A, right). Consistent with this, an opposing trend was observed under the same conditions in an inducible MPK38 shRNA system (Fig. 8B, right). We also compared the effect of STRAP Ser188 phosphorylation by MPK38 on STRAP-dependent cell death in STRAP-null MEF cells vs. STRAP-intact MEF cells. Cell death in the STRAP-null MEF cells was much lower than in the STRAP-intact MEF cells (Fig. 8C), again demonstrating that MPK38-mediated STRAP Ser188 phosphorylation plays a pivotal role in STRAP-mediated cell death in vivo.
Figure 8.

A critical role for MPK38-mediated STRAP Ser188 phosphorylation in STRAP-dependent cell death through ASK1/TGF-β/p53 signaling pathways. (A) HEK293 cells were treated with 1-chloro-2,4-dinitrobenzene (DNCB) (50 μM) for 30 min and further incubated with (+) or without (−) the following stimuli: H2O2 (5 mM, 30 min), TGF-β1 (100 ng/ml, 20 h), or 5-fluorouracil (5FU: 0.38 mM, 30 h). STRAP or Trx immunoprecipitates (IP) were subjected to immunoblot analyses with anti-MPK38, anti-phospho-STRAP(S188), and anti-phospho-Trx(T76) antibodies to assess the association between endogenous MPK38 and STRAP (or Trx) and the level of STRAP Ser188 (or Trx Thr76) phosphorylation. The kinase activity of MPK38 was determined by in vitro kinase assays with ZPR9 as a substrate (IP:α-MPK38). Results were expressed as mean SEM (*P < 0.05, **P < 0.01). (B) HEK293 cells expressing inducible MPK38 or scrambled shRNA were incubated with (+) or without (−) the stimuli described above in the presence or absence of doxycycline (Dox: 1 μg/ml, 72 h). To determine the endogenous association between MPK38 and STRAP (or Trx) and the level of STRAP Ser188 (or Trx Thr76) phosphorylation, STRAP or Trx immunoprecipitates were subjected to immunoblot analyses with anti-MPK38, anti-phospho-STRAP(S188), and anti-phospho-Trx(T76) antibodies. Sc, scrambled. Results were expressed as mean SEM (*P < 0.05). (C) STRAP-null or intact MEF cells were treated with DNCB and further incubated with (+) or without (−) the stimuli described in Fig. 8A. The levels of STRAP Ser188 phosphorylation and MPK38 kinase activity were determined by immunoblot analyses and in vitro kinase assays. Apoptotic cell death under the same conditions was determined using the green fluorescent protein expression system (A–C). The relative levels of association between MPK38 and STRAP (or Trx), STRAP Ser188 (or Trx Thr76) phosphorylation, and MPK38 kinase activity were quantified by densitometric analyses. The fold increase relative to untreated parental HEK293, scrambled shRNA HEK293, or STRAP-intact MEF cells was calculated. Results were expressed as mean SEM (**P < 0.01, ***P < 0.001 versus STRAP-intact MEF). Sc, scrambled; P-ZPR9, phosphorylated ZPR9; P-MPK38, autophosphorylated MPK38.
MPK38-mediated STRAP Ser188 phosphorylation induces cell death by modulating key steps in ASK1, TGF-β, p53, and PI3K/PDK1 signaling pathways. Because STRAP was shown to interact with ASK1, Smad3, p53, and PDK1 and to regulate their respective signaling pathways,1,2,6,8 we performed in vivo binding assays to examine whether MPK38-mediated STRAP Ser188 phosphorylation influenced the association between STRAP and ASK1, Smad3, p53, and PDK1. The association between STRAP and ASK1, Smad3, and PDK1 was decreased in the presence of WT STRAP compared to the S188A STRAP mutant, whereas WT STRAP contributed to the increase in STRAP-p53 association (Fig. 9A, upper). These results indicate that STRAP Ser188 phosphorylation by MPK38 plays an important role in inducing STRAP-mediated cell death through ASK1, TGF-β, p53, and PI3K/PDK1 signaling pathways. To define in more detail the mechanism by which STRAP-dependent signaling through ASK1, TGF-β, p53, and PI3K/PDK1 is regulated, we also examined the effect of the S188A STRAP mutant on the association between ASK1 and MKK3, the type I TGF-β receptor (TβR1) and Smad3, p53 and Mdm2, and PDK1 and AKT1, which are key steps in their respective signaling pathways. The association between ASK1 and MKK3, as well as between TβR1 and Smad3, was considerably increased by WT STRAP compared to the S188A mutant. On the other hand, WT STRAP decreased the binding between both p53 and Mdm2 and PDK1 and AKT1 (Fig. 9A, lower). These results indicate that STRAP Ser188 phosphorylation by MPK38 contributes to the stimulation of ASK1, TGF-β, and p53 signaling pathways as well as to the inhibition of the PI3K/PDK1 signaling pathway, leading to STRAP-mediated cell death. Together, these data provide evidence that phosphorylation of STRAP Ser188 by MPK38 regulates STRAP-dependent ASK1, TGF-β, p53, and PI3K/PDK1 signaling at the level of either complex formation between STRAP and ASK1, Smad3, p53, and PDK1 or at respective key signaling steps, such as ASK1-MKK3, TβR1-Smad3, p53-Mdm2, and PDK1-AKT1 complex formation.
