

Abstract
Human p21Waf1 protein is well known for being transcriptionally induced by p53 and activating the cell cycle checkpoint arrest in response to DNA breaks. Here we report that p21Waf1 protein undergoes a bimodal regulation, being upregulated in response to low doses of DNA damage but rapidly and transiently degraded in response to high doses of DNA lesions. Responsible for this degradation is the checkpoint kinase Chk1, which phosphorylates p21Waf1 on T145 and S146 residues and induces its proteasome-dependent proteolysis. The initial p21Waf1 degradation is then counteracted by the ATM-Chk2 pathway, which promotes the p53-dependent accumulation of p21Waf1 at any dose of damage. We also found that p21Waf1 ablation favors the activation of an apoptotic program to eliminate otherwise irreparable cells. These findings support a model in which in human cells a balance between ATM-Chk2-p53 and the ATR-Chk1 pathways modulates p21Waf1 protein levels in relation to cytostatic and cytotoxic doses of DNA damage.
Keywords: apoptosis, cell survival, DNA damage response, double strand breaks
Abbreviations
- BLM
bleomycin
- DDR
DNA damage response
- DSBs
double strand breaks
- FCS
fetal calf serum
- IF
immunofluorescence
Introduction
The DNA damage response (DDR) is a sensitive and complex network of pathways protecting the integrity of the genome.1 The apical kinases of the DDR cascade are ATM and Chk2 for the response to DSBs, and ATR and Chk1 for other lesions like pyrimidine dimers or replication fork arrest, while one of the main common effectors is the transcription factor p53.1 DDR-dependent post-translational modifications enable p53 to escape degradation and direct this protein on specific targets.2
A key transcription target of p53 is the p21Waf1 gene, whose encoded protein was initially described as a negative regulator of CDKs, with a role in G1 and G2 arrest maintenance after DNA damage.3,4 In addition, p21Waf1 has been found to repress apoptosis,5 an observation well correlating with p21Waf1 accumulation detectable in several tumors.6 Furthermore, p21Waf1 was described as an important factor in DNA damage-induced senescence7 and, more recently, as a regulator of DSBs repair.8
It is generally assumed that, in response to DNA damage, p53 induces the transcription and accumulation of the p21Waf1 protein, thus promoting cell cycle arrest and, eventually, apoptosis repression. However, p21Waf1 protein poorly accumulates in S phase cells also in presence of DSBs, as a consequence of proteasome-dependent p21 degradation.9 Yet, conditions like replication forks progression impairment or exposure to high UV doses cause a reduction, rather than an increase, in the steady state levels of p21Waf1. Particularly, p21Waf1 is reduced in human cells after DNA replication arrest, in an ATR- and Chk1-dependent manner and through the repression of p21Waf1 mRNA elongation.10 Furthermore, in response to UV irradiation, the decrease of p21Waf1 protein level occurs through a proteasome-dependent pathway. However, contrasting data have been reported about the correlation with the UV dose,11,12 the role of ubiquitin12,13 and p21Waf1 translocation to cytoplasm.13,14 Other DNA damaging agents are supposed to reduce p21Waf1 levels, but a precise correlation with the presence of DSBs is still lacking, since ionizing radiation was described not to affect11 or reduce p21Waf1 in a dose-dependent manner.14,15 The biological consequence of a p21Waf1 decrease remains to be defined.
Here we show that p21Waf1 transiently decreases at any cell cycle phase above a certain level of DNA damage. The degree and duration of this decrease is dose-dependent and cell type-specific, although more evident and irreversible in the absence of a functional p53. p21Waf1 downregulation in response to DBSs is due to protein degradation and specifically it is mediated by the 26S proteasome. This degradation is not linked to the ATM-Chk2 signaling, that actually counteracts this pathway. Instead, p21Waf1 is targeted for degradation by Chk1, which phosphorylates p21Waf1 on residues known to be involved in its stability. The outcome of this transient reduction of basal p21Waf1 seems linked to p21Waf1 anti-apoptotic activity.
Results
Since p21Waf1 levels strongly decrease in UV-irradiated cells, while contrasting results have been reported for treatments with DSB-inducing agents, we analyzed the p21Waf1 protein in U2OS cells treated with different doses of bleomycin (BLM), a chemotherapeutic drug that primarily generates DSBs. We found that the basal levels of p21Waf1, while steadily increase by 3 hrs onwards after treatment with 6 μM BLM, transiently decrease by 50% and 75% after 3 and 6 hrs of treatment with 120 μM BLM and re-accumulate at 24 hrs (Fig. 1A top). To quantify the cytotoxic effect of the treatments, we determined by FACS analysis the percentage of subdiploid apoptotic cells at 24 hrs and cell viability by trypan blue assay at 48 hrs. Whereas treatment with 6 μM BLM had a minor effect on cell survival (Fig. S1A left and middle), exposure to 120 μM BLM resulted in 14% of cell death at 24 hrs and about 50% of cell viability at 48 hrs (Fig. S1A left and middle). Both conditions activated the G2/M checkpoint, as evaluated by FACS analysis (data not shown). Long-term survival and reversibility of cell-cycle arrest were tested by colony-formation assay (Fig. S1A right) and it was found that cells pre-treated for 3 hrs with 6 μM BLM were able to form colonies when cultured in the absence of drug (>80% compared to the untreated cells, Fig. S1A right), whereas cells pre-treated with 120 μM BLM only yielded <10% colonies, Fig. S1A right). These results suggest a cytostatic effect by 6 μM and cytotoxic effect by 120 μM BLM exposure.
Figure 1.

(See previous page). p21Waf1 downregulation in U2OS cells following treatment with DNA damaging agents. (A) Western blot analysis of p21Waf1, p53 and cleaved PARP in U2OS cells exposed to increasing doses of bleomycin (BLM), neocarzionostatin (NCS) and etoposide (Eto). The histograms for p21Waf1 and p53 proteins were obtained from densitometric analysis of the bands, normalized against actin, from 3 independent western blot experiments. (B) p21Waf1 protein detected by immunofluorescence in cells exposed for 3 hrs to genotoxic agents. The intensity of single cell signal was assessed by ImageJ software analysis on microscope images. Cells with p21Waf1 signal intensity lower than 10% of the average signal detectable in the positive control (undamaged cells), were considered as p21Waf1 negative. (C) EdU and p21Waf1 double staining. To mark DNA replication, EdU was added to cell cultures 15 minutes before BLM treatment. p21Waf1 protein levels were evaluated as described in (B), in untreated and treated cells (120 μM BLM, 3 hrs) and in EdU-positive (cells that transit in S-phase during the time of the experiment) and negative populations.
