

Abstract
CDC25 dual-specificity phosphatases play a central role in cell cycle control through the activation of Cyclin-Dependent Kinases (CDKs). Expression during mitosis of a stabilized CDC25B mutant (CDC25B-DDA), which cannot interact with the F-box protein βTrCP for proteasome-dependent degradation, causes mitotic defects and chromosome segregation errors in mammalian cells. We found, using the same CDC25B mutant, that stabilization and failure to degrade CDC25B during mitosis lead to the appearance of multipolar spindle cells resulting from a fragmentation of pericentriolar material (PCM) and abolish mitotic Plk1-dependent phosphorylation of Kizuna (Kiz), which is essential for the function of Kiz in maintaining spindle pole integrity. Thus, in mitosis Kiz is a new substrate of CDC25B whose dephosphorylation following CDC25B stabilization leads to the formation of multipolar spindles. Furthermore, endogenous Kiz and CDC25B interact only in mitosis, suggesting that Kiz phosphorylation depends on a balance between CDC25B and Plk1 activities. Our data identify a novel mitotic substrate of CDC25B phosphatase that plays a key role in mitosis control.
Keywords: CDC25B, centrosome, Kizuna, mitosis
Abbreviations
- Kiz
Kizuna protein
- PCM
pericentriolar material
- DMSO
dimethyl-sulfoxyde
- DSP
Dithiobis [succinimidyl propionate]
Introduction
CDC25 dual-specificity phosphatases govern cell cycle progression notably through the spatially and temporally regulated activation of Cyclin-Dependent Kinase (CDK)/Cyclin complexes.1,2 In higher Metazoans, each CDC25 isoform (CDC25A, B and C) is generally considered to play a specific role at different phases of the cell cycle.3 Although there is still some debate over the possible redundancy of the 3 isoforms, particularly based on the observations made in CDC25 knockout mice,4-6 CDC25B is considered to play a major role in initiating entry into mitosis and is often called the starter of mitosis.7-9 At the G2/M transition, a pool of CDC25B is phosphorylated and activated by Aurora-A kinase at centrosomes,10 where the initial activation of CDK1/Cyclin B complexes takes place,11 suggesting that CDC25B might locally participate in the control of the onset of mitosis. Furthermore, CDC25B also participates in the control of γ-Tubulin localization to the centrosomes, is involved in the centrosome duplication cycle,12 and regulates proteasome-mediated degradation of centrin 2.13
Centrosomes are the major microtubule organizing centers. They are composed of a pair of centrioles surrounded by a network of scaffold proteins called pericentriolar material (PCM). They play a central role during mitosis in guiding spindle formation and therefore in maintaining genomic integrity.14 Indeed, loss of the correct coordination of centrosome duplication and defects in centrosome function result in the formation of multipolar spindles that can lead to abnormal cell divisions.15-17 The centrosome cycle (duplication, separation and maturation) is under the control of 2 major types of protein kinases: CDK2 associated with either Cyclin E or Cyclin A 18-20 and the Polo-like kinase family (Plk).21 Among the Plk family members, Plk4 is considered as a master regulator of centriole biogenesis,22-24 whereas Plk1 initiates centrosome maturation 25 and participates in the formation of the bipolar mitotic spindle.26 At the onset of mitosis, centrosome maturation requires a critical reorganization in which the PCM expands through rapid recruitment of additional proteins and is stabilized to form stable spindle poles.27 Loss of PCM factors, such as Kiz,28 Astrin,29 and Tastin,30 leads to centriole disengagement and PCM fragmentation. Among these proteins, Kiz plays a critical role in PCM stabilization via its association with Pericentrin. The scaffold Kiz function at spindles poles to prevent PCM fragmentation and the appearance of multipolar spindle depends on its Plk1-mediated phosphorylation on residue Thr379.28,31
We previously showed that the expression of a CDC25B mutant (CDC25B-DDA), which cannot interact with the F-box protein βTrCP (the substrate-recognition component of the SCFβTrCP ubiquitin-ligase complex) and thus is not targeted for proteasome-dependent degradation, stabilizes CDC25B in mitosis. This mutant induces important mitotic defects, as indicated by the appearance of lagging and misaligned chromosomes and the presence of cells with multipolar spindles, without affecting the activity of CDK1/Cyclin B complexes.32 In this work, we show that the presence of cells presenting multipolar spindles, during mitotic phases, is not due to centrosome amplification but to PCM fragmentation as a consequence of untimed CDC25B-dependent Kiz dephosphorylation. This, in turn, demonstrates that Kiz is a novel substrate for CDC25B.
Results and Discussion
In agreement with our previous results,32 immunofluorescence analysis with anti-α-Tubulin antibodies showed that the fraction of cells with multipolar spindles, present at different stages of mitosis, was significantly higher in U2OS Tet-off cells following induced expression of a stabilized CDC25B mutant (CDC25B-DDA) (24%) than a wild-type CDC25B (U2OS-CDC25B-Wt) (3%) (Fig. 1A). As this phenotype is frequently due to amplification of centrosomes,33 we quantified the number of centrosomes in U2OS-CDC25B-Wt and U2OS-CDC25B-DDA cells by labeling them with anti-γ-Tubulin antibodies, a centrosome marker (Fig. 1B-C). More than 95 % of mitotic U2OS-CDC25B-Wt cells (n = 200) had 2 γ-Tubulin dots (i.e., bipolar spindles), while 26.3 % of mitotic U2OS-CDC25B-DDA cells (n = 180) displayed more than 2 γ-Tubulin dots (i.e., multipolar spindles) (Fig. 1B and C, upper panel). Conversely, during interphase, the number of centrosomes was comparable in U2OS-CDC25B-DDA, U2OS-CDC25B-Wt (Fig. 1C, lower panel) and parental U2OS cells (data not shown). Indeed, only 2.75 % of U2OS-CDC25B-DDA cells in interphase contained more than 2 γ-Tubulin dots (i.e., cells with centrosome amplification), a percentage similar to that found in U2OS-CDC25B-Wt cells (2.65 %) (Fig. 1C, lower panel). This finding contradicts a previous report showing that forced expression of CDC25B induces centrosome over-duplication in S phase.12 This discrepancy could be linked to different levels of ectopic CDC25B expression relative to the endogenous protein. As shown previously,32 the expression levels of HA-tagged CDC25B-Wt or -DDA in asynchronous stable inducible U2OS cells were similar. However, during mitosis, the level of HA-tagged CDC25B-DDA is higher than that of HA-tagged CDC25B-Wt (Fig. 5A). Importantly, while ectopic expression of mCherry-CDC25B-Wt or -DDA (under a strong promoter) induced a similar multipolar spindle phenotype in 27.5 % and 28.5 % of mitotic cells, respectively (Fig. 2), overexpression of mCherry-CDC25B-C/S, a catalytic inactivated mutant of CDC25B, did it at a lower percentage (13%) comparable to mCherry alone (13%). These results indicate that the phosphatase activity of CDC25B is absolutely required to induce multipolar spindle.
Figure 1.
