

In mammals, the transcriptional activity of genes is often attenuated by epigenetic silencing mechanisms involving DNA hypermethylation and/or inhibitory histone modifications. Alterations in epigenetic regulation contribute to a number of developmental anomalies, genetic disorders and diseases, including cancer. Therefore, a detailed understanding of the mechanisms involved in epigenetic silencing is expected to shed light on these human disease states and suggest new therapeutic approaches.
Inhibitory histone modifications are promoted by a variety of epigenetic regulators including the Polycomb repressive complexes PRC1 and PRC2, which function to silence specific sets of genes.1 In the canonical PRC1/2-mediated silencing pathway, PRC2 first interacts with the promoter and catalyzes histone H3 lysine 27 (H3K27) trimethylation. This repressive mark is then recognized by PRC1, which consists of a histone H2A E3 ubiquitin ligase that mono-ubiquitinates H2A on lysine 119 (Fig. 1, left). In mammalian cells, the PRC1 component RNF2 is the major H2A ubiquitin ligase involved in gene regulation.
Figure 1.
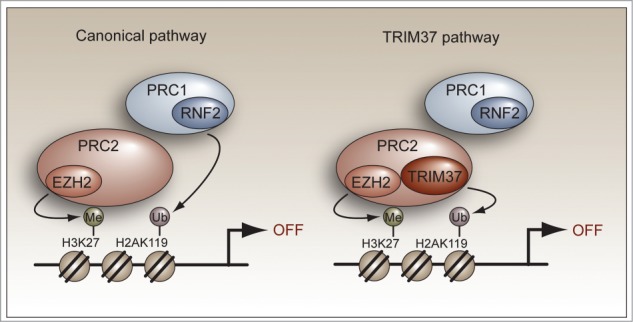
TRIM37 represses gene expression through a non-canonical PRC1/2-mediated silencing pathway. (Left) The canonical model for PRC1/2-mediated gene silencing. PRC2 first interacts with the promoter and catalyzes H3K27 trimethylation using the PRC2 subunit and histone methyltransferase EZH2. The H3K27 trimethylation mark is then recognized by PRC1 followed by RNF2-catalyzed H2A mono-ubiquitination. (Right) TRIM37-mediated target gene silencing. TRIM37 associates with PRC2 and promotes extensive changes in gene expression that include the silencing of multiple tumor suppressors. TRIM37 is also required for PRC1 occupancy, which presumably is mediated by PRC2 as in the canonical pathway.
We recently characterized a new epigenetic regulator, called TRIM37, that is involved in PRC1/2-mediated epigenetic repression.2 TRIM37 contains an RING finger domain that is a hallmark of E3 ubiquitin ligases, but its substrate(s) and function(s) remained unidentified. We demonstrated that TRIM37 functions as an H2A E3 ubiquitin ligase that, like RNF2, mono-ubiquitinates histone H2A at lysine 119. Notably, the TRIM37 gene is located in 17q23, a chromosomal region that is amplified in almost 40% of breast cancers. Interestingly, in breast cancer cell lines harboring 17q23 amplification, RNF2 levels are greatly reduced, suggesting TRIM37 might functionally replace RNF2 in 17q23-amplified breast cancer cells. Unexpectedly, however, a variety of biochemical experiments demonstrated that unlike RNF2, TRIM37 is associated with PRC2, not PRC1.
To identify TRIM37 target genes, we performed genome-wide chromatin immunoprecipitation (ChIP)-chip and identified 9412 gene promoters that are bound by TRIM37 and enriched for ubiquitinated H2A; importantly, this list of TRIM37 target genes includes many putative tumor suppressors. Moreover, we found that TRIM37 was co-bound with PRC2 and PRC1 to specific target genes, resulting in their transcriptional silencing. Analysis of a published comprehensive dataset of human breast cancer samples revealed a statistically significant correlation between increased TRIM37 levels and decreased expression of TRIM37 target genes, emphasizing the clinical relevance of TRIM37.