The transient decrease of p21Waf1 was also seen in response to other DSB-inducing agents like neocarzinostatin (40 nM), H2O2 (800 μM) and IR (50 Gy) (Fig. S1B). An equitoxic dose of etoposide (10 μM, Fig. S1C), a topoisomerase II inhibitor yielding limited amounts of DSBs as secondary lesions,16 caused instead p21Waf1 accumulation already at 3 hrs (Fig. 1A bottom and Fig. S1B). All these treatments invariably enhanced p53 accumulation (Fig. 1A; Fig. S1B) and, except for etoposide, increased the γ-H2AX signal linked to DSBs production (Fig. S1D). Furthermore, 120 μM BLM elicited the accumulation of the p53 transcriptional target Mdm2, confirming full p53 activation under such experimental conditions (Fig. S1E). Immunofluorescence (IF) analysis showed decreased p21Waf1 protein after BLM treatment in most of the cell population (Fig. 1B) and, given the absence of significant changes in cell cycle distribution at 6 hrs of treatment with each BLM doses or etoposide (Fig. S1F), we can exclude that p21Waf1 reduction could be linked to cell cycle phase redistribution. To further confirm that p21Waf1 decrease is independent of cell cycle phase, cells replicating DNA were marked by EdU incorporation for 3 hrs in presence or absence of 120 μM BLM. Immunostaining analyses showed that a fraction of EdU positive cells are negative for p21Waf1 also before drug addition, as expected since this protein is at very low level during S-phase.9 However, a significant decrease of p21Waf1 protein levels is detectable in both EdU-positive and -negative cells in presence of BLM (Fig. 1C), supporting the observation that this event is not limited to S-phase cells. The independence of p21Waf1 decrease from cell cycle phase was further confirmed by cyclin B/p21Waf1 co-immunostaining (Fig. S1G), since cyclin B is a positive marker of S-G2 cells.
The genotoxic dose-dependent p21Waf1 decrease was detectable also in a normal lymphoblastoid cell line (LCL) as well as in several cancer cell lines carrying wild type p53 (HCT116, SH-SY5Y) and mutant p53 (T47D, Hela and Saos-2) (Fig. 2A). No p21Waf1 decrease was detectable in BJ-hTERT fibroblasts and IGROV-1 cancer cells treated for 6 hrs with up to 360 μM BLM (Fig. 2A; Fig. S2A), and actually, in the case of BJ-hTERT, IR and H2O2 strongly induced p21Waf1 protein accumulation (Fig. S2B). Notably, in HCT116 cells, loss of p53 resulted in a lower basal expression of p21Waf1, strongly reduced after BLM treatment and that did not recover at 24 hrs (Fig. 2B). A similar behavior was observed in a time course analyses of the p53 null Saos-2 cells (Fig. S2A), confirming that p53 is important for p21Waf1 recovery at later time points after BLM exposure.
Figure 2.
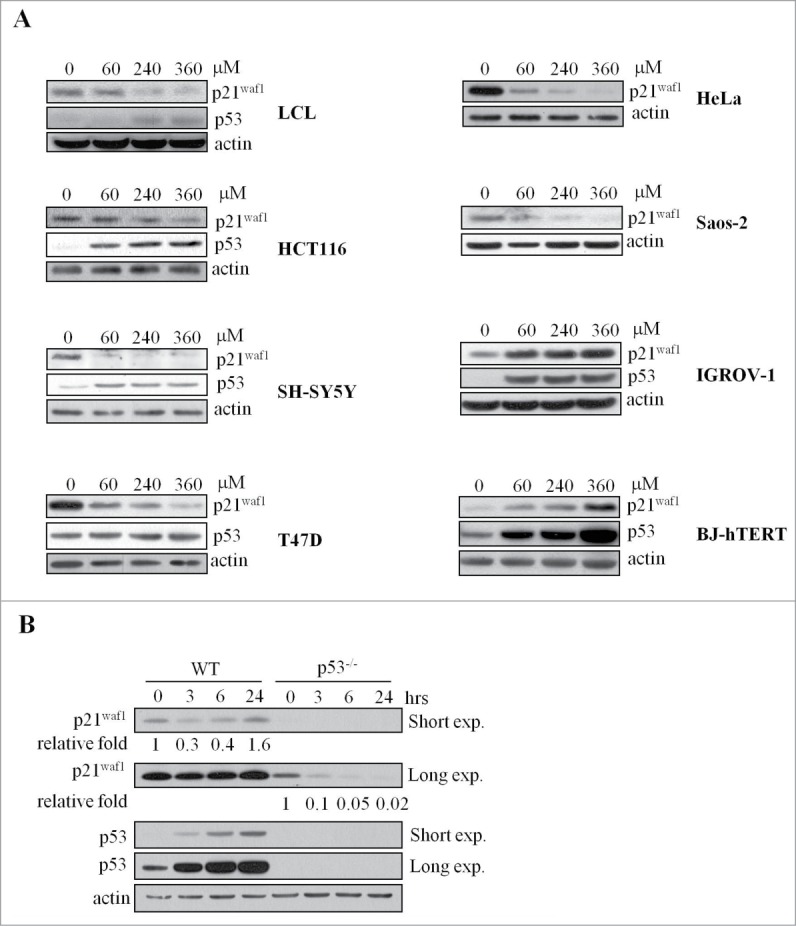
p21Waf1 regulation in normal and cancer cell lines. (A) Human normal lymphoblastoid cells (LCL), normal fibroblast h-TERT immortalized (BJ-hTERT), ovarian carcinoma (IGROV-1), colon carcinoma (HCT116), neuroblastoma (SH-SY5Y), breast cancer (T47D), cervix adenocarcinoma (HeLa) and osteosarcoma (Saos-2) cell lines were tested by immunoblot for p21Waf1 and p53 before and after 3 hrs of treatment with increasing concentrations of BLM. HeLa and Saos-2 are negative for p53, T47D cells have a stabilized, but not inducible form of p53. (B) p53 wt or p53 KO HCT116 cells were analyzed by protein gel blot for p21Waf1 and p53 before and after treatment for up to 24 hrs with 240 μM BLM. Relative quantification of band intensities, obtained by densitometric analyses, normalized to actin for loading, is shown.
Stalled forks can induce a Chk1-dependent reduction of p21Waf1 mRNA owing to repression of transcription elongation.10 We thus quantified p21Waf1 mRNA by RT-PCR and observed an increased signal in response to all dose of BLM tested, and after 10 μM etoposide treatment (Fig. 3A), excluding that p21Waf1 protein downregulation could be due to decreased transcription. To determine the involvement of protein degradation or impaired translation in these events, cells were pre-incubated with the 26S proteasome inhibitor MG132 or the translation elongation inhibitor cycloheximide (CHX) prior to treatment with 120 μM BLM. As expected, CHX treatment reduced p21Waf1 protein in untreated cells, but an additional decrease was detectable in presence of BLM. On the contrary, MG132 increased p21Waf1 accumulation in untreated cells but prevented its decrease after BLM treatment (Fig. 3B). To confirm that p21Waf1 decrease was due to protein degradation, CHX was added to U2OS cells to block the de novo protein synthesis and the remaining p21Waf1 levels were monitored at different time points in presence or absence of 120 μM BLM. The half life of p21Waf1 was reduced to <30 min in presence of 120 μM BLM (Fig. 3C and S3). It has been suggested that p21Waf1 degradation correlates with its nuclear/cytoplasmic localization,13 but in BLM-treated cells the nuclear localization of p21Waf1 was not affected by pre-incubation with MG132 (Fig. 3D).
Figure 3.

p21Waf1 downregulation is due to protein degradation. (A) p21Waf1 transcript and protein levels following BLM or Eto treatments were analyzed on the same U2OS samples by semi-quantitative RT-PCR and western blot. Relative quantification of band intensities was carried out considering the untreated sample as 1. The experiment shown is representative of 3 independent experiments. (B) U2OS were pre-treated for 30 min with 10 μM MG132 or 10 μg/ml cycloheximide (CHX) before addition of 120 μM BLM for 3 hrs. Total lysates were analyzed by protein gel blotting for p21Waf1. Relative quantification of band intensities, obtained by densitometric analyses normalized to actin loading is shown. (C) p21Waf1 protein half life was assessed in presence of CHX alone or CHX and 120 μM BLM. The data plotted in the graph were obtained by western blot and densitometric analysis of the bands. Untreated samples were considered as 1. (D) Immunofluorescence analysis of p21Waf1 protein localization in U2OS cells pretreated for 30 min with 10 μM MG132 and treated for 3 hrs with 120 μM BLM. Nuclei were visualized by DAPI staining. Data in the graph were obtained analyzing the microscope images as in Fig. 1B.