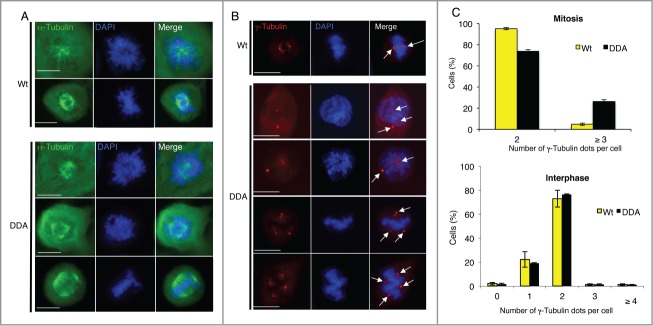
Stabilization of CDC25B causes centrosome aberration only in mitosis. (A). Twenty-4 hours after induction of CDC25B-Wt (Wt) or CDC25B-DDA (DDA) expression in U2OS Tet-off cell lines stably transfected with the respective plasmids, asynchronous cells were fixed, and incubated with an anti-α-Tubulin antibody (green). DNA was stained with DAPI. Images are from fluorescence microscopy of prophasic and metaphasic cells. Scale bar: 10 μm. (B). Experiments were performed as in A, except that cells were stained with an anti-γ-Tubulin antibody (red). Positive γ-Tubulin dots are highlighted by arrows. Images are from fluorescence microscopy of representative prophasic and metaphasic cells (3 independent experiments). Scale bar: 10 μm. (C). Quantification of the number of γ-Tubulin-positive dots per cell in mitotic (upper panel) or interphase (lower panel) U2OS cells that express either CDC25B-Wt or CDC25B-DDA. Error bars indicate ± SD of 3 independent experiments.
Figure 5.
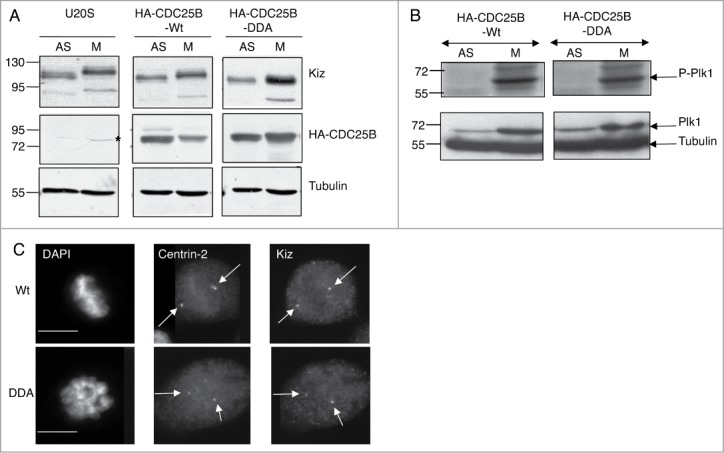
Stabilization of CDC25B during mitosis modifies Kiz phosphorylation without affecting Plk1 activity and centrosomal localization of Kiz. (A). Asynchronously growing (AS) and nocodazole treated (M) parental U2OS cells or that express CDC25B-Wt or CDC25B-DDA were lysed and immunoprobed as indicated. *: non-specific band. (B). Experiments were performed as in A, except that cell extracts were immunoprobed with anti-Plk1 and anti-Phopho-Plk1 (Thr120). (C). Twenty-4 hours after the induction of CDC25B-Wt or CDC25B-DDA expression in U2OS Tet-off cells, cells were fixed and incubated with anti-Centrin-2 and -Kiz antibodies. DNA was stained with DAPI. Images of mitotic cells are shown. Scale bar, 10 μm.
Figure 2.
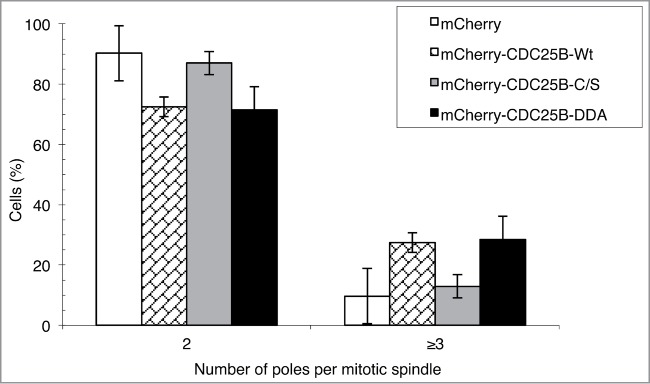
CDC25B phosphatase activity is required to induce multipolar spindles in cells. HeLa cells were transiently transfected with pmCherry-CDC25B (-Wt, -C/S or -DDA) or pmCherry expressing vectors. Fourteen hours post-transfection, cells expressing mCherry-tagged CDC25B-Wt, CDC25B-C/S, CDC25B-DDA, or mCherry alone were imaged by time-lapse microscopy for 24 hours. The number of poles per mitotic spindle was counted. Data represent the percentage of cells with 2 or more than 2 poles and values are the mean ± SD of 3 independent experiments. Total number of counted cells: mCherry, n = 161; mCherry-CDC25B-Wt, n = 106; mCherry-CDC25B-C/S, n = 126; mCherry-CDC25B-DDA, n = 144.
Since the abnormal number of γ-Tubulin dots was only observed in mitosis, with spindle poles that fragment, a consequence resulting from aberrant splitting and fragmentation of centrioles,34-35 we co-stained asynchronous U2OS-CDC25B-Wt and U2OS-CDC25B-DDA cells with antibodies against γ-Tubulin and Centrin-2, a centriole marker, to investigate spindle pole composition (Fig. 3). In mitotic U2OS-CDC25B-Wt cells (n = 120), γ-Tubulin and Centrin-2 co-localized (Fig. 3A) in all, but 2.38 % of cells (Fig. 3B). Conversely, in mitotic U2OS-CDC25B-DDA cells (from prophase to metaphase) (n = 130), γ-Tubulin and Centrin-2 dots did not co-localize in 18.7 % of cells (Fig. 3A-B). In some U2OS-CDC25B-DDA cells with more than 2 γ-Tubulin dots, the distance between centrin-2 spots was significantly greater than expected for a normal diplosome, a phenotype corresponding to a disengagement of centriolar pairs (Fig. 3A). Moreover only one centrin-2 spot was present on the other pole. Quantification of the proportion of cells with disengaged centriolar pairs showed that it was not an artifact of CDC25B-DDA expression as a similar percentage of cells with this phenotype (Fig. 3A and 4A) was also observed in U2OS-CDC25B-Wt and parental U2OS cells, either in cells containing 2 or more than 2 γ-Tubulin dots (Fig. 4B). Similarly, the absence of Centrin-2 spot on the opposite spindle pole was also observed in the same proportion in the 3 cell lines. Taken together, these results demonstrate that the presence of extra γ-Tubulin dots in U2OS-CDC25B-DDA cells (Fig. 3A) is neither a consequence of centrosome over-duplication nor a disengagement of centriolar pairs, but rather the result of PCM fragmentation. The results in Figures 1 and 3 show that high level CDC25B expression (due to its stabilization) during mitosis weakens PCM cohesion, leading to its fragmentation and consequently to the formation of multipolar spindles.
Figure 3.