Figure 9.

Modulation of key signaling complexes in ASK1/TGF-β/p53/PDK1 signaling pathways by MPK38-mediated STRAP Ser188 phosphorylation. (A) To examine the effect of MPK38-mediated STRAP Ser188 phosphorylation on the interaction between STRAP and ASK1, Smad3, p53, or PDK1, HEK293 cells were transfected with the indicated combinations of expression vectors for ASK1, Smad3, p53, and PDK1, together with wild-type (WT) and mutant (S188A) STRAP. Cell lysates were subjected to immunoprecipitation with an anti-HA antibody (IP:α-HA) or glutathione-Sepharose beads (GST purification), and complex formation between STRAP and its binding partners (ASK1, Smad3, p53, and PDK1) was assessed by immunoblot analysis with an anti-FLAG antibody (upper, top panels). To determine the effect of MPK38-mediated STRAP Ser188 phosphorylation on in vivo complex formation between ASK1 and MKK3, the type I TGF-β receptor (TβR1) and Smad3, p53 and Mdm2, and PDK1 and AKT1, WT and mutant (S188A) STRAP were transfected into HEK293 cells, and the cell lysates were subjected to immunoprecipitation using the indicated antibodies. Immune complexes were analyzed for the presence of ASK1, Smad3, Mdm2, and AKT1 by immunoblot analyses using anti-ASK1, anti-Smad3, anti-Mdm2, and anti-AKT1 antibodies (lower, top panels). The amount of immunoprecipitated MKK3, TβR1, p53, and PDK1 was analyzed by immunoblot using the indicated antibodies (lower, 2nd panels). (B) The effect of MPK38-mediated phosphorylation-defective mutants (T838A of ASK1, S204A of Smad3, S15A of p53, and T354A of PDK1) on complex formation between ASK1 and MKK3, activated type I TGF-β receptor [TβR1(TD)] and Smad3, p53 and Mdm2, and PDK1 and AKT1. HEK293 cells were transfected with the indicated combinations of expression vectors for MKK3, TβR1(TD), Mdm2, and AKT1, together with WT and MPK38-mediated phosphorylation-defective mutants of ASK1, Smad3, p53, and PDK1. Cell lysates were subjected to precipitation with glutathione-Sepharose beads (GST purification), and complex formation was assessed by immunoblot analysis using an anti-FLAG antibody (top panels). The experiments were performed at least 2 times with similar results. (C) A proposed model for the role of MPK38-mediated STRAP Ser188 phosphorylation in cell death signaling through ASK1, TGF-β, p53, and PI3K/PDK1.
STRAP Ser188 phosphorylation induced by adenoviral delivery of MPK38 increases hepatic cell death signaling through ASK1, TGF-β, p53, and PI3K/PDK1 in mice. To demonstrate the role of MPK38-mediated STRAP Ser188 phosphorylation in STRAP-mediated cell death through ASK1, TGF-β, p53, and PI3K/PDK1 signaling pathways in vivo, we performed adenoviral delivery of MPK38 in mice. Adenoviruses for GFP, WT MPK38, and kinase-dead (K40R) MPK38 were injected via the tail vein. Ad-MPK38 infection, but not Ad-K40R infection, significantly stimulated ASK1, TGF-β, and p53 signaling pathways and inhibited the PI3K/PDK1 signaling compared to mice injected with GFP (vector alone) or control WT (-) mice (Fig. 10A). Concomitantly, there was a significant increase in the levels of STRAP Ser188 phosphorylation in mice expressing WT MPK38, but not in those injected with kinase-dead MPK38, compared to mice injected with GFP (V) or control WT (-) mice (Fig. 10B, 4th panel). These findings demonstrate a critical role for MPK38-mediated STRAP Ser188 phosphorylation in inducing hepatic cell-death signaling through ASK1, TGF-β, p53, and PI3K/PDK1. Western blot analysis clearly indicated that both adenoviral MPK38 and K40R were successfully delivered to the mouse liver in this analysis (Fig. 10B, top panel). We also observed elevated levels of serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST), which act as biochemical markers of hepatocellular injury, in WT MPK38 mice, reflecting enhanced apoptotic signaling in mouse livers infected with Ad-MPK38 (Fig. 10C). Consistent with this, adenoviral delivery of MPK38 resulted in a significant increase in hepatic cell death/nuclei DNA fragmentation, as judged by in situ cell-death detection (TUNEL) assay, in a kinase-dependent manner, probably resulting from the effect of MPK38 on STRAP-dependent ASK1, TGF-β, p53, and PI3K/PDK1 signaling via phosphorylation of STRAP at Ser188 (Fig. 10D). Taken together, these data indicate that STRAP Ser188 phosphorylation by MPK38 leads to cell death in vivo by stimulating STRAP-dependent signaling through ASK1, TGF-β, and p53, as well as by inhibiting STRAP-dependent PI3K/PDK1 signaling.