The main regulator of the response to DSBs is the ATM-Chk2 pathway, backed-up by the ATR-Chk1 pathway.17,18 To verify the involvement of these kinases in p21Waf1 expression, we pre-treated U2OS cells with KU55933 and VRX0466617, the chemical inhibitors respectively of ATM19 and Chk2.20 Both compounds enhanced p21Waf1 degradation (Fig. 4A), indicating that the ATM-Chk2 pathway promotes p21Waf1 accumulation at any dose of BLM. Similar results were obtained after Chk2 silencing (data not shown). These observations suggest that the ATM-Chk2-p53 pathway is active in presence of severe DNA damage and partially counteracts p21Waf1 decrease, perhaps by modulating p21Waf1 transcription. To confirm this possibility and according to the previously reported attenuation of the ATM-Chk2 pathway in U2OS cells,21 we observed that Chk2 overexpression significantly counteracts p21Waf1 reduction after severe DNA damage (Fig. 4B). This finding is consistent with previous observations implicating the ATM-Chk2-p53 pathway in p21Waf1 accumulation after DSBs induction.22
Figure 4.

Chk1-dependent degradation of p21Waf1 by bleomycin. (A) U2OS cells were pre-treated for 1 hr with inhibitors of ATM (KU55933, KU, 10mM) or Chk2 (VRX0466617, VRX, 10 μM), prior to treatment with BLM for 3 hrs. Cell lysates were analyzed by protein gel blot. pS966-Smc1 was used as a reporter of the ATM/ATR kinase activity. Relative quantification of band intensities, obtained by densitometric analyses normalized to actin loading sample, is shown. The samples with no inhibitors and no BLM were considered as 1. (B) U2OS were transfected with either mock or HA-Chk2 vector and 48 hrs later treated or not with 120 μM BLM for 3 hrs. (C) U2OS cells were pre-treated for 1 hr with 300 nM of the Chk1 inhibitor UCN-01, then treated for 3 hrs with BLM and analyzed by western blot. (D) U2OS cells were pre-treated with UCN-01 (300 nM, 1 hr) or PF-477736 (400 nM, 1 hr) and then exposed to 120 μM BLM for the indicated times. Total Chk1 and the pS296 fraction, p21Waf1, p53 and actin were evaluated by protein gel blotting. (E) p21Waf1 mRNA levels in presence of 120 μM BLM and PF-477736 (PF). Relative quantification of band intensities, obtained by densitometric analyses normalized to actin mRNA is shown. (F) Edu and p21Waf1 double staining in presence of PF-477736. p21Waf1 positivity was evaluated in EdU-positive and negative cells as described in Fig. 1B. PF-477736 was added 1 hr before BLM treatment, EdU 15 min before BLM. The image is representative of 3 independent experiments. (G) The ATR inhibitor VE-821 (1 μM) was added 1 hr before 120 μM BLM. Total lysates were obtained at the indicated time point after BLM addition. Total Chk1 and the pS345 fraction, p21Waf1 and actin were evaluated by western blotting. Relative quantification of band intensities, obtained by densitometric analyses normalized to actin loading is shown. Untreated samples were considered as 1. (H) Time course analysis of p21Waf1 degradation and Chk1 autophosphorylation in U2OS cells exposed to 120 μM BLM. (I) Pre-treatment with PF-477736 (PF, 400 nM, 1 hr) of T47D, HeLA and LCL cells partially or completely represses p21Waf1 downregulation 3 hrs following 120 μM BLM.
Notably, the Chk1 inhibitors UCN-01 and PF-47773623 prevented p21Waf1 degradation in cells treated with 120 μM BLM (Figs. 4C and D) and neither of these inhibitors alone had any significant effect on cell cycle phase distribution, p53 and p21Waf1 accumulation and γ-H2AXγ phosphorylation up to 6 hrs after treatment (Figs. S4A and B and data not shown). However, markers of DDR were detectable at 24 hrs, probably as a consequence of DNA replication stress,24 but no increase in DNA damage, evaluated by γ-H2AXγ staining was evident for up to 6 hrs of treatment with PF-477736 and BLM combination (Fig. S4C). Furthermore, no changes in p21Waf1 mRNA were induced by Chk1 inhibitors alone or in combination with BLM (Fig. 4E), excluding a significant activity of Chk1 on p21Waf1 mRNA synthesis, stability and translation, under these experimental conditions, and also excluding the contribution of p53 transcriptional activity. Additionally, Chk1 silencing prevented p21Waf1 degradation by BLM (Fig. S4D), even though markedly increased the basal levels of p53 and p21Waf1 (Fig. S4D), possibly as a consequence of cell cycle perturbation24 or DNA damage occurrence, as also detectable 24 hrs after Chk1 inhibition (Fig. S4A). Moreover, the inhibitory effect of UCN-01 on p21Waf1 degradation was detectable at all stages of the cell cycle, as shown by EdU (Fig. 4F, compare with Fig. 1C) and cyclin B (Fig. S4E) stainings. Since Chk1 activation in response to UV or fork stalling occurs through ATR phosphorylation, we tested the role of this kinase in p21Waf1 degradation. Before the addition of BLM we treated U2OS cells with VE-821, a specific inhibitor of ATR activity25 and we evaluated p21Waf1 protein levels. As a reporter of ATR inhibition we evaluated, with a phosphospecific antibody, Chk1 phosphorylation on S345, a known ATR target site.26 A robust but not complete reduction of S345 phosphorylation was detectable in VE-821 treated cells, suggesting a residual ATR activity or, alternatively, that this event and therefore Chk1 activation, is partially independent by ATR in presence of 120 μM BLM. However, a reduction of p21Waf1 degradation was detectable in VE-821 treated cells both 3 and 6 hrs after BLM addition (Fig. 4G).
The role of Chk1 on p21Waf1 degradation was further investigated by measuring its autophosphorylation on S296 (a marker of Chk1 activity). We observed a time-dependent Chk1-pS296 increase already at 1 hr of treatment with 120 μM BLM, which inversely correlated with p21Waf1 degradation (Fig. 4H). Similar data were obtained by IF analysis of Chk1-pS345 (Fig. S4F), demonstrating the presence of Chk1 activity throughout the cell cycle after treatment with 120 μM BLM, since more than 90% of the cells was positive for pS345. Accordingly, 24 hrs after BLM treatment, when p21Waf1 reaccumulates (Fig. 1A), Chk1 is dephosphorylated and partially degraded (Fig. S4G). Furthermore, while p53 accumulates similarly at 3hrs of treatment with 6 μM and 120 μM BLM, Chk1 phosphorylation strongly increases at the 120 μM dose (Fig. S4H). Altogether, these observations support a correlation between Chk1 activation and p21Waf1 degradation after BLM exposure. However, since the Chk1 inhibitor PF-477736 prevented p21Waf1 degradation more effectively in T47D and LCL than in HeLa (Fig. 4I) or HCT116 p53−/− cells (Fig. S4I), it is possible that alternative pathways might contribute to p21Waf1 degradation in some cell lines.