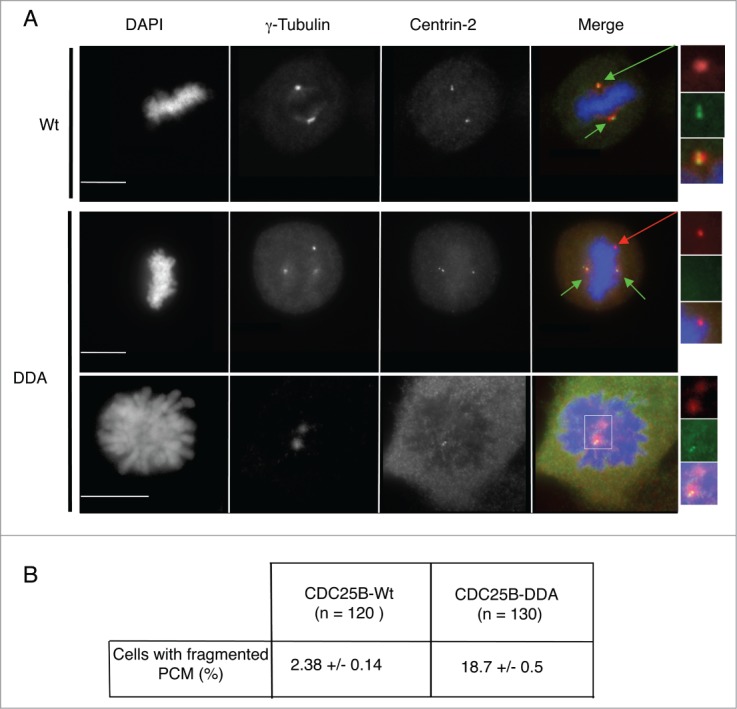
Multipolar spindles in U2OS-CDC25B-DDA cells are the result of PCM fragmentation. (A). Representative fluorescent microscopy images of asynchronously growing U2OS cells in metaphase or prophase that express CDC25B-Wt or CDC25B-DDA fixed and stained with anti-γ-Tubulin and anti-Centrin-2 antibodies. DNA was stained with DAPI. Centrosomes are highlighted by green arrows, and PCM fragmentation by a red arrow (magnified view in insets). In the merged images, yellow shows co-localization of Centrin-2 and γ-Tubulin. Scale bar: 10 μm. (B). The number of cells with fragmented PCM (i.e., cells with an excess of γ-Tubulin dots) was quantified in U2OS-CDC25B-Wt and U2OS-CDC25B-DDA cells. Values are the mean ± SD of 3 independent experiments.
Figure 4.
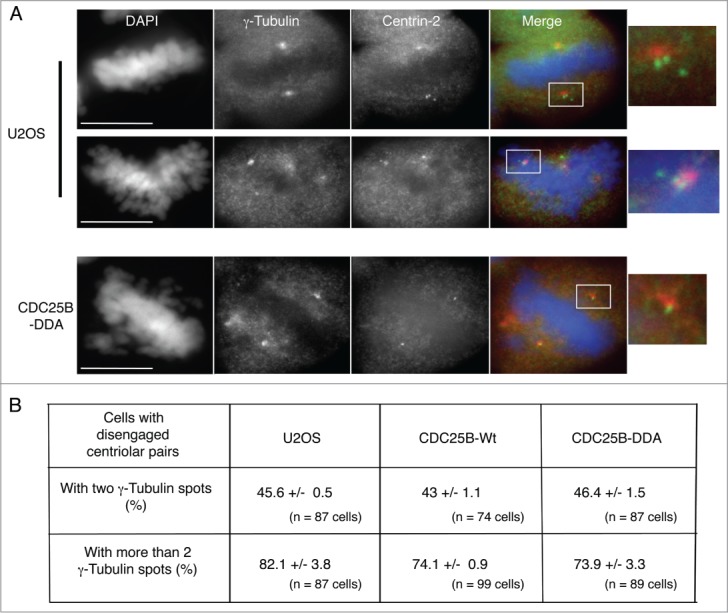
Stabilization of CDC25B does not induce a disengagement of centriolar pairs. (A). Representative fluorescent microscopy images of mitotic cells from an asynchronous population of parental or CDC25B-DDA U2OS cells fixed and stained with anti-γ-Tubulin and anti-Centrin-2 antibodies. DNA was stained with DAPI. In the merged images, yellow shows co-localization of Centrin-2 and γ-Tubulin. Scale bar: 10 μm. (B). The number of cells with disengaged centriolar pairs (i.e., cells in which the distance between centriols is greater than expected for a diplosome) in cells presenting either 2 or more than 2 γ-Tubulin dots was quantified in U2OS, U2OS-CDC25B-Wt and U2OS-CDC25B-DDA cells. Values are the mean ± SD of 3 independent experiments.
During mitosis, the activity of CDK1/Cyclin B complexes is not affected in U2OS-CDC25B-DDA cells,32 suggesting that multipolar phenotype is directly linked to CDC25B. We know that inhibition of Plk1-mediated Kiz protein phosphorylation at Thr379, a phosphorylation that positively controls Kiz function in the stabilization of PCM during mitosis, causes PCM fragmentation.28 In addition we show that the expression of non-phosphorylable Kiz mutant (Kiz-T379A) in cells depleted for Kiz, did not rescue PCM fragmentation induced by the depletion of Kiz,28 but rather increased it at a level comparable to that observed in U2OS-CDC25B-DDA cells (Fig. S1). Thus, we investigated whether CDC25B-DDA could affect Kiz phosphorylation. Kiz electrophoretic mobility was reduced in mitotic parental U2OS and U2OS-CDC25B-Wt cells in comparison to asynchronously growing cells (Fig. 5A), showing that Kiz was phosphorylated in mitosis as previously described.28 Conversely, in nocodazole treated U2OS-CDC25B-DDA cells, Kiz electrophoretic mobility was not reduced (Fig. 5A), indicating that CDC25B stabilization affects Kiz phosphorylation. CDC25B stabilization could impair Kiz phosphorylation indirectly by affecting either the level and/or activity of Plk1 kinase, or Kiz centrosomal localization, which depends on Kiz interaction with Cep72.36 To explore these possibilities, the expression level and activity of Plk1 kinase were assessed by immunoblotting. We found that neither Plk1 expression level nor its activation, visualized by its phosphorylation on Thr120 were affected in mitotic U2OS-CDC25B-DDA cells in comparison to mitotic U2OS-CDC25B-Wt cells, in which Kiz phosphorylation was not impaired (Fig. 5B). Also, immunofluorescence analysis showed that Kiz localized normally at centrosomes in mitotic U2OS-CDC25B-DDA cells (Fig. 5C).
These findings suggest a potential enzyme-substrate relationship between CDC25B and Kiz at centrosomes that could affect Plk1-mediated phosphorylation of Kiz. To confirm this possibility, we performed an in vitro kinase and phosphatase assay with recombinant GST-Kizuna (GST-Kiz), His6-Plk1 and MBP-CDC25B (Fig. 6A, left panel). As a specificity control, we used CDC25A, which is closely related to CDC25B and shares CDK/Cyclin substrates with CDC25B. We first verified that recombinant CDC25A and B had a similar phosphatase activity, i.e., were able to activate to a similar extent the initially inhibited (phosphorylated) CDK1/Cyclin B complexes (Fig. 6A, right panel). When GST-Kiz, previously phosphorylated by Plk1, was incubated with MBP-CDC25B for 20 min,33 P incorporation in Kiz was significantly reduced to 30 to 35 % of the initial phosphorylation level, showing that Plk1-phosphorylated Kiz can be a CDC25B substrate (Fig. 6A-B). Conversely, under the same conditions, MBP-CDC25A did not dephosphorylate Kiz as efficiently as CDC25B (3 to 11 % reduction). Analysis of a time-course dephosphorylation of Plk1-phosphorylated Kiz by both phosphatases showed that Kiz-dephosphorylation was more efficient with MBP-CDC25B than with MBP-CDC25A (Fig. 6C), and when MBP-CDC25B was inactivated by a specific CDC25-inhibitor (NSC-663284), the level of Kiz dephosphorylation was similar to that obtained with MBP-CDC25A alone (20 and 30 min) (Fig. 6C). The 35% remaining Kiz-phosphorylation observed in the case of MBP-CDC25B treatment could indicate that either residual phosphorylation corresponds to off-target phosphorylation (see below) or in vitro Plk1 phosphorylates also Kiz, on sites not targeted by CDC25B but maybe by other phosphatases, including CDC25C. Surprisingly, we never obtained a total inhibition of Kiz dephosphorylation in presence of CDC25 inhibitor possibly due to the presence of contaminating phosphatases. As the 3 CDC25 phosphatases bear a catalytic domain highly conserved but greatly differ in their N-terminal regulatory domain, our results strongly support the idea that CDC25B effect on Kiz is highly specific and is not the result of a promiscuous in vitro interaction. Since Kiz Thr379 is the only phosphorylation site for Plk1 in cellulo,28 we also used in the same assay the Kiz-T379A mutant (GST-Kiz-TA) as a control of in vitro off-target phosphorylation. As with GST-Kiz-Wt, CDC25B did not modify the residual phosphorylation level of GST-Kiz-TA (Fig. 6A-B). Altogether, these results demonstrate that CDC25B, but not CDC25A, can specifically reverse Plk1-dependent Kiz phosphorylation. Thus, occurrence of multipolar spindles caused by PCM fragmentation in cells where CDC25B is stabilized could be the consequence of Kiz dephosphorylation by this phosphatase. To support these in vitro experiments, we expressed a phosphomimetic mutant of Kiz (Kiz-T379E) to counteract the appearance of multipolar spindles in U2OS-CDC25B-DDA cells. Unfortunately, all our attempts resulted in cell death (Fig. S2, upper panel) or the total absence of mitotic cells (Fig. S2, lower panel), depending of expression vector used.