Our results raised the possibility that in 17q23-amplified breast cancers, the elevated levels of TRIM37 contribute to oncogenesis by promoting silencing of tumor suppressor genes. In support of this model, we showed that RNA interference (RNAi)-mediated knockdown of TRIM37 in breast cancer cells containing 17q23 amplification reduced tumor growth in xenograft mouse model studies. Conversely, ectopic expression of TRIM37 in non-malignant and immortalized pre-malignant breast epithelial lines not only transformed the cells but also induced tumor formation in xenograft mice.
Collectively, our findings provide crucial insight into how TRIM37 functions and uncover a non-canonical PRC1/2-mediated silencing pathway (Fig. 1, right). Upon overexpression, TRIM37 associates with and alters the specificity of PRC2, thereby promoting extensive changes in gene expression that include the silencing of multiple tumor suppressors. The promoter-bound TRIM37–PRC2 complex carries out both H3K27 trimethylation and H2A mono-ubiquitination in a mechanism that is distinct from the canonical PRC1/2-mediated silencing pathway.
How PRC2 and PRC1 are selectively recruited to specific genomic loci in mammalian cells represents a major unanswered question in the field. Unlike in Drosophila, where so-called Polycomb response elements (PREs) direct PRC1/2 recruitment, such elements appear to be lacking in mammalian genomes.1 Some models propose that mammalian PRCs are directly recruited either by transcription factors or long non-coding RNAs, whereas other models hypothesize that PRCs are indirectly recruited to genes that are already transcriptionally inactive.3,4 The TRIM37-mediated epigenetic silencing model that we have derived and characterized in breast cancers harboring 17q23 amplification provides a powerful tool to investigate the mechanisms of PRC1/2 recruitment.
Our study identified TRIM37 as an oncogenic counterpart of RNF2, the main H2A E3 ubiquitin ligase functioning in normal mammalian cells. Inhibition of TRIM37 activity is expected to selectively prevent the growth of 17q23 amplicon-positive tumors without affecting normal cells. Therefore, TRIM37 represents an attractive potential target for the development of novel therapies for breast cancers harboring 17q23 amplification.
The characterization of TRIM37 as an oncogene may also provide insight into an autosomal recessive growth disorder called mulibrey nanism. A number of truncating frame-shift mutations in TRIM37 have been identified in individuals with the disease.5,6 In most cases, the truncated TRIM37 protein is predicted to retain an intact and likely functional RING domain, raising the possibility that the oncogenic activity of TRIM37 might contribute to the high frequency of tumors associated with mulibrey nanism.7 However, whether aberrant TRIM37 E3 ubiquitin ligase activity is an underlying cause of the growth defects observed in mulibrey nanism is an interesting question that merits further study.
References
- 1.Margueron R, Reinberg D. Nature 2011; 469:343–9; PMID:21248841; http://dx.doi.org/ 10.1038/nature09784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhatnagar S, et al.. Nature 2014; 516:116–20; PMID:25470042; http://dx.doi.org/ 10.1038/nature13955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klose RJ, et al.. PLoS Genet 2013; 9:e1003717; PMID:23990804; http://dx.doi.org/ 10.1371/journal.pgen.1003717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brockdorff N. RNA 2013; 19:429–42; PMID:23421328; http://dx.doi.org/ 10.1261/rna.037598.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Avela K, et al.. Nat Genet 2000; 25:298–301; PMID:10888877; http://dx.doi.org/ 10.1038/77053 [DOI] [PubMed] [Google Scholar]
- 6.Hamalainen RH, et al.. Hum Mutat 2004; 23:522; PMID:15108285; http://dx.doi.org/ 10.1002/humu.9233 [DOI] [PubMed] [Google Scholar]
- 7.Karlberg N, et al.. J Pathol 2009; 218:163–71; PMID:19334051; http://dx.doi.org/ 10.1002/path.25383 [DOI] [PubMed] [Google Scholar]