We additionally investigated the role of REGγ, which regulates p21Waf1 half life in unstressed cells,27 and of caspases, known to cleave p21Waf1 in response to genotoxic agents.28 For this, we analyzed cells that were silenced for REGγ or pre-treated with the pan-caspase inhibitor z-VAD-fmk29 and found that REGγ depletion increases the basal level of p21Waf1, but nevertheless BLM induced p21Waf1 degradation in siREGγ as in siCtrl cells (Fig. S5A). Similarly, z-VAD-fmk had no effect on p21Waf1 protein levels in our experimental conditions (Fig. S5B). Moreover, since GSK3β is involved in p21Waf1 degradation after UV damage,12 we inhibited this kinase with LiCl,30 but no effect on p21Waf1 protein decrease after BLM was observed (Fig. S5C). Altogether, our data suggest that high doses of DSBs elicit a specific Chk1-dependent degradation of p21Waf1.
Chk1 kinase might promote p21Waf1 degradation by direct phosphorylation. Chk1 (and Chk2) preferentially phosphorylates proteins carrying an RXXS/T motif31, which in p21Waf1 is present on T97, T145, S146. We thus performed in vitro kinase assays and found that Chk1, but not Chk2, was able to phosphorylate recombinant GST-p21Waf1 (Fig. 5A). Moreover, using specific antibodies against the phosphorylated RXXS/T consensus motif or against phospho-S146, we were able to demonstrate that Chk1 in vitro phosphorylates these residues (Fig. 5B), but unfortunately, these antibodies did not work on immunoprecipitated p21Waf1, also in presence of MG132 (data not shown). However, T145, S146, were of interest because of their involvement in p21Waf1 protein stability,32,33 and therefore Ala substitutions were introduced in these residues to test their effect in p21Waf1 degradation. While the exogenous wild type p21Waf1 protein was reduced after BLM and Chk1 inhibitor repressed this decrease (Fig. 5C), the p21Waf1-T145A/S146A double mutant was only slightly degraded and unaffected by the Chk1 inhibitor (Fig. 5C). These data strongly suggest that the phosphorylation of p21Waf1 on T145, S146 by Chk1 promotes its degradation.
Figure 5.
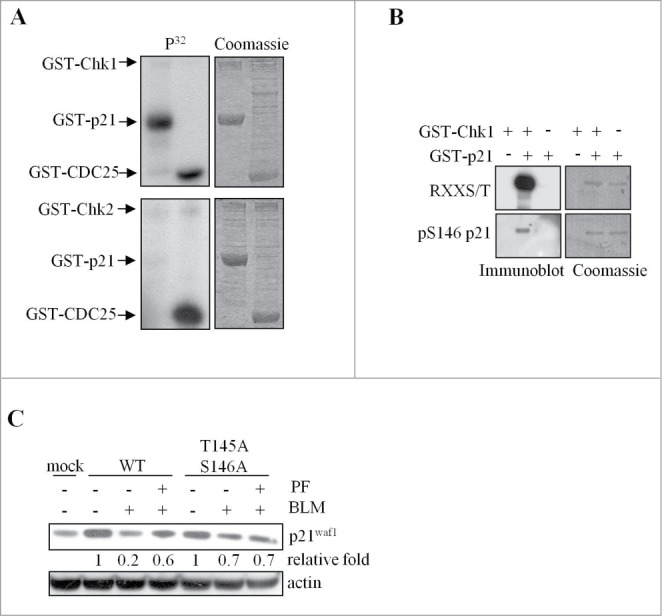
Chk1 phosphorylates in vitro p21Waf1 at sites involved in p21Waf1 degradation. (A) In vitro kinase assays performed with recombinant GST-Chk1 (upper) or GST-Chk2 (lower) kinases and GST-p21Waf1 or GST-Cdc25C fragment as substrates. Low amounts of kinases were added to increase the specificity of the assay. Left panel: autoradiography of incorporated 32P; right panel: Coomassie staining. (B) The non radioactive in vitro kinase assay containing GST-Chk1 and GST-p21Waf1 proteins was immunoblotted with an anti-RXXpS/T specific antibody or an anti-pS146- p21Waf1 antibody. Normalization was done by Coomassie staining. (C) U2OS cells were transfected with wt or T145A/S146A p21Waf1 double mutant. Tests were conducted 72 hrs after transfection, when the expression levels of the exogenous proteins were reduced to less than fold2- of the endogenous p21Waf1 and to avoid possible effects of the overexpression on cell cycle progression. Cells were pre-treated or not with 400 nM PF-477736 for 1 hr and then exposed to 120 μM BLM for 3 hrs. Whole cell extracts were analyzed by protein gel blot. The densitometric analyses of the experiment, normalized to actin and to mock value, are shown.
To investigate the biological impact of p21Waf1 protein degradation, we initially evaluated the effects on the p21Waf1-regulated G1/S and G2/M checkpoints in the LCL cells. Enumerating EdU-labeled S-phase cells 3 and 6 hrs after genotoxic treatments, we observed G1/S checkpoint activation, independent of the used doses, inducing (360 μM BLM or 50 Gy IR) or not (30 μM BLM or 5 Gy IR) p21Waf1 degradation (Fig. 6A). Therefore p21Waf1 degradation does not impair the G1/S checkpoint, at high dose of damage. The G2/M checkpoint was analyzed in cells pre-incubated with nocodazole (used as a mitotic trap to evaluate the G2/M transition rate avoiding M phase exit) and exposed to BLM or etoposide for 3 and 6 hrs. Mitotic cells were then enumerated and whereas control untreated cells showed a time-dependent increase in mitotic figures (Fig. 6B), cells treated with genotoxic agents showed less mitosis irrespective of dose and type of damage (Fig. 6B), indicative of a G2 arrest. Since G1 and G2 checkpoints were fully active at all doses of BLM, inducing or not p21Waf1 degradation, it is clear that the decrease of p21Waf1 is not involved in DNA damage checkpoints regulation.
Figure 6.

p21Waf1 degradation impacts on apoptosis. (A) To assess G1 to S transition, LCL replicating cells were labeled with EdU and evaluated before and 3 and 6 hrs after the indicated DNA damaging treatments. The fraction of EdU positive cells before drugs addition or IR exposure was subtracted from each time point. (B) The G2 to M transition was assessed in U2OS cells pre-treated with 100 ng/ml nocodazole before exposure to genotoxic agents to trap cells in M phase to correctly enumerate cells entering in M phase avoiding mitosis exit. (C) U2OS cells were transfected with siRNA to knock down p21Waf1 (sip21) or a control sequence (sictrl). 48 hrs after transfection, cells were treated for 24 hrs with the genotoxic agents, stained with propidium iodide and the subdiploid apoptotic fraction quantitated by DNA flow cytofluorimetry. (D) The same cells were also lysed and tested for PARP cleavage by immunoblotting. The densitometric analysis of the cleaved form of PARP is shown after normalization on the 6 μM BLM sample. (E) U2OS cells transiently transfected with wild type (OP WT) or mutated (OP S-T/A) forms of p21Waf1, were exposed (72 hrs after transfection to reduce the amount of exogenous p21Waf1 protein and limit the impact of overexpression on cell cycle progression) for 24 hrs to BLM. The subdiploid apoptotic fraction quantified by DNA flow cytofluorimetry was evaluated as above. (F) The same cells were also lysed and tested for PARP cleavage by immunoblotting. The densitometric analyses of the cleaved form of PARP, normalized with vinculin and relative to the wt sample, are shown.