Figure 6.

Kiz is dephosphorylated by CDC25B, not CDC25A. (A). Recombinant GST-Kiz-Wt or GST-Kiz-TA (T379A) immobilized on Glutathione beads was used in a phosphorylation/dephosphorylation assay in the presence of recombinant His6-Plk1 and MBP-CDC25A or MBP-CDC25B, as described in “Materials and methods." The phosphorylation level of GST-Kiz was then determined by autoradiography. Incorporation of33 P into GST-Kiz, was normalized to the protein amount of each band based on Coomassie staining (left panel). The relative activity of recombinant MBP-CDC25 phosphatases was indirectly determined by measuring the activation of inactive CDK1 associated with Cyclin B (i.e., CDK1 phosphorylation on Thr14 and Tyr15), in a kinase assay using histone H1 as substrate. For this 50 ng of recombinant MBP-CDC25A or MBP-CDC25-B were incubated with immunoprecipitated inactive CDK1/Cyclin B complexes. Activity of CDK1/Cyclin B complexes was then tested in an in vitro histone H1 kinase assay, as described in.51 Equal loading was shown by Coomassie staining. The incorporation of33 P into histone H1 reflects the activity of CDK1/Cyclin B complexes (right panel). (B). Quantification of33 P incorporation in recombinant GST-Kiz obtained in A. Values from 3 independent experiments were normalized to those of GST-Kiz-Wt incubated only with His6-Plk1. Data are the mean ± SEM of 3 independent experiments (*, P = 0.0318). (C). Quantification of33P incorporation in recombinant GSt-Kiz-Wt during a kinetic of dephosphorylation by recombinant MBP-CDC25B, in presence or not of NSC-663284 (500 nM), or MBP-CDC25A. Values were normalized to those of GST-Kiz-Wt incubated only with His6-Plk1.
To determine if CDC25B could directly regulate the phosphorylation level of Kiz, we investigated whether Kiz-CDC25B complexes exist in cells. Immunoprecipitation experiments (Fig. 7A) showed a weak interaction between co-expressed HA-CDC25B and Myc-Kiz in asynchronously growing HeLa cells. The weak signal probably reflects a transient enzyme-substrate interaction that occurs in a precise cell compartment and cell cycle phase. Indeed, while endogenous Kiz is localized at the centrosome throughout the cell cycle,28 only a subset of endogenous CDC25B is located there.10,37 We next assessed a potential differential interaction between endogenous Kiz and CDC25B in mitosis relative to interphase. CDC25B and Kiz were immunoprecipitated from asynchronous or mitotic HeLa cells, and the immunoprecipitates analyzed by immunoblotting. While no interaction could be detected between CDC25B and Kiz in extracts from asynchronous cells (Fig. 7B, left panel), hypophosphorylated forms of Kiz were co-immunoprecipitated with CDC25B from mitotic cell extracts (Fig. 7B, middle panel). These forms co-migrate with the hypo-phosphorylated Kiz bands following λ phosphatase treatment of the immunoprecipitates obtained with anti-Kiz antibodies. These results demonstrate that CDC25B can interact with Kiz and dephosphorylates it in mitosis.
Figure 7.
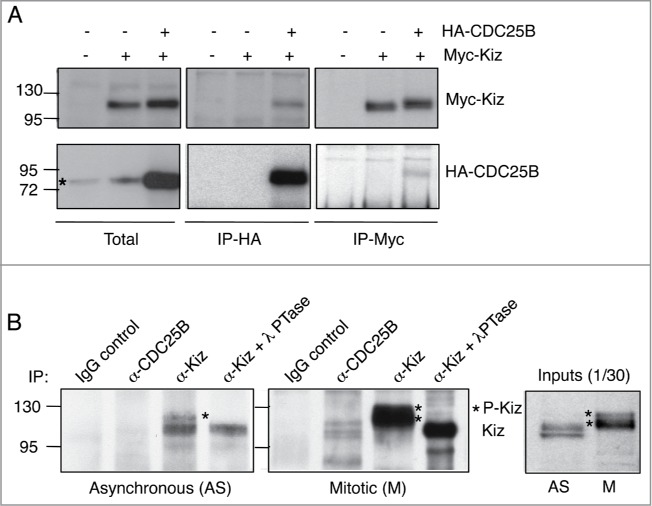
CDC25B interacts with Kiz during mitosis. (A). HeLa cells were transfected or not with HA-CDC25B-Wt or Myc-Kiz (pRK5) and lysed 24 h after transfection. Cell extracts were incubated with control IgG or anti-Myc/anti-HA antibodies, and immunocomplexes probed for the indicated proteins. *: non-specific band. (B). Asynchronous (left panel) or mitotic (right panel) HeLa cells were lysed in the presence of a cross-linking agent (DSP), immunoprecipitated with anti-CDC25B or anti-Kiz antibodies and immunoprobed with anti-Kiz antibodies. In indicated cases, immunoprecipitates obtained with anti-Kiz antibodies were treated with lambda phosphatase (λPTase, 200 units) 15 min at 30°C before SDS-PAGE (α-Kiz + λPTase).
To dissect the mechanism governing Kiz regulation by CDC25B during mitosis, we investigated in cellulo the precise timing of Kiz phosphorylation and the effects of Plk1 and CDC25B inhibitors. Immunoblotting analyses of extract from asynchronous, G2/M transition block or pro-metaphase arrested cells indicated that Kiz was neither phosphorylated in interphase nor at the G2/M transition, but fully phosphorylated in pro-metaphase resulting in a mobility shift in SDS-PAGE (Fig. 8A). We then examined Kiz mobility in mitotic cells during a time-course after release from G2/M block. Probing for Kiz revealed that the change in electrophoretic mobility of Kiz occurred already 12 min after G2/M-block release (when cells enter mitosis), but no reduction of the shift was observed in the presence of Plk1 inhibitor (Fig. S3). These results suggest that: i) Kiz is phosphorylated as soon as cells enter mitosis and ii) under our condition the mitosis duration (40-50 min) is too short to see in early phases of mitosis the effect of Plk1 inhibitor or alternatively Kiz is phosphorylated by other kinases in addition to Plk1. Entrance into mitosis is induced by the activation of CDK1/Cyclin B complexes, which phosphorylate many substrates, phosphorylation that can prime for Plk1 phosphorylation in some cases. Performing an in vitro kinase assay with CDK1/Cyclin B, we could show that Kiz is a substrate for CDK1/Cyclin B in vitro (Fig. 8B). Next we used a pro-metaphase cell extract in an in vitro kinase assay with recombinant GST-Kiz-Wt in presence or not of Plk1,38 CDK1 and CDC2539 inhibitors (Fig. 8C). As shown in Figure 8C, the pro-metaphase cell extract was able to phosphorylate GST-Kiz-Wt in vitro, and the pre-incubation of the same extract with either Plk1 or CDK1 inhibitors decreased GST-Kiz phosphorylation of 30% and 40% after 30 min, respectively. Moreover, a pre-incubation of the extract with both inhibitors had an additive inhibition of Gst-Kiz phosphorylation. These results are in agreement with the hypothesis that CDK1 is involved in the priming phosphorylation of Kiz. Noteworthily, phosphorylation of GST-Kiz was inhibited by 50% when CDC25 inhibitor was added to the pro-metaphasic cell extract. Knowing that CDK1 contributes to the phosphorylation of Kiz and CDC25B positively controls CDK1 kinase activity (Fig. 8D) our result appears consistent. Altogether these results support the involvement of CDC25B in the complex regulation of Kiz activity in PCM stabilization through the control of its phosphorylation.