Figure 10.
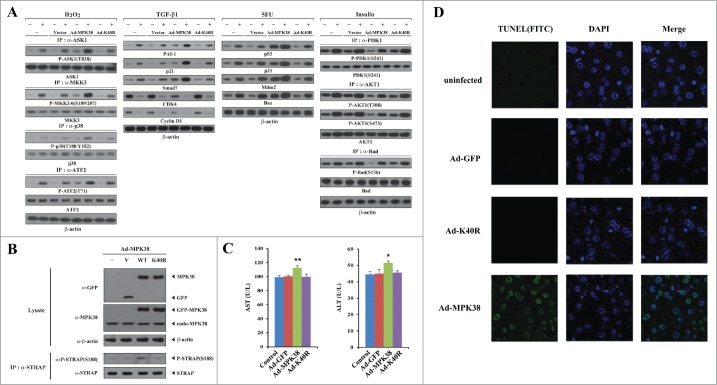
Stimulation of hepatic death signaling through ASK1, TGF-β, p53, and PI3K/PDK1 by adenoviral delivery of MPK38 in mice. At 5–6 days after injection of adenovirus-expressing green fluorescent protein (GFP) (V) (n = 3), wild-type (WT) MPK38 (n = 3), or kinase-dead (K40R) MPK38 (n = 3), mice were sacrificed and primary hepatocytes were prepared from livers as described in “Materials and methods." Immunoblot analyses were performed using the indicated antibodies to examine the effect of adenoviral delivery of MPK38 on hepatic cell death through ASK1/TGF-β/p53/PDK1 signaling pathways (A), as described in Figs. 3–6. The expression levels of GFP and MPK38, as well as the phosphorylation level of STRAP at Ser188, were also determined by immunoblot analysis using the indicated antibodies (B). The levels of serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were measured in mice injected with Ad-GFP, Ad-MPK38, or Ad-K40R (C). Results were expressed as mean SEM (*P < 0.05, **P < 0.01 vs. control). Nuclei DNA fragmentation was determined by TUNEL staining, as described in “Materials and methods," in mouse liver 5–6 days after injection of Ad-GFP, Ad-MPK38, or Ad-K40R (D). Control, WT mice; V, vector; Ad, adenovirus.
Discussion
MPK38 has been shown to stimulate the TGF-β signaling pathway by interacting with and phosphorylating Smad proteins, suggesting that cross-talk occurs between MPK38 and TGF-β signaling via Smad proteins.16 It has been reported that STRAP directly interacts with Smad proteins and suppresses TGF-β signaling.1 STRAP has also been shown to stimulate PDK1 and p53 activity via direct interaction.2,8 STRAP was recently shown to interact with ASK1 and inhibit its activity and function.6 In addition, we have shown that MPK38 stimulates ASK1 and p53 signaling pathways and inhibits PI3K/PDK1 signaling through direct interaction and phosphorylation, leading to cell death.10,14,15 Therefore, it is likely that STRAP functionally links MPK38 and ASK1, TGF-β, p53, and PI3K/PDK1 signaling pathways.
In the present study, we demonstrated that MPK38 directly associates with, and phosphorylates, STRAP at Ser188 (Fig. 2), suggesting that STRAP is a substrate for MPK38. STRAP has been shown to interact with ASK1 and p53 in a redox-dependent fashion through their cysteine residues, thereby regulating ASK1 and p53 function.6,8 In addition, a critical role for the redox potential in the regulation of MPK38 activity and function was recently demonstrated.17 These observations prompted us to investigate the role of cysteine residues in the interaction between MPK38 and STRAP using cysteine-to-serine amino acid-substitution mutants of MPK38 and STRAP. Our results revealed that a disulfide linkage involving Cys339 and Cys377 of MPK38 and Cys152 and Cys270 of STRAP mediated complex formation (Fig. 1C), indicating a redox dependency of the interaction between MPK38 and STRAP. This result also suggests that STRAP competes with Trx for binding of MPK38 because both STRAP and Trx utilize the same binding sites of MPK38, namely, Cys339 and Cys377. Moreover, the kinase activity of MPK38 was required for its ability to regulate STRAP-mediated transcription and apoptosis (see Figs. 3–6), implying that phosphorylation of STRAP Ser188 by MPK38 plays a crucial role in the regulation of STRAP-mediated ASK1, TGF-β, p53, and PDK1 function.