Since p21Waf1 exerts an anti-apoptotic effect against genotoxic agents, we analyzed this phenomenon in response to BLM. As determined by cytofluorimetric analyses, knockdown of p21Waf1 increased of about threefold the amount of sub-diploid cells at 6 μM BLM doses, but less at 120 μM BLM (Fig. 6C). Similar observations on the anti-apoptotic activity of p21Waf1 were obtained with a biochemical approach testing PARP cleavage (Fig. 6D). However, in sip21Waf1 cells treated with 6 μM BLM, apoptosis does not reach the level obtained with 120 μM in both assays, thus demonstrating that besides p21Waf1 downregulation other pro-apoptotic signals are necessary to conduct cells to suicide. Moreover, cells expressing p21Waf1 T145A/S146A, which is not degraded after damage, showed reduced apoptosis, as evaluated by cytofluorimetric analysis and PARP protein cleavage, compared to cells expressing wild type p21Waf1(Figs. 6E and F). Data about apoptosis were not influenced by cell cycle perturbations since p21Waf1 silencing does not significantly alter the cell cycle distribution of the cellular population (Fig. S6A). Furthermore, also cells expressing low levels of the wild type or mutant form of p21Waf1 show comparable cell cycle profiles (Fig. S6B). These results suggest that the early downregulation of p21Waf1 protein following severe DNA damage might facilitate apoptosis.
Discussion
The notions that the human ATR-Chk1 pathway is solely involved in the response to stalled replication forks or UV-induced DNA damage and that p21Waf1 is up-regulated after damage have been recently re-evaluated.11,15,17,18
Here, we demonstrate that p21Waf1 is transiently degraded in response to high yields of DNA DSBs induced by BLM. BLM is an anticancer drug used at high doses (10-27 mg/m2, peak in the blood = 10 μg/ml; t1/2 = 21 hrs) in combination with other chemotherapeutic drugs or radiotherapy, or at 0.25-1 mg/cm2 in local electrochemotherapy for skin metastases of different tumor types. It is important to note that the highest dose of BLM used in this study induces in U2OS less than 50% of cell death at 48 hrs, with about 5-10% survival in long-term colony assays.
Transient degradation of p21Waf1 is cell line-specific and more pronounced in cells with reduced activation of the ATM-Chk2 pathway, and somewhat dependent on the type of DNA lesion since this phenomenon is not observed in response to the topoisomerase 2 inhibitor etoposide, which strongly activates DDR and apoptosis. We found that p21Waf1 downregulation is not restricted to S phase, as previously demonstrated after HU treatment10 or as suggested by PCNA-dependent degradation upon UV irradiation.34 Indeed, a relation with cell cycle is unlikely since p21Waf1 downregulation occurs in the whole population, in cells with an active (LCL) or impaired G1 checkpoint (i.e. U2OS), and only in response to a severe damage, although the transition through S phase in the latter is similar at high or low doses of BLM early after damage (data not shown).
Furthermore, we found that in U2OS p21Waf1 mRNA induction after DNA damage is detectable at all doses of BLM tested and after etoposide, excluding mRNA synthesis or stability events in p21Waf1 downregulation. Therefore, we can rule out the possibility that specific post-translational modifications on p53 could repress p21Waf1 transcription. Regulation of p21Waf1 at the translational level was also described, but we found that in the presence of a translational repressor p21Waf1 decrease was still detectable. Indeed, we demonstrated that the expression of p21Waf1 is regulated after BLM treatment by 26S proteasome-mediated degradation and not by transcription or translation, but with a mechanism different from that described for UV treatment35 as it does not involve significant translocation of p21Waf1 to the cytoplasm.
Since the main regulators of the DDR are the ATM-Chk2 and ATR-Chk1 pathways, we determined their impact on BLM-induced p21Waf1 degradation. We found that inhibition of ATM or Chk2 does not prevent, but rather enhance, p21Waf1 degradation, in accordance with previous findings demonstrating that the ATM-Chk2 pathway promotes p53-dependent transcription of p21Waf1 upon DSBs.22
Unexpectedly, selective inhibition of Chk1 kinase prevented p21Waf1 degradation in several human cell lines, with a more evident effect in cells with active p53. However, in cells with inactive p53 and low p21Waf1 protein levels (e.g. HeLa or HCT116 p53−/−), Chk1 inhibition cannot completely rescue p21Waf1 degradation. Two factors could explain these findings: i) other pathways could contribute to p21Waf1 downregulation; ii) impaired p53 transcriptional activity cannot promote p21Waf1 accumulation. Notably ATR inhibition was also able to significantly reduce p21Waf1 degradation, suggesting that Chk1 activation, in presence of a severe amount of DSBs, is mainly imputable to ATR.
It was previously demonstrated that Chk1 activation is restricted to S and G2 cells upon IR.36 Furthermore, previous studies indicate that in S phase cells, experiencing DSBs or stalled forks, p21Waf1 protein levels are low but stable and that, in Chk1−/− cells, p21Waf1 activates a strong S phase entry block.37 Differently, our data demonstrate that in presence of a severe damage, specifically DSBs, Chk1 is activated at all cell cycle phases and directly targets, by phosphorylation, p21Waf1 for degradation. We also found that Chk1 phosphorylates p21Waf1 in vitro, possibly on the T145 and S146 consensus sites, and that a p21Waf1-T145A-S146A double mutant was more stable than the ectopically expressed wt counterpart in cells treated with high doses of BLM. Notably, these sites have been previously implicated in p21Waf1 protein turnover,32,33 and furthermore, in murine cells treated with a mitogenic factor, Chk1 phosphorylates p21Waf1on T140 and S141 (homologues of human T145 and S146), leading to its degradation.38 Since these Chk1-phosphorylated residues on p21Waf1 map on the region of interaction with PCNA, a role for PCNA in p21Waf1 degradation after DSBs is predictable, as already suggested.34
The observations that the p21Waf1-T145A/S146A is slightly degraded in a Chk1-independent way, and that p21Waf1 is partially decreased in p53−/− cells treated with a Chk1 inhibitor and BLM, suggest that besides Chk1 activity, other mechanisms can promote p21Waf1 downregulation. Since the ATR-GSK3β pathway mediates the degradation of p21Waf1 in response to UV irradiation,12,35 we inhibited GSK3β but no differences in p21Waf1 degradation were detected. Notably, UCN-01 and PF-477736 not only do not inhibit GSK3β but, in some conditions, they indirectly activate this kinase.39 In some cell types, p21Waf1 is induced by DNA damage but rapidly cleaved by caspases.40 Inhibition of caspases in our cells did not affect the p21Waf1 response to BLM. Finally, we found that REG-γ, an activator of the 20S proteasome, was able to reduce p21Waf1 protein levels in undamaged cells,27 but no after BLM treatment.
Collectively, our data suggest that the ATM-Chk2-p53 and the ATR-Chk1 pathways contribute to modulate the response of human cells to different yields of DNA breaks. Chk1 is particularly active in response to high doses of BLM, while p53 accumulates independently by the amount of damage. At the same time, p53 protein increases with the time up to 24 hrs, while Chk1 at 24 hrs is downregulated. These 2 observations could explain the dose dependence and transient nature of Chk1 mediated p21Waf1 downregulation. Therefore, the presence of DSBs generally favors the activation of the ATM-Chk2-p53 pathway, resulting in p21Waf1 mRNA and protein accumulation. Yet, when the number of DSBs is high and/or in conditions where the ATM-Chk2-p53 pathway is partially impaired, as frequently seen in cancer cells, the ATR-Chk1 pathway could be transiently prominent. The transient nature of p21Waf1 degradation could reflect the necessity to preserve cells that are able to repair the majority of DNA lesions. However, we cannot exclude the role of a factor acting as Chk1 activity regulator toward p21Waf1.