Figure 8.
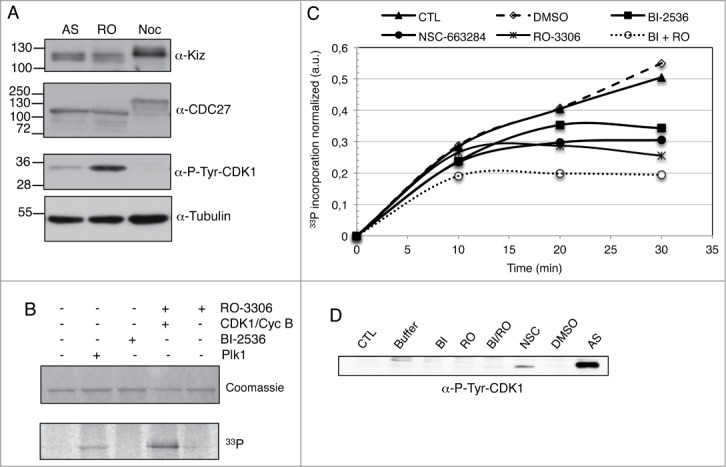
CDK1/Cyclin B, Plk1 and CDC25B regulate Kiz-phosphorylation level during mitosis. (A). Total cell extract (30 μg) of asynchronous (AS), RO-3306 (RO) or nocodazole treated (Noc) HeLa cells were analyzed on SDS-PAGE and immunoprobed as indicated. Anti-CDC27 and anti-P-Tyr-CDK1 were used to check mitosis progression. Anti-Tubulin served as a loading control. (B). Recombinant GST-Kiz-Wt immobilized on Glutathione beads was used in an in vitro kinase assay in the presence of recombinant His6-Plk1 or GST-CDK1/GST-Cyclin B kinases, pre-incubated or not with BI-2536 (250 nM) and RO-3306 (1 μM), respectively. The phosphorylation level of GST-Kiz was then determined by autoradiography. (C). Recombinant GST-Kiz-Wt immobilized on Glutathione beads was used in a phosphorylation assay in the presence of pro-metaphase HeLa cell extract, as described in “Materials and methods," in presence or not of BI-2536 (250 nM), RO-3306 (1 μM), NSC-663284 (1 μM) or DMSO. The phosphorylation level of GST-Kiz was then determined by autoradiography. Incorporation of33 P into GST-Kiz, was normalized to the protein amount of each band based on Coomassie staining. Values from 3 independent experiments were normalized to those of GST-Kiz-Wt incubated only with pro-metaphase extract. Data are the mean of 3 independent experiments. (D). Pro-metaphase HeLa cell extract was incubated for 30 min at 30°C with kinase buffer in presence of the indicated kinases or phosphatase inhibitors, as in C, analyzed on SDS-PAGE and immunoprobed with anti-P-Tyr-CDK1. Extract from asynchronous HeLa cells was used as control for CDK1 phosphorylation on Tyr-15.
In summary, our experiments show that: i) in mitosis, expression of a CDC25B mutant (CDC25B-DDA) whose proteasomal degradation is impaired32 leads to Kiz dephosphorylation and PCM fragmentation, ii) in vitro, CDC25B, but not its close relative CDC25A, can reverse Plk1-dependent phosphorylation of Kiz, a modification that is critical for PCM stability,28 iii) in cellulo, CDC25B interacts with Kiz during mitosis, when its phosphorylation is required for PCM stability. Based on these results, we propose a model in which, during mitosis (from prophase to the end of metaphase), the phosphorylation level of Kiz at centrosomes is controlled by a balance between Plk1 kinase and CDC25B phosphatase activities (Fig. 9). When cells enter mitosis, unphosphorylated Kiz would be first phosphorylated by CDK1/Cyclin B (on site(s) that remain to be characterized), which would prime it for Plk1 phosphorylation. Until metaphase, a precise balance between Plk1 kinase and CDC25B (and potentially CDC25C) phosphatase activities could allow at the same time expansion and stabilization of the PCM necessary for a proper establishment of the mitotic spindle. Under normal conditions, the abrupt, proteasome-dependent degradation of CDC25B at the metaphase-anaphase transition, tilts the balance in favor of Plk1. Thus ensuring the complete phosphorylation of Kiz that is required for its function to stabilize and maintain a robust PCM architecture (Fig. 9A). When CDC25B is stabilized, i.e., U2OS-CDC25B-DDA cells, this results in its high expression level throughout mitosis, which exceeds the CDC25B threshold required to maintain a proper balance between Kiz-phosphorylation and -dephosphorylation. As a consequence, Kiz is maintained under an hypo-phosphorylated status, and PCM is structurally weakened during all mitotic phases (Fig. 9B).
Figure 9.

Proposed model for the regulation of Kiz phosphorylation during mitosis. (A). During mitosis, phosphorylation level of Kiz results from CDK1/Cyc B kinase activity and a balance between Plk1 kinase and CDC25B phosphatase activities. At the entrance into mitosis Kiz is phosphorylated by CDK1/Cyc B, a priming phosphorylation that facilitates its phosphorylation on Thr379 by Plk1. Until metaphase-anaphase transition, phosphorylation state of Thr379 results from a balance between activities of Plk1 and CDC25B (and maybe CDC25C) to finely control the equilibrium between expansion and stabilization of the PCM around the centrosomes. At the metaphase-anaphase transition, when CDC25B is degraded by the proteasome, Kiz is fully phosphorylated on Thr379 by Plk1. (B). During mitosis, if a defect in proteasome-dependent degradation of CDC25B occurs, such as when the CDC25B-DDA mutant is expressed, the high level of CDC25B maintains Kiz in an hypo-phosphorylated form that hinders its ability to stabilize PCM around the centrosomes during all phases of mitosis, resulting into the formation of multipolar spindles.
Furthermore, these data identify Kiz as a new substrate of the CDC25B phosphatase other than CDK1/Cyclin B during mitosis. Thus, CDC25B participates not only in the control of centrosome duplication12 through the regulation of CDK/Cyclin complexes and centrin 2 stability,13 but also in the control of the stability of the PCM around centrosomes during mitosis by dephosphorylating Kiz. This finding allows us to extend the list of substrates for the members of CDC25 phosphatase family. Some of them require the catalytic activity of CDC25, such as the transcription factor Cut,40 Raf kinase41 that in turn controls CDC25A activity,42 the epidermal growth factor receptor (EGFR),43 or the extracellular signal-regulated kinase (ERK)44 for CDC25A. Others, such as steroid receptors for CDC25B, are independent of the phosphatase activity,45 and CDC25B is considered as a co-activator. Our result suggests that, during the cell cycle, a defect in the degradation of CDC25 phosphatases will affect differently their various substrates, ultimately leading to genomic instability. It will be interesting to determine whether such defects exist in cancers cells overexpressing CDC25B, as it was shown for CDC25A,46 and to identify other new CDC25 substrates that might participate in chromosomal instability during carcinogenesis.