When considering the possible mechanism by which MPK38-mediated STRAP Ser188 phosphorylation contributes to cell death through ASK1, TGF-β, p53, and PI3K/PDK1 signaling pathways, it would be interesting to investigate whether STRAP Ser188 phosphorylation can influence complex formation between ASK1 and MKK3, the TGF-β receptor and Smad3, p53 and Mdm2, or PDK1 and AKT1, because a key interaction between the TGF-β receptor and Smad proteins, which is essential for propagating TGF-β signaling, was previously shown to be modulated by MPK38.16 p53-Mdm2 and PDK1-AKT1 complex formation, key steps in the activation of p53 and PI3K/PDK1 signaling pathways, respectively, were decreased in cells expressing WT STRAP (Fig. 9A) compared to cells expressing the S188A STRAP mutant, which is defective in MPK38-mediated phosphorylation (see Fig. 2). These observations indicate that phosphorylation of STRAP Ser188 by MPK38 modulates complex formation between both p53 and Mdm2 and PDK1 and AKT1, leading to the stimulation of p53-induced cell death as well as the inhibition of PI3K/PDK1-mediated cell growth. Conversely, compared to the S188A STRAP mutant, WT STRAP considerably increased complex formation between the TGF-β receptor and Smad3, resulting in the stimulation of TGF-β signaling. In addition, WT STRAP stabilized the interaction between ASK1 and MKK3, leading to the stimulation of ASK1-mediated apoptosis (Fig. 9A). These findings provide evidence that STRAP Ser188 phosphorylation by MPK38 plays a pivotal role in inducing STRAP-mediated cell death through ASK1, TGF-β, p53, and PI3K/PDK1 signaling pathways. Because MPK38 has been demonstrated to directly phosphorylate ASK1, Smad3, p53, and PDK1 to promote its pro-apoptotic activity,10,14-16 it is possible that direct phosphorylation of ASK1, Smad3, p53, and PDK1 by MPK38 may be part of the mechanism that controls MPK38-induced cell death through ASK1/TGF-β/p53/PDK1 signaling pathways. To demonstrate this, we analyzed the effect of MPK38-mediated phosphorylation-defective mutants (T838A of ASK1, S204A of Smad3, S15A of p53, and T354A of PDK1) on ASK1-MKK3, activated type I TGF-β receptor [TβR1(TD)]-Smad3, p53-Mdm2, and PDK1-AKT1 complex formation using in vivo binding assays. As shown in Fig. 9B, all mutants that were defective for MPK38-mediated phosphorylation showed decreased cell-death signaling through ASK1, TGF-β, p53, and PI3K/PDK1. This suggests a model in which, in addition to STRAP Ser188 phosphorylation by MPK38, MPK38-mediated phosphorylation of ASK1 at Thr838, Smad3 at Ser204, p53 at Ser15, and PDK1 at Thr354 may serve as key events for MPK38-dependent cell death through ASK1, TGF-β, p53, and PI3K/PDK1 signaling pathways in the presence of various apoptotic stresses (Fig. 9C).
Multisite phosphorylation can modulate protein function. There are a number of situations in which one phosphorylation event agonizes and another antagonizes the activity of the same protein. For example, previous studies have shown that multisite phosphorylation of Smad proteins by various intracellular protein kinases plays an important role in controlling Smad-mediated signaling.22 We previously suggested the involvement of ASK1-mediated phosphorylation of STRAP at Thr175 and Ser179 in the suppression of ASK1-induced apoptosis in the resting state.6 In addition, PDK1-mediated phosphorylation of STRAP at Ser219 was shown to potentiate STRAP-mediated inhibition of TGF-β signaling, contributing to the cell survival (data not shown). On the other hand, our present results provide evidence that STRAP Ser188 phosphorylation by MPK38 plays a pivotal role in the induction of STRAP-mediated cell death through ASK1, TGF-β, p53, and PI3K/PDK1 signaling pathways. These findings indicate that different STRAP phosphorylations by various intracellular protein kinases, including MPK38, ASK1, and PDK1, appear to have different roles in the regulation of STRAP-mediated functions, such as cell survival and death. Therefore, we hypothesize that multisite phosphorylation of STRAP by various intracellular protein kinases plays a key role in determining the cellular decision between the pro-apoptotic and anti-apoptotic functions of STRAP.
In summary, our present results suggest a possible mechanism by which STRAP stimulates ASK1, TGF-β, and p53 signaling and inhibits PI3K/PDK1 signaling, leading to cell death. STRAP was phosphorylated at Ser188 by MPK38, and this, in turn, resulted in STRAP-dependent cell death through the modulation of complex formation between key signaling components essential for ASK1/TGF-β/p53/PDK1 signaling pathways in cells. In this respect, it is possible that STRAP may serve as a medium of bonding between MPK38 and ASK1, TGF-β, p53, and PI3K/PDK1 signaling pathways. The discovery that MPK38-mediated STRAP Ser188 phosphorylation can trigger STRAP-mediated cell death, together with STRAP phosphorylation at Thr175 and Ser179 by ASK1 for STRAP-mediated inhibition of ASK1-induced cell death,6 may be a possible explanation for how the pro- or anti-apoptotic function of STRAP is determined. Therefore, further studies to identify STRAP phosphorylation sites linked to cell growth or death will be necessary to clarify the importance of multisite phosphorylation of STRAP in the differential regulation of STRAP-mediated functions.