The G1/S and G2/M checkpoints were activated by low and high doses of BLM, thus excluding an essential role for p21Waf1 downregulation in activating these checkpoints. In contrast, apoptosis, was repressed by overexpression of p21Waf1 and even more of p21Waf1-T145A-S146A, and enhanced by p21Waf1 knockdown. A transient p21Waf1 downregulation, together with other molecular signals, could be important to start an apoptotic program in presence of irreparable DSBs. Probably, it is not a coincidence that cells with an intact ATM-Chk2 pathway and resistant to apoptosis, like BJ-hTERT fibroblasts, showed no p21Waf1 degradation in response to any dose of DSBs-inducing agents. However, p21Waf1 should not be considered a general apoptosis inhibitor, since U2OS cells treated with etoposide are prone to cell death although they start to accumulate p21Waf1 early after drug exposure and p21Waf1 silencing at low dose of damage is sufficient to significantly, but not extensively, increase apoptosis.
Therefore, it is also important to underline that the biological effect of p21Waf1 degradation could derive from a decrease under the basal level of this protein, which is known to be active, for example on cell cycle control, also in unstressed conditions. On the other side, downstream events could derive from a time window where p21Waf1 does not accumulate and that correlate with the intensity of the damage.
Collectively, our work highlights, i) The presence of a balance between the ATM-Chk2 and ATR-Chk1 pathways during DDR, influenced by the doses and nature of DNA damaging agents, as well as by the cellular background and presence of specific mutations (i.e., in ATM or p53). ii) The role of Chk1 in DSBs response, not limited to S phase. iii) The complexity of p21Waf1 regulation at transcriptional and translational level, with mechanisms that could be relevant at different doses and types of damage and at different times during DDR. iiii) The importance of the basal levels of p21Waf1.
These observations prompt to reconsider the role of some DDR components during tumorigenesis and cancer treatment, and may have implications for the use of radiation or chemotherapeutic agents inducing DSBs in combination with Chk1 inhibitors. Indeed, Chk1 inhibitors, which are now in clinical trials, could affect not only cell cycle arrest, but also apoptosis and DNA repair, depending on DNA lesion and cellular background.
Materials and Methods
Cell culture and treatments
Osteosarcoma cells (U2OS and Saos-2), ovarian carcinoma (IGROV-1), colon carcinoma (HCT116), neuroblastoma (SH-SY5Y), breast cancer (T47D) and cervix adenocarcinoma (HeLa) cells were cultured in DMEM (BioWhittaker) plus 10% FCS. The EBV-immortalized normal lymphoblastoid cell line (LCL) was cultured in RPMI1640 plus 15% FCS. hTERT-immortalized normal human foreskin fibroblasts BJ-hTERT were cultured in DMEM plus M199 (4:1 ratio) with 10% FCS and 10 μg/ml hygromycin B. Irradiations were performed with a 137Cs source providing 675 cGy/min. KU-55933 (R&D Chemicals) and VRX046661720 was used at 10 μM. BLM (Nippon Kayaku) was dissolved in PBS at a concentration of 10 mg/ml (equivalent to 10 U/ml or 7 M), stored at −20°C and used by 1 month. Etoposide (TEVA) treatments were performed using a final 10 μM concentration unless otherwise indicated. MG132 (Sigma) and CHX (Sigma) were used respectively at 10 μM and 100 μg/ml to evaluate the role of proteasome and mRNA translation in p21waf1 protein decrease. To test p21waf1 half-life, we treated U2OS cells with 40 μg/ml CHX 20 minutes before the addition of BLM. UCN-01 (Sigma-Aldrich), PF-477736 (Sigma-Aldrich) and VE-821 (SelleckChem) were added 1 hr before BLM exposure, at a concentration of 300 nM, 400 nM and 1 μM, respectively.
Plasmids and site-directed mutagenesis
p21waf1 gene cloned in pCEP plasmid was a kind gift of S. Soddu. T145 and S146 mutants to Alanine were obtained using the QuikChange II XL Site Directed Mutagenesis Kit (StrataGene, Agilent Technologies). Chk2 cloning in pCDNA3-HA vector was previously described.41
Cells transfection and siRNAs
Cells were transfected using Lipofectamine 2000 (Invitrogen) according to the protocol and lysed in Laemmli buffer for western blot analysis. siRNAs against p21Waf1 were Flexi Tube siRNA (Qiagen). siRNA against luciferase (siLUC) was custom made by Thermo Scientific Dharmacon.
Western blots and antibodies
Immunoblots were performed on total cell extracts as described42 and the antibodies used were: p21 (H164, Santa Cruz), p21-pS146 (Santa Cruz), Phospho-Akt Substrate (RXXS*/T*) (110B7E, Cell Signaling Technology, Beverly, MA) Chk2-pT68 (2661; Cell Signaling Technology), ATM-pS1981 (AF1655, R&D Systems), Smc1-pS966 (ab1276, Abcam), Chk2,43 p53 (DO7, Zymed), cyclin B1 (BD-Biosciences), γ-H2AX (Upstate, Millipore), cleaved PARP-1 (Cell Signaling Technology), β-actin (Sigma), Chk1-pS345 and Chk1-pS296 (Cell Signaling Technology), hemagglutinin (HA) tag (12CA5; Roche, Mannheim, Germany). Densitometric analyses of protein gel blot signals were done with ImageQuant TL software (GE Healthcare Life Sciences).
Immunofluorescence and flow cytometry analysis
To stain p21Waf1 in U2OS, cells were grown on coverslips, rinsed with PBS and fixed with paraformaldehyde. Then cells were permeabilized with 10 mM Hepes pH 7.5, 100 mM NaCl, 1 mM MgCl2, 1 mM EGTA, 0.32 M sucrose, 0.5% Triton X-100, blocked with 3% BSA and incubated at RT for 1 hr with primary antibodies. Following extensive washing, coverslips were incubated with AlexaFluor488-conjugated anti-rabbit antibodies for 1 hr at RT. DNA was stained with 0.2 μg/ml DAPI. Images were captured with a Nikon Eclipse E1000 microscope equipped with a DXM1200F digital camera and signal intensity evaluated by ImageJ software. Click-iT-Edu Alexa Fluor 488 (Life Technologies) staining was performed as suggested by the manufacturer's instruction, with the addition of an immunostaining step to reveal p21Waf1, using an Alexa Fluor 555-conjugated anti-rabbit as secondary antibody. Obtained signals were quantified with ImageJ software. Flow cytometry analyses to evaluate DNA content were conducted as previously described.41
Semi-quantitative PCR
After treatment, cells were collected and subjected to the simultaneous extraction of RNA and proteins by the Illustra™ triplePrep Kit (GE Healthcare, USA), following the manufacturer's instructions. One μg of RNA was reverse-transcribed into cDNA by the Cloned AMV First-Strand cDNA Synthesis Kit (Invitrogen, USA), according to the manufacturer's instruction. PCR was carried out under conditions of linear phase amplification, using primers specific for p21Waf1 and for the housekeeping gene β-actin (p21-FW 5′-GCGCTAATGGCGGGCTGCAT-3′ and p21-REV 5′-GCCGGCGTTTGGAGTGGTAG-3′; β-actin-FW 5′-GCTCGTCGTCGACAACGGCTC-3′ and β-actin-REV 5′-CAAACATGATCTGGGTCATCTTCTC-3′). Thirty cycles at 94°C × 15′′ 64°C × 15′′ and 72°C × 30′′ for p21Waf1 and 29 cycles at 94°C × 15′′ 64°C × 15′′ and 72°C × 30′′ for β-actin were performed. The PCR products (364 bp for p21Waf1 and 352 bp for β-actin) were run on a 2% agarose gel stained with ethidium bromide and photographed. Finally, densitometry analysis was performed and the ratio p21Waf1/β-actin calculated.