Materials and Methods
Plasmids
Human Kizuna cDNA was PCR-amplified from pDONR223 (provided by the Montpellier Genomic Collections facility, Institut de Génétique Moléculaire de Montpellier, Montpellier, France) and cloned into pRK5-Myc (provided by G. Bompard, Institut de Génétique Humaine, Montpellier, France), and pGEX-2T (GE Healthcare). PCR-based site-directed mutagenesis (Thr379-Ala) was performed using the pGEX-Kiz plasmid as a template. pcDNA-6Myc-Kizr (Wt, T379E and T379A) were provided by M. Ohsugi and T. Yamamoto (Institute of Medical Science, university of Tokyo, Tokyo, Japan). CDC25B1-Wt and CDC25B1-DDA subcloned into pcDNA3-HA and pMAL-C2 were described elsewhere.47,48 Both CDC25B-Wt and -DDA open reading frames were cloned into pmCherry-C1. Point mutation in CDC25B catalytic domain (Cyst473Ser) was generated by PCR mutagenesis on the pmCherry-CDC25B.
Antibodies and compounds
The following antibodies were used for immunoprecipitation or immunoblotting: rabbit polyclonal anti-CDC25B (sc-326, Santa Cruz Biotechnology, Inc..), anti-Kizuna (A300-946A, Bethyl Laboratories, Inc..), anti-Phospho-Plk1 (Thr210) (# 5472, Cell Signaling Technology), anti-P-Tyr-CDK1 (# 9111, Cell Signaling Technology), anti-CDC27 (a gift of T. Lorca (Center de Recherche de Biochimie Macromoléculaire, Montpellier, France), monoclonal anti-α−Tubulin (clone DM1A, Sigma-Aldrich), anti-Plk1 (F8, sc-17783, TEBU), anti-HA (clone 12CA5, Roche Diagnostic), anti-Myc (clone 9E10, Abcam), and GFP-Trap agarose beads (ChromoTek). Secondary HRP-conjugated antibodies were purchased from Bio-Rad SA.
The following antibodies were used for indirect immunofluorescence: rabbit polyclonal anti-γ-Tubulin and anti-Centrin-2 were a gift of B. Raynaud-Messina (Center de Biologie du Développement, Toulouse, France) and J. Piette (Center de Recherche de Biochimie Macromoléculaire, Montpellier, France), respectively. Secondary antibodies were Alexa Fluor 488 or 594 conjugates (Interchim).
BI-2536 (Plk1 inhibitor, S1109, Selleckchem), RO-3306 (CDK1 inhibitor, SML0569, Sigma-Aldrich), NSC-663284 (CDC25 inhibitor, N7537, Sigma-Aldrich) and MG132 (Calbiochem) were dissolved in DMSO and used at the indicated concentrations.
Cell culture and transfections
HeLa cells, U2OS cells and U2OS Tet-off cells were grown in DMEM (Lonza) containing 4.5 g/liter glucose, 10% heat inactivated fetal bovine serum (Biowest), 2mM glutamine, 100 U/ml penicillin and 10 μg/ml streptomycin (Lonza). The U2OS Tet-off cell lines that stably express HA-CDC25B1-Wt or HA-CDC25B1-DDA were previously described;47,32 culture medium for U2OS Tet-off cell was supplemented with 250 μg/ml G418 (Sigma-Aldrich), 200 μg/ml hygromycin B (Calbiochem) and 2 μg/ml tetracycline (Sigma-Aldrich). For induction of HA-CDC25B1 expression, tetracycline was removed from the medium and cells were analyzed 24 hours after induction. HeLa and U2OS cells were arrested in pro-metaphase by adding 200 ng/ml or 40 ng/ml nocodazole (Sigma-Aldrich), respectively, for 14 h. Mitotic cells were recovered by shake-off. To obtain U2OS cells arrested at G2/M transition, cells were first blocked at G1/S transition by a treatment with 2.5 mM Thymidine (Sigma-Aldrich) for 24 h, then released for 6 h before adding 9 μM RO-3306 (Sigma-Aldrich) for 11 h. To induce entrance and progression into mitosis, cells were extensively washed with fresh media. Transient transfections with expression vectors (Myc-Kiz, HA-CDC25B or mCherry-CDC25B) in asynchronous HeLa or U2OS cells were performed using JetPEITM (Ozyme), according to the manufacturer's instructions.
Indirect immunofluor-escence and time-lapse analysis
Cells grown on coverslips were either fixed with 3.7% paraformaldehyde and then permeabilized with 0.25% Triton X100 and cold methanol, or only fixed/permeabilized with cold methanol at -20°C. Cells were washed with PBS and blocked with 1% FCS/PBS for 15 min. Incubation with primary antibodies was carried out at 37°C for 1 h and with secondary antibodies at room temperature for 40 min. DNA was stained with DAPI (D9542, Sigma-Aldrich). Microscopic examinations were performed with 63X/1.4 NA or 100X/1.4 NA oil immersion objective lenses using a DM 6000 microscope (Leica). Microphotographs were taken with a 12-bit Coolsnap HQ2 1 camera. Images were acquired as TIF files using the MetaMorph imaging software (Molecular Devices).
HeLa cells, transfected with various pmCherry expression vectors, were imaged 14 hours after transfection, using a DMIRE2 microscope (Leica) with a 40X N PLAN L 0.55 LMC objective. Images were collected every 10 minutes using a Micromax YHS 1300 camera (Roper Scientific Princeton Instruments) monitored by MetaMorph software.
Expression and purification of recombinant proteins
Recombinant MBP-CDC25A and MBP-CDC25B1 were expressed in the E. coli JM109 strain in the presence of 0.2% glucose, induced with 100 μM isopropyl ß-D-thiogalactoside (IPTG) (EU0008, Euromedex) at 25°C for 3 hours, and affinity purified with amylose beads (New England Biolabs, Inc.) as described previously.47 Recombinant GST-Kizuna-Wt (GST-Kiz-Wt) and GST-Kizuna-T379A (GST-Kiz-TA) were expressed in the E. coli TOP10 strain and induced with 1 mM IPTG at 37°C for 3 hours. After lysis in a buffer containing 20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM EDTA, 0.5% NP40 and 1X complete protease inhibitor cocktail (11873580001, Roche Diagnostic), recombinant proteins were purified on Glutathione-Sepharose beads (17-0756-01, GE Healthcare). Recombinant His6-Plk1, a gift of Dr. S. Rouquier (Institut de Génétique Humaine, Montpellier, France), was purified under native conditions as described previously.49 Recombinant CDK1-Cyclin B (# 7518) was purchased from Cell Signaling Technology.
Co-immunoprecipitation
For co-immunoprecipitation of ectopically expressed proteins, HeLa cells were co-transfected with the corresponding constructs. Cells were homogenized in lysis buffer (50 mM Tris-HCl, pH 7.5, 250 mM NaCl, 0.1 % Triton X100, 1 mM DTT, 5 mM EDTA, 50 mM NaF and 0.1 mM Na3VO4) in the presence of 1X complete protease inhibitor cocktail and 25 μM MG132 (BML-PI102-0005, ENZO life) at 4°C for 30 min. After centrifugation at 13.200 rpm for 10 min, 2 μg anti-HA or anti-Myc antibodies were added to the pre-cleared supernatants. Proteins were immunoprecipitated at 4°C for 2 hours and collected by addition of 10 μl protein-A or -G Sepharose. After extensive washes, beads were boiled in 2X loading sample buffer and analyzed by SDS-PAGE and immunoblotting.