Materials and Methods
Cell culture, transfection, plasmids, and antibodies. HEK293, 293T, HepG2, MCF7, and HaCaT cells were maintained in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% fetal bovine serum (FBS; Invitrogen) and antibiotics (Invitrogen), and transiently transfected using WelFect-ExTM Plus according to the manufacturer's instructions.15 STRAP-null MEF cells were a kind gift from Dr. B. Stefanovic (Florida State University, Tallahassee, USA). The WT and kinase-dead (K40R) MPK38 plasmids, p3TP-Lux reporter, p53-Luc reporter, activator protein 1 (AP-1)-Luc reporter, FLAG-tagged WT STRAP and 8 STRAP deletion constructs (WD1–3, WD4–7, WD4, WD4(C152S), WD6, WD7, WD7(C270S), and CT) have been described previously.6 The cysteine-to-serine single and double amino acid-substitution mutants of STRAP (C152S, C270S, and C152S/C270S) and MPK38 (C286S, C339S, C377S, and C339S/C377S) have been described previously.6,17 Anti-FLAG (M2), anti-His, anti-Myc, anti-STRAP, anti-MPK38, anti-GST, anti-hemagglutinin (HA), anti-CDK4, anti-Cyclin D1, anti-Smad3, anti-Smad7, anti-p53(DO-1), anti-Mdm2, anti-Bax, anti-PAI-1, anti-p21, anti-β-actin, anti-phospho-ASK1(T845), anti-ASK1, anti-phospho-MKK3/6(S189/207), anti-MKK3, anti-phospho-p38(T180/Y182), anti-p38, anti-phospho-ATF2(T71), anti-ATF2, anti-phospho-PDK1(S241), anti-PDK1, anti-phospho-AKT1(T308), anti-phospho-AKT1(S473), anti-AKT1, anti-phospho-Bad(S136), anti-Bad, anti-phospho-Trx(T76), anti-Trx, and anti-phospho-Ser/Thr antibodies have been described elsewhere.2,6,10,15,17,23,24 The anti-rabbit phospho-STRAP(S188) antibody was raised against a synthetic phosphopeptide antigen NMSVSSMEYIP, where S represents phosphoserine (Young In Frontier, Seoul, Korea).
Inducible MPK38 shRNA cell line and in vitro interaction. The inducible MPK38 shRNA HEK293 cell line has been described previously.15,16 To determine the in vitro interaction between MPK38 and STRAP proteins, autophosphorylated recombinant MPK38 (∼2 μg) was incubated with unlabeled recombinant GST or GST-STRAP proteins (∼3 μg each) at 4°C for 1 h,8 and then analyzed using native polyacrylamide gel electrophoresis (PAGE).
Construction of STRAP mutants. The serine (or threonine) to alanine amino acid-substitution mutants of STRAP used for the in vitro kinase assays were produced by polymerase chain reaction (PCR) and cloned into the pGEX4T-1 (Amersham Biosciences) vector, as described previously.10 The following primers for STRAP, forward 5′-GCAAGCTTATGGCAATGAGACAGACG-3′, reverse 5′-GCCTCGAGGGCCTTAACATCAG GAGC-3′ (HindIII and XhoI sites, respectively, are underlined), were used in conjunction with one of the following mutant primers: for STRAP(S188A), forward 5′-AATATGTCTGTT AGTGCTATGGAATATATTCCT-3′ and reverse 5′-AGGAATATATTCCATAGCACTAACAGA CATATT-3′; for STRAP(S210A), forward 5′-TCTATTGCTTTTCATGCTGCAGTAAGTTTGG AC-3′ and reverse 5′-GTCCAAACTTACTGCAGCATGAAAAGCAATAGA-3′; and for STRAP(T326A), forward 5′-GGTTTTCCAGAGACAGCAGAAGAGGAGCTAGAA-3′ and reverse 5′-TTCTAGCTCCTCTTCTGCTGTCTCTGGAAAACC-3′.
Recombinant proteins and MPK38 kinase assay. Recombinant GST-tagged WT and mutant forms of STRAP or His-tagged MPK38 were purified by affinity chromatography on glutathione-Sepharose 4B or His columns (Amersham Bio Sciences). The recombinant MPK38 proteins or MPK38 proteins purified from HEK293 cells transfected with GST-MPK38 using glutathione-Sepharose beads were incubated at 37°C for 15 min with the recombinant GST-tagged WT or mutant forms of STRAP (∼3 μg each) or ZPR9 (∼500 ng) substrates in kinase buffer (50 mM HEPES, pH 7.4, 1 mM DTT, and 10 mM MgCl2) and 5 μCi of [γ-32P]-ATP, as described previously.15 The reaction mixtures were separated by SDS-PAGE and analyzed by autoradiography. Protein concentration was determined by the Bradford assay.