Kinase assays
In vitro Chk2 and Chk1 kinase assays were performed as previously described44 using Cdc25C fragment and full length p21Waf1 and Chk2 cloned in pGEX-4T-1 vector and produced in E. coli. Chk1 was purchased from Millipore.
G1/S and G2/M transition analysis
To test G1/S transition DNA replication was measured using a Click-it EdU assay kit (Invitrogen). To evaluate G2/M transition, nocodazole (100 ng/ml) was added 30 min before bleomycin exposure to trap checkpoint defective cells. Cells were successively fixed and stained with an Alexa Fluor 488-conjugated anti-phospho-Histone H3 (Ser10) to reveal mitotic cells.
Funding Statement
This work was financially supported by the Italian Association for Cancer Research (AIRC) (grant IG10248).
Disclosure of Potential Conflicts of Interest
Supplemental Materials
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 2010; 40:179-204; PMID:20965415; http://dx.doi.org/ 10.1016/j.molcel.2010.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Menendez D, Inga A, Resnick MA. The expanding universe of p53 targets. Nat Rev Cancer 2009; 9:724-37; PMID:19776742; http://dx.doi.org/ 10.1038/nrc2730 [DOI] [PubMed] [Google Scholar]
- 3. Waldman T, Kinzler KW, Vogelstein B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res 1995; 55:5187-90; PMID:7585571 [PubMed] [Google Scholar]
- 4. Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998; 282:1497-501; PMID:9822382; http://dx.doi.org/ 10.1126/science.282.5393.1497 [DOI] [PubMed] [Google Scholar]
- 5. Gartel AL, Tyner AL. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer Ther 2002; 1:639-49; PMID:12479224. [PubMed] [Google Scholar]
- 6. Abbas T, Dutta A. P21 in cancer: intricate networks and multiple activities. Nat Rev Cancer 2009; 9:400-14; PMID:19440234; http://dx.doi.org/ 10.1038/nrc2657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fang L, Igarashi M, Leung J, Sugrue MM, Lee SW, Aaronson SA. p21Waf1Cip1Sdi1 induces permanent growth arrest with markers of replicative senescence in human tumor cells lacking functional p53. Oncogene 1999; 18:2789-97; PMID:10362249; http://dx.doi.org/ 10.1038/sj.onc.1202615 [DOI] [PubMed] [Google Scholar]
- 8. Mauro M, Rego MA, Boisvert RA, Esashi F, Cavallo F, Jasin M, Howlett NG. p21 promotes error-free replication-coupled DNA double-strand break repair. Nucleic Acids Res 2012; 40:8348-60; PMID:22735704; http://dx.doi.org/ 10.1093/nar/gks612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ciznadija D, Zhu XH, Koff A. Hdm2- and proteasome-dependent turnover limits p21 accumulation during S phase. Cell Cycle 2011; 10:2714-23; PMID:21768776; http://dx.doi.org/ 10.4161/cc.10.16.16725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Beckerman R, Donner AJ, Mattia M, Peart MJ, Manley JL, Espinosa JM, Prives C. A role for Chk1 in blocking transcriptional elongation of p21 RNA during the S-phase checkpoint. Genes Dev 2009; 23:1364-77; PMID:19487575; http://dx.doi.org/ 10.1101/gad.1795709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bendjennat M, Boulaire J, Jascur T, Brickner H, Barbier V, Sarasin A, Fotedar A, Fotedar R. UV irradiation triggers ubiquitin-dependent degradation of p21(WAF1) to promote DNA repair. Cell 2003; 114:599-610; PMID:13678583; http://dx.doi.org/ 10.1016/j.cell.2003.08.001 [DOI] [PubMed] [Google Scholar]
- 12. Lee JY, Yu SJ, Park YG, Kim J, Sohn J. Glycogen synthase kinase 3beta phosphorylates p21WAF1CIP1 for proteasomal degradation after UV irradiation. Mol Cell Biol 2007; 27:3187-98; PMID:17283049; http://dx.doi.org/ 10.1128/MCB.01461-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hwang CY, Kim IY, Kwon KS. Cytoplasmic localization and ubiquitination of p21(Cip1) by reactive oxygen species. Biochem Biophys Res Commun 2007; 358:219-25; PMID:17477906; http://dx.doi.org/ 10.1016/j.bbrc.2007.04.120 [DOI] [PubMed] [Google Scholar]
- 14. Savio M, Coppa T, Cazzalini O, Perucca P, Necchi D, Nardo T, Stivala LA, Prosperi E. Degradation of p21CDKN1A after DNA damage is independent of type of lesion, and is not required for DNA repair. DNA Repair (Amst) 2009; 8:778-85; PMID:19321391; http://dx.doi.org/ 10.1016/j.dnarep.2009.02.005 [DOI] [PubMed] [Google Scholar]
- 15. Stuart SA, Wang JY. Ionizing radiation induces ATM-independent degradation of p21Cip1 in transformed cells. J Biol Chem 2009; 284:15061-70; PMID:19332548; http://dx.doi.org/ 10.1074/jbc.M808810200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Muslimovic A, Nystrom S, Gao Y, Hammarsten O. Numerical analysis of etoposide induced DNA breaks. PLoS One 2009; 4:e5859; PMID:19516899; http://dx.doi.org/ 10.1371/journal.pone.0005859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhou XY, Wang X, Hu B, Guan J, Iliakis G, Wang Y. An ATM-independent S-phase checkpoint response involves CHK1 pathway. Cancer Res 2002; 62:1598-603; PMID:11912127 [PubMed] [Google Scholar]
- 18. Landsverk KS, Patzke S, Rein ID, Stokke C, Lyng H, De Angelis PM, Stokke T. Three independent mechanisms for arrest in G2 after ionizing radiation. Cell Cycle 2011; 10:819-29; PMID:21325885; http://dx.doi.org/ 10.4161/cc.10.5.14968 [DOI] [PubMed] [Google Scholar]
- 19. Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res 2004; 64:9152-9; PMID:15604286; http://dx.doi.org/ 10.1158/0008-5472.CAN-04-2727 [DOI] [PubMed] [Google Scholar]
- 20. Carlessi L, Buscemi G, Fontanella E, Delia D. A protein phosphatase feedback mechanism regulates the basal phosphorylation of Chk2 kinase in the absence of DNA damage. Biochim Biophys Acta 2010; 1803:1213-23; PMID:20599567; http://dx.doi.org/ 10.1016/j.bbamcr.2010.06.002 [DOI] [PubMed] [Google Scholar]
- 21. Lossaint G, Besnard E, Fisher D, Piette J, Dulic V. Chk1 is dispensable for G2 arrest in response to sustained DNA damage when the ATMp53p21 pathway is functional. Oncogene 2011; 30:4261-74; PMID:.21532626; http://dx.doi.org/ 10.1038/onc2011.135 [DOI] [PubMed] [Google Scholar]