For co-immunoprecipitation of endogenous proteins, cell extracts were obtained as described in MacKay et al.50 Briefly, HeLa cells were homogenized in lysis buffer (40 mM Hepes-KOH, pH 7.5, 120 mM NaCl, 1% Triton X100, 1 mM EDTA), complete protease inhibitor cocktail and 2 mg/ml of Dithiobis [succinimidy propionate] (DSP) (22585, Pierce) at 4°C for 30 min. DSP was neutralized by addition of 1M Tris-HCL, pH 7.5, to a final concentration of 200 mM, 25 μM MG132 was then added and the mix incubated at 4°C for another 30 min. After centrifugation at 13.200 rpm for 10 min, 3 μg anti-CDC25B or anti-Kiz antibodies were added to the pre-cleared supernatants and proteins were immunoprecipitated as above. As control, random rabbit IgGs were used.
Kinase and phosphatase assays
1 μg of GST-Kiz bound to Glutathione-Sepharose beads was incubated in kinase buffer (20 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 1 mM EGTA, 5 mM DTT, 5 mM NaF, 50 μM ATP) supplemented with 5 μCi of γ-33 P-ATP (Perkin Elmer) and 150 ng of recombinant His6-Plk1 were added to the mix that was incubated at 30°C for 15 min. After 2 washes with TNE buffer (20 mM Tris-HCl, pH 7.5, 125 mM NaCl, 2 mM EGTA, 0.5 % NP40) and one wash with phosphatase buffer (50 mM Tris-HCl, pH 8, 50 mM NaCl, 1 mM DTT, 1 mM EDTA), GST-Kiz beads were incubated with 72 ng MBP-CDC25A or 78 ng of MBP-CDC25B1 at 30°C for 20 min, washed twice with phosphatase buffer and boiled in 2X loading sample buffer before analysis by SDS-PAGE.
For in vitro phosphorylation assay using cell extract, HeLa cells blocked in pro-metaphase were lysis in NET buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.25% IGEPAL, 1 mM EDTA) containing 10 mM PMSF, 25 μM MG132, complete protease inhibitor cocktail, during 20 min at 4°C. After centrifugation, concentration of clarified cell extract was determined. 1 μg of GST-Kiz bound to Glutathione-Sepharose beads was incubated in kinase buffer (50 mM Tris-HCl, pH 7.5, 5 mM MgCl2, 1 mM DTT, 5 μM MG132, 50 μM ATP) supplemented with 5 μCi γ-33P-ATP and 30 μg of cell extract was added to the mix that was incubated at 30°C for indicated time. After 2 washes with TNE buffer, beads were boiled in 2X loading sample buffer before analysis by SDS-PAGE.
Statistical analysis
Statistical analyses were performed using the non-parametric Mann-Whitney test and the Prism software (GraphPad Software). Statistical significance was defined as P ≤ 0.05.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank S. Rouquier for recombinant purified His6-Plk1, B. Messina-Raynaud for polyclonal anti-γ-Tubulin antibodies, J. Piette for anti-Centrin-2 antibodies, M. Ohsugi and T. Yamamoto for pcDNA-6Myc-Kizuna plasmids, and Montpellier RIO Imaging for the service quality. We are grateful to S. Boulon, C. Bonne-Andréa for their helpful discussions and comments on our manuscript. We also thank R. Hipskind for critical reading of the manuscript.
Funding
This work was funded by fellowships from the Fondation ARC to Y.T. and a grant from the Ligue Régionale Languedoc-Roussillon Contre le Cancer (N°078211) to V.B.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website. http://www.tandfonline.com/kccy
References
- 1. Morgan DO. Principles of CDK regulation. Nature 1995; 374:131-4; PMID:7877684; http://dx.doi.org/ 10.1038/374131a0 [DOI] [PubMed] [Google Scholar]
- 2. Boutros R, Lobjois V, Ducommun B. CDC25 phosphatases in cancer cells: key players? Good targets? Nat Rev Cancer 2007; 7:495-507; PMID:17568790; http://dx.doi.org/ 10.1038/nrc2169 [DOI] [PubMed] [Google Scholar]
- 3. Boutros R, Dozier C, Ducommun B. The when and wheres of CDC25 phosphatases. Curr Opin Cell Biol 2006; 18:185-91; PMID:16488126; http://dx.doi.org/ 10.1016/j.ceb.2006.02.003 [DOI] [PubMed] [Google Scholar]
- 4. Ferguson AM, White LS, Donovan PJ, Piwnica-Worms H. Normal cell cycle and checkpoint responses in mice and cells lacking Cdc25B and Cdc25C protein phosphatases. Mol Cell Biol 2005; 25:2853-60; PMID:15767688; http://dx.doi.org/ 10.1128/MCB.25.7.2853-2860.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee G, White LS, Hurov KE, Stappenbeck TS, Piwnica-Worms H. Response of small intestinal epithelial cells to acute disruption of cell division through CDC25 deletion. Proc Natl Acad Sci U S A 2009; 106:4701-6; PMID:19273838; http://dx.doi.org/ 10.1073/pnas.0900751106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee G, Origanti S, White LS, Sun J, Stappenbeck TS, Piwnica-Worms H. Contributions made by CDC25 phosphatases to proliferation of intestinal epithelial stem and progenitor cells. PLoS One 2011; 6:e15561; PMID:21283624; http://dx.doi.org/ 10.1371/journal.pone.0015561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lammer C, Wagerer S, Saffrich R, Mertens D, Ansorge W, Hoffmann I. The cdc25B phosphatase is essential for the G2/M phase transition in human cells. J Cell Sci 1998; 111(Pt 16):2445-53; PMID:9683638; . [DOI] [PubMed] [Google Scholar]
- 8. Karlsson C, Katich S, Hagting A, Hoffmann I, Pines J. Cdc25B and Cdc25C differ markedly in their properties as initiators of mitosis. J Cell Biol 1999; 146:573-84; PMID:10444066; http://dx.doi.org/ 10.1083/jcb.146.3.573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lindqvist A, Kallstrom H, Lundgren A, Barsoum E, Rosenthal CK. Cdc25B cooperates with Cdc25A to induce mitosis but has a unique role in activating cyclin B1-Cdk1 at the centrosome. J Cell Biol 2005; 171:35-45; PMID:16216921; http://dx.doi.org/ 10.1083/jcb.200503066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dutertre S, Cazales M, Quaranta M, Froment C, Trabut V, Dozier C, Mirey G, Bouché JP, Theis-Febvre N, Schmitt E, et al. Phosphorylation of CDC25B by Aurora-A at the centrosome contributes to the G2-M transition. J Cell Sci 2004; 117:2523-31; PMID:15128871; http://dx.doi.org/ 10.1242/jcs.01108 [DOI] [PubMed] [Google Scholar]
- 11. Jackman M, Lindon C, Nigg EA, Pines J. Active cyclin B1-Cdk1 first appears on centrosomes in prophase. Nat Cell Biol 2003; 5:143-8; PMID:12524548; http://dx.doi.org/ 10.1038/ncb918 [DOI] [PubMed] [Google Scholar]
- 12. Boutros R, Lobjois V, Ducommun B. CDC25B involvement in the centrosome duplication cycle and in microtubule nucleation. Cancer Res 2007; 67:11557-64; PMID:18089784; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-2415 [DOI] [PubMed] [Google Scholar]