Small interfering RNA (siRNA) experiments. RNA interference was carried out in HEK293, HepG2, or MCF7 cells using the MPK38-specific siRNAs oligonucleotide #1 (5′-CAGGCAGACAAUGGAGGAUTT-3′), targeting amino acids 297–303 and oligonucleotide #2 (5′-AACCCAAGGGUAACAAGGATT-3′), targeting amino acids 156–162 of MPK38 (GenBankTM accession number NM010790), as well as a control scrambled siRNA (5′-GCGCGGGGCACGUUGGUGUTT-3′), as described previously.10
Luciferase reporter assay. MCF7, HepG2, or HEK293 cells were transfected with p53-Luc, p3TP-Lux, or AP-1-Luc reporter plasmids, along with each expression vector as indicated, using WelFect-ExTM Plus (WelGENE, Daegu, Korea), according to the manufacturer's instructions. The cells were lysed and luciferase activity was detected using a dual luciferase assay kit (Promega), as described previously.2 The data were normalized to the expression levels of a co-transfected β-galactosidase reporter control.
Green fluorescent protein-based apoptosis assay and in situ cell-death detection (TUNEL). GFP-based cell-death experiments were carried out in 293T, HaCaT, or MCF7 cells transfected with the indicated expression vectors, as described previously.10,16 The nuclei of GFP-positive cells were stained with 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI) and analyzed for apoptotic morphology under a fluorescence microscope. The percentage of apoptotic cells was calculated as the number of GFP-positive cells with apoptotic nuclei divided by the total number of GFP-positive cells. Apoptosis in paraffin-embedded tissue sections obtained from mouse liver infected with Ad-GFP, Ad-MPK38, or Ad-K40R was assessed by detection of DNA fragmentation using the in situ cell-death detection kit, POD (Roche Diagnostics), according to the manufacturer's instructions. Mounting solution containing DAPI was used to counterstain nuclei. Sections were visualized under a fluorescence microscope (Zeiss, Oberkochen, Germany).
Assays for protein stability and ubiquitination. For measuring the stability of STRAP, HEK293 cells transfected with an empty vector or the WT or mutant forms of MPK38 (C286S, C339S, C377S, and C339S/C377S), as indicated, or HEK293 cells treated with or without DNCB were used for immunoblot analysis with an anti-STRAP antibody. Time intervals indicate the number of minutes after cycloheximide (CHX) treatment (20 μg/ml). β-actin was used as a loading control. The ubiquitination assay was performed using HEK293 cells transfected with expression plasmids encoding STRAP, WT and mutant forms of MPK38 (C286S, C339S, C377S, and C339S/C377S), and HA-tagged ubiquitin, as described previously.7
Cell-cycle analysis. Assays were performed using HaCaT cells (2 × 105/60 mm dish) transfected with the indicated combinations of plasmid vectors (empty vector, STRAP, S188A, and MPK38), as described previously.16 The fraction of cells in each stage of the cell cycle was analyzed after 10% serum treatment for 24 h in the presence or absence of TGF-β1 (2 ng/ml). Cell-cycle analysis was performed using a FACSCalibur-S system (BD Biosciences).
Isolation of primary mouse hepatocytes. Primary hepatocytes were prepared from the livers of 8-10-week-old male C57BL/6 mice by the collagenase perfusion method, as described previously,25 with some modifications. Mice were anesthetized and the abdomen was surgically opened. Each liver was perfused at a flow rate of 25 ml/min for 10 min via the hepatic portal vein with pre-warmed (37°C) Krebs-Henseleit buffer without Ca2+ and SO42- (115 mM NaCl, 25 mM NaHCO3, 5.9 mM KCl, 1.18 mM MgCl2, 1.23 mM NaH2PO4, and 6 mM glucose), followed by perfusion at a flow rate of 20 ml/min for 10 min with pre-warmed (37°C) collagenase type II solution (20 mg collagenase type II in 100 ml Krebs-Henseleit buffer without SO42− but containing 0.1 mM CaCl2 and 3% BSA). The liver was transferred into a petri dish containing 5 ml collagenase type II solution and then cut into small pieces, and finally flushed with ice-cold Krebs-Henseleit buffer containing 1.2 mM Na2SO4 and 1.25 mM CaCl2 using a household sieve. The cell suspension was then filtrated through a cell strainer (70 μm nylon filter) into a 50 ml tube. Approximately 10 ml ice-cold DMEM was added to the filtrated cell suspension, which was subsequently centrifuged at 500 rpm for 2 min at 4°C. The supernatant was aspirated, and the remaining hepatocyte pellet was washed with 10 ml ice-cold DMEM 3 times. The cell suspension was finally adjusted to a density of 17% iodixanol in Krebs-Henseleit buffer containing 1.25 mM CaCl2 and 1.2 mM Na2SO4. Hepatocytes were isolated after centrifugation at 1,400 rpm for 20 min at 4°C.