- 22. Takai H, Naka K, Okada Y, Watanabe M, Harada N, Saito S, Anderson CW, Appella E, Nakanishi M, Suzuki H, et al. Chk2-deficient mice exhibit radioresistance and defective p53-mediated transcription. EMBO J 2002; 21:5195-205; PMID:12356735; http://dx.doi.org/ 10.1093/emboj/cdf506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Blasina A, Hallin J, Chen E, Arango ME, Kraynov E, Register J, Grant S, Ninkovic S, Chen P, Nichols T, et al. Breaching the DNA damage checkpoint via PF-00477736, a novel small-molecule inhibitor of checkpoint kinase 1. Mol Cancer Ther 2008; 7:2394-404; PMID:18723486; http://dx.doi.org/ 10.1158/1535-7163.MCT-07-2391 [DOI] [PubMed] [Google Scholar]
- 24. Syljuasen RG, Sorensen CS, Hansen LT, Fugger K, Lundin C, Johansson F, Helleday T, Sehested M, Lukas J, Bartek J. Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol Cell Biol 2005; 25:3553-62; PMID:15831461; http://dx.doi.org/ 10.1128/MCB.25.9.3553-3562.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Reaper PM, Griffiths MR, Long JM, Charrier JD, Maccormick S, Charlton PA, Golec JM, Pollard JR. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol 2011; 7:428-30; PMID:21490603; http://dx.doi.org/ 10.1038/nchembio.573 [DOI] [PubMed] [Google Scholar]
- 26. Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, et al. Chk1 is an essential kinase that is regulated by atr and required for the G(2)M DNA damage checkpoint. Genes Dev 2000; 14:1448-59; PMID:10859164; http://dx.doi.org/ 10.1101/gad.840500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li X, Amazit L, Long W, Lonard DM, Monaco JJ, O’Malley BW. Ubiquitin- and ATP-independent proteolytic turnover of p21 by the REGgamma-proteasome pathway. Mol Cell 2007; 26:831-42; PMID:17588518; http://dx.doi.org/ 10.1016/j.molcel.2007.05.028 [DOI] [PubMed] [Google Scholar]
- 28. Zhang Y, Fujita N, Tsuruo T. Caspase-mediated cleavage of p21Waf1Cip1 converts cancer cells from growth arrest to undergoing apoptosis. Oncogene 1999; 18:1131-8; PMID:10022118; http://dx.doi.org/ 10.1038/sj.onc.1202426 [DOI] [PubMed] [Google Scholar]
- 29. Wang Z, Watt W, Brooks NA, Harris MS, Urban J, Boatman D, McMillan M, Kahn M, Heinrikson RL, Finzel BC, et al. Kinetic and structural characterization of caspase-3 and caspase-8 inhibition by a novel class of irreversible inhibitors. Biochim Biophys Acta 2010; 1804:1817-31; PMID:20580860; http://dx.doi.org/ 10.1016/j.bbapap.2010.05.007 [DOI] [PubMed] [Google Scholar]
- 30. Stambolic V, Ruel L, Woodgett JR. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr Biol 1996; 6:1664-8; PMID:8994831; http://dx.doi.org/ 10.1016/S0960-9822(02)70790-2 [DOI] [PubMed] [Google Scholar]
- 31. O’Neill T, Giarratani L, Chen P, Iyer L, Lee CH, Bobiak M, Kanai F, Zhou BB, Chung JH, Rathbun GA. Determination of substrate motifs for human Chk1 and hCds1Chk2 by the oriented peptide library approach. J Biol Chem 2002; 277:16102-15; PMID:11821419; http://dx.doi.org/ 10.1074/jbc.M111705200 [DOI] [PubMed] [Google Scholar]
- 32. Scott MT, Ingram A, Ball KL. PDK1-dependent activation of atypical PKC leads to degradation of the p21 tumour modifier protein. EMBO J 2002; 21:6771-80; PMID:12485998; http://dx.doi.org/ 10.1093/emboj/cdf684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cornils H, Kohler RS, Hergovich A, Hemmings BA. Human NDR kinases control G(1)S cell cycle transition by directly regulating p21 stability. Mol Cell Biol 2011; 31:1382-95; PMID:21262772; http://dx.doi.org/ 10.1128/MCB.01216-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Soria G, Gottifredi V. PCNA-coupled p21 degradation after DNA damage: the exception that confirms the rule? DNA Repair (Amst) 2010; 9:358-64; PMID:20060369; http://dx.doi.org/ 10.1016/j.dnarep.2009.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lei X, Liu B, Han W, Ming M, He YY. UVB-induced p21 degradation promotes apoptosis of human keratinocytes. Photochem Photobiol Sci 2010; 9:1640-8; PMID:20931139; http://dx.doi.org/ 10.1039/c0pp00244e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, Jackson SP. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol 2006; 8:37-45; PMID:16327781; http://dx.doi.org/ 10.1038/ncb1337 [DOI] [PubMed] [Google Scholar]
- 37. Rodriguez R, Meuth M. Chk1 and p21 cooperate to prevent apoptosis during DNA replication fork stress. Mol Biol Cell 2006; 17:402-12; PMID:16280359; http://dx.doi.org/ 10.1091/mbc.E05-07-0594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ullah Z, de Renty C, DePamphilis ML. Checkpoint kinase 1 prevents cell cycle exit linked to terminal cell differentiation. Mol Cell Biol 2011; 31:4129-43; PMID:21791608; http://dx.doi.org/ 10.1128/MCB.05723-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sato S, Fujita N, Tsuruo T. Interference with PDK1-akt survival signaling pathway by UCN-01 (7-hydroxystaurosporine). Oncogene 2002; 21:1727-38; PMID:11896604; http://dx.doi.org/ 10.1038/sj.onc.1205225 [DOI] [PubMed] [Google Scholar]
- 40. Gervais JL, Seth P, Zhang H. Cleavage of CDK inhibitor p21(Cip1Waf1) by caspases is an early event during DNA damage-induced apoptosis. J Biol Chem 1998; 273:19207-12; PMID:9668108; http://dx.doi.org/ 10.1074/jbc.273.30.19207 [DOI] [PubMed] [Google Scholar]
- 41. Buscemi G, Carlessi L, Zannini L, Lisanti S, Fontanella E, Canevari S, Delia D. DNA damage-induced cell cycle regulation and function of novel Chk2 phosphoresidues. Mol Cell Biol 2006; 26:7832-45; PMID:16940182; http://dx.doi.org/ 10.1128/MCB.00534-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zannini L, Buscemi G, Kim JE, Fontanella E, Delia D. DBC1 phosphorylation by ATMATR inhibits SIRT1 deacetylase in response to DNA damage. J Mol Cell Biol 2012; 4:294-303; PMID:22735644; http://dx.doi.org/ 10.1093/jmcb/mjs035 [DOI] [PubMed] [Google Scholar]
- 43. Zannini L, Buscemi G, Fontanella E, Lisanti S, Delia D. REGgammaPA28gamma proteasome activator interacts with PML and Chk2 and affects PML nuclear bodies number. Cell Cycle 2009; 8:2399-407; PMID:19556897; http://dx.doi.org/ 10.4161/cc.8.15.9084 [DOI] [PubMed] [Google Scholar]
- 44. Buscemi G, Zannini L, Fontanella E, Lecis D, Lisanti S, Delia D. The shelterin protein TRF2 inhibits Chk2 activity at telomeres in the absence of DNA damage. Curr Biol 2009; 19:874-9; PMID:19375317; http://dx.doi.org/ 10.1016/j.cub.2009.03.064 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.