

AMP-activated protein kinase (AMPK) consists of a catalytic α subunit and regulatory β and γ subunits, and is activated in response to an increase in the AMP/ATP ratio upon metabolic stresses. Two AMPK kinases have been identified to mediate activating phosphorylation of AMPKα at Thr172, namely liver kinase B1 (LKB1) and Ca2+/calmodulin-dependent protein kinase kinase β (CaMKKβ) [reviewed in 1]. Increased AMP:ATP can activate AMPK allosterically by binding to the AMPKγ subunit and inhibiting dephosphorylation of Thr172 due to constitutive LKB1 activity. In cells expressing CaMKKβ, increased intracellular Ca2+ concentrations activate AMPK independent of changes in AMP:ATP. Once activated, AMPK phosphorylates multiple downstream catabolic targets that promote ATP generation, whereas anabolic processes including fatty acid synthesis and protein translation tend to be suppressed.1
During tumorigenesis and related depletion of cellular energy, AMPK activation is thought to favor energy-producing catabolic processes. The role of AMPK in prostate cancer (PC) remains to be fully characterized, with significant discrepancies in the literature. We have recently observed that AMPK Thr172 phosphorylation was significantly elevated in human prostate cancer (PC) cells and clinical PC samples. In clinical PC, while not statistically significant, there was a trend for increasing phospho-AMPK Thr172 with increasing Gleason sum score (or increasingly less differentiated disease). The increased AMPK Thr172 phosphoryation was associated with phosphorylation of the AMPK substrate, acetyl CoA-carboxylase (ACC; p = 0.0023), suggesting enhanced AMPK and inhibited ACC activities in PC.2 These findings support earlier reports of increased phospho-ACC levels in clinical PC.3
Functional significance of the observed upregulation of AMPK/ACC status in PC progression remains unclear. In DU145 cells, an androgen receptor (AR) negative human PC line with activated Akt and upregulated glycolysis, 5-aminoimidazole-4-carboxamide riboside (AICAR)-mediated AMPK activation has been reported to suppress proliferation without evidence of increased apoptosis.4 Our recent comparative analysis of the paired PC3 and PC3M cells as models of progressively invasive (androgen-independent) PC also supported the notion of tumor suppressing effects upon activation of AMPK (Fig. 1). Treatment with AICAR or an alternative direct AMPK activator, A-769662 suppressed proliferation, migration and invasion in both cell lines, with down-regulation of mTOR and P70S6Ki levels regardless of the Akt status. Involvement of AMPK was confirmed with the AMPK inhibitor, Compound C and siRNA-mediated AMPK silencing.2
Figure 1.
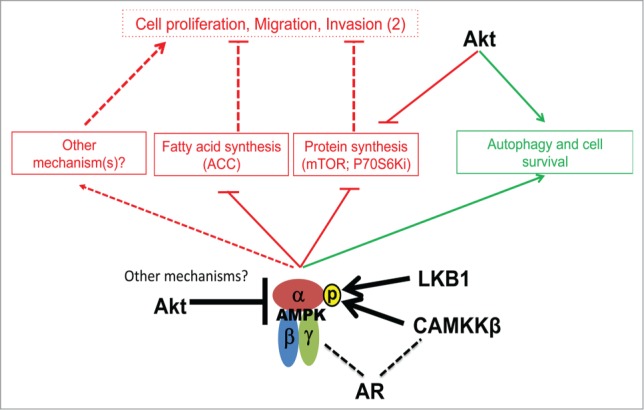
A schematic representation of signaling network involving AMPK and other key signaling nodes in prostate carcinogenesis. The AMPK-mediated anti-tumor effects (proliferative/migratory/invasive) are considered downstream of fatty acid and protein synthesis. Akt has both AMPK-dependent and -independent functions, with its effects on autophagy having potential pro-survival effect (highlighted in green). Androgen Receptor (AR) interacts with CAMKKβ (or CAMKK2) and AMPK to drive both hormone naïve and castration independent prostate cancer.
Despite similar functional responses in PC3 and PC3M cells, AMPK activation also resulted in sustained phospho-Akt activation in PC3M cells, but not PC3 cells. However, combining LY-294002 with AICAR to simultaneously suppress phosphatidylinositol 3′-kinase (PI3K) and activate AMPK did not produce any additional anti-proliferative effects in PC3M cells. This suggests that any efforts to exploit AMPK as a potential therapeutic target may be considered independent of PI3K/Akt signaling. This is a surprising finding as there is significant evidence for cross talk between AMPK and AKT/mTOR pathways. Indeed, the PI3K signaling network exhibits extensive cross-regulatory interactions with the intermediate metabolism network, including AMPK, to fuel resistance to stress and uncontrolled growth.5 Akt has also been reported to dramatically reduce the AMP/ATP ratio and suppress AMPK activity in cells overexpressing a constitutive Akt mutant.6 Hence, while the effects of AMPK activation was not influenced by a PI3K inhibitor in our study, this may be context and cell type specific, and a detailed understanding of AMPK/PI3K/mTOR crosstalk will have important implications in cancer therapies.
Popovics et al.7 have recently hypothesized that AMPK activators used in pre-clinical studies are likely to promote autophagy and apoptosis since they mediate their effects, at least in part, by an inhibitory action on the mTORC1 complex. They proposed that modulators of the CaMKKβ-AMPK pathway may be used in combination with mTOR-specific inhibitors, such as the rapamycin analogs, to maximise anti-tumor effects, thus avoiding the the potential of stimulating diverse pro-tumor processes in parallel with antitumor effects when mTOR inhibitor is used alone.
Ongoing research into the role of AMPK in the followings research priorities will provide important insight into its potential for targeted therapy: (1) The in vivo impact of AMPK-mediated signaling in the context of primary and metastatic diseases; (2) The complex interplay between CAMKKβ, androgen receptor and AMPK (Fig. 1); (3) Factors that modulate AMPK-induced effects on cell fate (including survival, apoptosis and autophagy) and motility; and (4) Factors influencing the metabolic consequences of AMPK functions, including glucose and lipid metabolism.
References
- 1.Hardie DG. Biochem Soc Trans 2011; 39(1):1–13; PMID:21265739; http://dx.doi.org/ 10.1042/BST0390001 [DOI] [PubMed] [Google Scholar]
- 2.Choudhury Y. Oncoscience 2014; 1:446–456; PMID:25594043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park HU. Mol Cancer Ther 2009; 8(4):733–41; PMID:19372545; http://dx.doi.org/ 10.1158/1535-7163.MCT-08-0631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jose C. Biochim Biophys Acta 2011; 1807(6):707–18; PMID:21692240; http://dx.doi.org/ 10.1016/j.bbabio.2010.12.002 [DOI] [PubMed] [Google Scholar]
- 5.Hahn-Windgassen A. J Biol Chem 2005; 280(37):32081–9; PMID:16027121; http://dx.doi.org/ 10.1074/jbc.M502876200 [DOI] [PubMed] [Google Scholar]
- 6.Tao R. J Mol Signal 2010; 5(1):1; PMID: 20167101; http://dx.doi.org/ 10.1186/1750-2187-5-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Popovics P. Expert Opin Ther Targets 2015; 1–16; PMID:25600663; http://dx.doi.org/ 10.1517/14728222.2015.1005603 [DOI] [PubMed] [Google Scholar]