Recombinant adenoviruses and animal experiments. To prepare adenoviruses for expression of WT and kinase-dead (K40R) MPK38, pEBG-MPK38 (WT and K40R) plasmids were used as templates for PCR with forward (5′-GTAACTATAACGGTCATGAAAGATTA TGACGAACTCCTCAAA-3′; the MPK38 sequence is underlined) and reverse (5′-ATTACCTCTTTCTCCTCACATCTTGCAGCCAGACAAGAT-3′; the MPK38 sequence is underlined) primers using Advantage HD polymerase mix (Clontech). The reaction parameters were 5 min at 95°C for 1 cycle; 45 s at 95°C, 1.45 min at 58°C, and 2 min at 72°C for 18 cycles; and 5 min at 72°C for 1 cycle. The amplified PCR products were purified from an agarose gel using the Nucleospin gel and PCR clean-up kit (Clontech) and then subcloned into a pAdenoX vector (Clontech). The recombinant adenoviral constructs were digested with PacI and used for transfection into HEK293 cells to produce recombinant adenoviruses. The viruses were purified by cesium chloride density centrifugation and dialyzed into PBS buffer. The virus titers were determined by Adeno-X Rapid Titer Kit (Clontech) according to the manufacturer's instructions. Adenovirus expressing GFP was used as a control. Male, 8-10-week-old C57BL/6 mice were purchased from Central Lab. Animal Inc. (Seoul, Korea) and caged under specific pathogen-free (SPF) conditions with pathogen-free water and food for maintenance. Recombinant adenovirus (1 × 109 plaque-forming units) was delivered into mice by tail vein injection. All animal treatment procedures were approved by the Institutional Animal Care and Use Committee of the veterinary school at Chungbuk National University.
Statistical analysis. Results are derived from at least 3 independent experiments and expressed as mean ± SEM. Statistical analysis was carried out by one-way ANOVA followed by Tukey multiple comparison test using the computer program GraphPad Prism (Graph-Pad Software).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by the National Research Foundation of Korea Grant (2012R1A2A2A01010541).
References
- 1. Datta PK, Moses HL. STRAP and Smad7 synergize in the inhibition of transforming growth factor beta signaling. Mol Cell Biol 2000; 20:3157-67; PMID:10757800; http://dx.doi.org/ 10.1128/MCB.20.9.3157-3167.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Seong H-A, Jung H, Choi H-S, Kim K-T, Ha H. Regulation of transforming growth factor-beta signaling and PDK1 kinase activity by physical interaction between PDK1 and serine-threonine kinase receptor-associated protein. J Biol Chem 2005; 280:42897-908; PMID:16251192; http://dx.doi.org/ 10.1074/jbc.M507539200 [DOI] [PubMed] [Google Scholar]
- 3. Halder SK, Anumanthan G, Maddula R, Mann J, Chytil A, Gonzalez AL, Washington MK, Moses HL, Beauchamp RD, Datta PK. Oncogenic function of a novel WD-domain protein, STRAP, in human carcinogenesis. Cancer Res 2006; 66:6156-66; PMID:16778189; http://dx.doi.org/ 10.1158/0008-5472.CAN-05-3261 [DOI] [PubMed] [Google Scholar]
- 4. Anumanthan G, Halder SK, Friedman DB, Datta PK. Oncogenic serine-threonine kinase receptor-associated protein modulates the function of Ewing sarcoma protein through a novel mechanism. Cancer Res 2006; 66:10824-32; PMID:17108118; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-1599 [DOI] [PubMed] [Google Scholar]
- 5. Brunen D, Willems SM, Kellner U, Midgley R, Simon I, Bernards R. TGF-β: an emerging player in drug resistance. Cell Cycle 2013; 12:2960-8; PMID:23974105; http://dx.doi.org/ 10.4161/cc.26034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jung H, Seong H-A, Manoharan R, Ha H. Serine-threonine kinase receptor-associated protein inhibits apoptosis signal-regulating kinase 1 function through direct interaction. J Biol Chem 2010; 285:54-70; PMID:19880523; http://dx.doi.org/ 10.1074/jbc.M109.045229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kashikar ND, Zhang W, Massion PP, Gonzalez AL, Datta PK. Role of STRAP in regulating GSK3β function and Notch3 stabilization. Cell Cycle 2011; 10:1639-54; PMID:21502811; http://dx.doi.org/ 10.4161/cc.10.10.15630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jung H, Seong H-A, Ha H. NM23-H1 tumor suppressor and its interacting partner STRAP activate p53 function. J Biol Chem 2007; 282:35293-307; PMID:17916563; http://dx.doi.org/ 10.1074/jbc.M705181200 [DOI] [PubMed] [Google Scholar]
- 9. Beullens M, Vancauwenbergh S, Morrice N, Derua R, Ceulemans H, Waelkens E, Bollen M. Substrate specificity and activity regulation of protein kinase MELK. J Biol Chem 2005; 280:40003-11; PMID:16216881; http://dx.doi.org/ 10.1074/jbc.M507274200 [DOI] [PubMed] [Google Scholar]
- 10. Jung H, Seong H-A, Ha H. Murine protein serine/threonine kinase 38 activates apoptosis signal-regulating kinase 1 via Thr 838 phosphorylation. J Biol Chem 2008; 283:34541-53; PMID:18948261; http://dx.doi.org/ 10.1074/jbc.M807219200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cohen P. The regulation of protein function by multisite phosphorylation – a 25 year update. Trends Biochem Sci 2000; 25:596-601; PMID:11116185; http://dx.doi.org/ 10.1016/S0968-0004(00)01712-6 [DOI] [PubMed] [Google Scholar]
- 12. Lin M-L, Park J-H, Nishidate T, Nakamura Y, Katagiri T. Involvement of maternal embryonic leucine zipper kinase (MELK) in mammary carcinogenesis through interaction with Bcl-G, a pro-apoptotic member of the Bcl-2 family. Breast Cancer Res 2007; 9:R17; PMID:17280616; http://dx.doi.org/ 10.1186/bcr1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Seong H-A, Kim K-T, Ha H. Enhancement of B-MYB transcriptional activity by ZPR9, a novel zinc finger protein. J Biol Chem 2003; 278:9655-62; PMID:12645566; http://dx.doi.org/ 10.1074/jbc.M207478200 [DOI] [PubMed] [Google Scholar]
- 14. Seong H-A, Jung H, Manoharan R, Ha H. PDK1 protein phosphorylation at Thr354 by murine protein serine-threonine kinase 38 contributes to negative regulation of PDK1 protein activity. J Biol Chem 2012; 287:20811-22; PMID:22544756; http://dx.doi.org/ 10.1074/jbc.M111.331827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Seong H-A, Ha H. Murine protein serine-threonine kinase 38 activates p53 function through Ser15 phosphorylation. J Biol Chem 2012; 287:20797-810; PMID:22532570; http://dx.doi.org/ 10.1074/jbc.M112.347757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Seong H-A, Jung H, Ha H. Murine protein serine/threonine kinase 38 stimulates TGF-beta signaling in a kinase-dependent manner via direct phosphorylation of Smad proteins. J Biol Chem 2010; 285:30959-70; PMID:20659902; http://dx.doi.org/ 10.1074/jbc.M110.138370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Manoharan R, Seong H-A, Ha H. Thioredoxin inhibits MPK38-induced ASK1, TGF-β, and p53 function in a phosphorylation-dependent manner. Free Radic Biol Med 2013; 63:313-24; PMID:23747528; http://dx.doi.org/ 10.1016/j.freeradbiomed.2013.05.020 [DOI] [PubMed] [Google Scholar]
- 18. Jakobsen SN, Hardie DG, Morrice N, Tornqvist HE. 5'-AMP-activated protein kinase phosphorylates IRS-1 on Ser-789 in mouse C2C12 myotubes in response to 5-aminoimidazole-4-carboxamide riboside. J Biol Chem 2001; 276:46912-6; PMID:11598104; http://dx.doi.org/ 10.1074/jbc.C100483200 [DOI] [PubMed] [Google Scholar]
- 19. Ichijo H, Nishida E, Irie K, ten Dijke P, Saitoh M, Moriguchi T, Takagi M, Matsumoto K, Miyazono K, Gotoh Y. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 1997; 275:90-4; PMID:8974401; http://dx.doi.org/ 10.1126/science.275.5296.90 [DOI] [PubMed] [Google Scholar]
- 20. Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J 1998; 17:2596-606; PMID:9564042; http://dx.doi.org/ 10.1093/emboj/17.9.2596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Seong H-A, Jung H, Kim K-T, Ha H. 3-Phosphoinositide-dependent PDK1 negatively regulates transforming growth factor-beta-induced signaling in a kinase-dependent manner through physical interaction with Smad proteins. J Biol Chem 2007; 282:12272-89; PMID:17327236; http://dx.doi.org/ 10.1074/jbc.M609279200 [DOI] [PubMed] [Google Scholar]
- 22. Wrighton KH, Lin X, Feng X-H. Phospho-control of TGF-beta superfamily signaling. Cell Res 2009; 19:8-20; PMID:19114991; http://dx.doi.org/ 10.1038/cr.2008.327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Seong H-A, Jung H, Ha H. NM23-H1 tumor suppressor physically interacts with serine-threonine kinase receptor-associated protein, a transforming growth factor-beta (TGF-beta) receptor-interacting protein, and negatively regulates TGF-beta signaling. J Biol Chem 2007; 282:12075-96; PMID:17314099; http://dx.doi.org/ 10.1074/jbc.M609832200 [DOI] [PubMed] [Google Scholar]
- 24. Jung H, Kim T, Chae HZ, Kim K-T, Ha H. Regulation of macrophage migration inhibitory factor and thiol-specific antioxidant protein PAG by direct interaction. J Biol Chem 2001; 276:15504-10; PMID:11297517; http://dx.doi.org/ 10.1074/jbc.M009620200 [DOI] [PubMed] [Google Scholar]
- 25. Chitraju C, Trötzmüller M, Hartler J, Wolinski H, Thallinger GG, Lass A, Zechner R, Zimmermann R, Kofeler HC, Spener F. Lipidomic analysis of lipid droplets from murine hepatocytes reveals distinct signatures for nutritional stress. J Lipid Res 2012; 53:2141-52; PMID:22872753; http://dx.doi.org/ 10.1194/jlr.M028902 [DOI] [PMC free article] [PubMed] [Google Scholar]