- 13. Boutros R, Mondesert O, Lorenzo C, Astuti P, McArthur G, Chircop M, Ducommun B, Gabrielli B. CDC25B overexpression stabilises centrin 2 and promotes the formation of excess centriolar foci. PLoS One 2013; 8:e67822; PMID:23840880; http://dx.doi.org/ 10.1371/journal.pone.0067822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nigg EA, Raff JW. Centrioles, centrosomes, and cilia in health and disease. Cell 2009; 139:663-78; PMID:19914163; http://dx.doi.org/ 10.1016/j.cell.2009.10.036 [DOI] [PubMed] [Google Scholar]
- 15. Fukasawa K. Oncogenes and tumour suppressors take on centrosomes. Nat Rev Cancer 2007; 7:911-24; PMID:18004399; http://dx.doi.org/ 10.1038/nrc2249 [DOI] [PubMed] [Google Scholar]
- 16. Ganem NJ, Godinho SA, Pellman D. A mechanism linking extra centrosomes to c hromosomal instability. Nature 2009; 460:278-82; PMID:19506557; http://dx.doi.org/ 10.1038/nature08136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Silkworth WT, Nardi IK, Scholl LM, Cimini D. Multipolar spindle pole coalescence is a major source of kinetochore mis-attachment and chromosome mis-segregation in cancer cells. PLoS One 2009; 4:e6564; PMID:19668340; http://dx.doi.org/ 10.1371/journal.pone.0006564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lacey KR, Jackson PK, Stearns T. Cyclin-dependent kinase control of centrosome duplication. Proc Natl Acad Sci U S A 1999; 96:2817-22; PMID:10077594; http://dx.doi.org/ 10.1073/pnas.96.6.2817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Meraldi P, Lukas J, Fry AM, Bartek J, Nigg EA. Centrosome duplication in mammalian somatic cells requires E2F and Cdk2-cyclin A. Nat Cell Biol 1999; 1:88-93; PMID:10559879; http://dx.doi.org/ 10.1038/10054 [DOI] [PubMed] [Google Scholar]
- 20. Hinchcliffe EH, Sluder G. Two for two: Cdk2 and its role in centrosome doubling. Oncogene 2002; 21:6154-60; PMID:12214244; http://dx.doi.org/ 10.1038/sj.onc.1205826 [DOI] [PubMed] [Google Scholar]
- 21. Barr FA, Sillje HH, Nigg EA. Polo-like kinases and the orchestration of cell division. Nat Rev Mol Cell Biol 2004; 5:429-40; PMID:15173822; http://dx.doi.org/ 10.1038/nrm1401 [DOI] [PubMed] [Google Scholar]
- 22. Bettencourt-Dias M, Rodrigues-Martins A, Carpenter L, Riparbelli M, Lehmann L, Gatt MK, Carmo N, Balloux F, Callaini G, Glover DM. SAK/PLK4 is required for centriole duplication and flagella development. Curr Biol 2005; 15:2199-207; PMID:16326102; http://dx.doi.org/ 10.1016/j.cub.2005.11.042 [DOI] [PubMed] [Google Scholar]
- 23. Habedanck R, Stierhof YD, Wilkinson CJ, Nigg EA. The Polo kinase Plk4 functions in centriole duplication. Nat Cell Biol 2005; 7:1140-6; PMID:16244668; http://dx.doi.org/ 10.1038/ncb1320 [DOI] [PubMed] [Google Scholar]
- 24. Holland AJ, Lan W, Cleveland DW. Centriole duplication: A lesson in self-control. Cell Cycle 2010; 9:2731-6; PMID:20647763; http://dx.doi.org/ 10.4161/cc.9.14.12184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee K, Rhee K. PLK1 phosphorylation of pericentrin initiates centrosome maturation at the onset of mitosis. J Cell Biol 2011; 195:1093-101; PMID:22184200; http://dx.doi.org/ 10.1083/jcb.201106093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sumara I, Gimenez-Abian JF, Gerlich D, Hirota T, Kraft C, de la Torre C, Ellenberg J, Peters JM. Roles of polo-like kinase 1 in the assembly of functional mitotic spindles. Curr Biol 2004; 14:1712-22; PMID:15458642; http://dx.doi.org/ 10.1016/j.cub.2004.09.049 [DOI] [PubMed] [Google Scholar]
- 27. Blagden SP, Glover DM. Polar expeditions–provisioning the centrosome for mitosis. Nat Cell Biol 2003; 5:505-11; PMID:12776127; http://dx.doi.org/ 10.1038/ncb0603-505 [DOI] [PubMed] [Google Scholar]
- 28. Oshimori N, Ohsugi M, Yamamoto T. The Plk1 target Kizuna stabilizes mitotic centrosomes to ensure spindle bipolarity. Nat Cell Biol 2006; 8:1095-101; PMID:16980960; http://dx.doi.org/ 10.1038/ncb1474 [DOI] [PubMed] [Google Scholar]
- 29. Thein KH, Kleylein-Sohn J, Nigg EA, Gruneberg U. Astrin is required for the maintenance of sister chromatid cohesion and centrosome integrity. J Cell Biol 2007; 178:345-54; PMID:17664331; http://dx.doi.org/ 10.1083/jcb.200701163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang S, Liu X, Yin Y, Fukuda MN, Zhou J. Tastin is required for bipolar spindle assembly and centrosome integrity during mitosis. FASEB J 2008; 22:1960-72; PMID:18218922; http://dx.doi.org/ 10.1096/fj.07-081463 [DOI] [PubMed] [Google Scholar]
- 31. Yuan J, Sanhaji M, Kramer A, Reindl W, Hofmann M, Kreis NN, Zimmer B, Berg T, Strebhardt K. Polo-box domain inhibitor poloxin activates the spindle assembly checkpoint and inhibits tumor growth in vivo. Am J Pathol 2011; 179:2091-9; PMID:21839059; http://dx.doi.org/ 10.1016/j.ajpath.2011.06.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thomas Y, Coux O, Baldin V. betaTrCP-dependent degradation of CDC25B phosphatase at the metaphase-anaphase transition is a pre-requisite for correct mitotic exit. Cell Cycle 2010; 9:4338-50; PMID:21051950; http://dx.doi.org/ 10.4161/cc.9.21.13593 [DOI] [PubMed] [Google Scholar]
- 33. Nigg EA. Origins and consequences of centrosome aberrations in human cancers. Int J Cancer 2006; 119:2717-23; PMID:17016823; http://dx.doi.org/ 10.1002/ijc.22245 [DOI] [PubMed] [Google Scholar]
- 34. Abal M, Keryer G, Bornens M. Centrioles resist forces applied on centrosomes during G2/M transition. Biol Cell 2005; 97:425-34; PMID:15898952; http://dx.doi.org/ 10.1042/BC20040112 [DOI] [PubMed] [Google Scholar]
- 35. Hut HM, Lemstra W, Blaauw EH, Van Cappellen GW, Kampinga HH, Sibon OC. Centrosomes split in the presence of impaired DNA integrity during mitosis. Mol Biol Cell 2003; 14:1993-2004; PMID:12802070; http://dx.doi.org/ 10.1091/mbc.E02-08-0510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Oshimori N, Li X, Ohsugi M, Yamamoto T. Cep72 regulates the localization of key centrosomal proteins and proper bipolar spindle formation. EMBO J 2009; 28:2066-76; PMID:19536135; http://dx.doi.org/ 10.1038/emboj.2009.161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Boutros R, Ducommun B. Asymmetric localization of the CDC25B phosphatase to the mother centrosome during interphase. Cell Cycle 2008; 7:401-6; PMID:18235220; http://dx.doi.org/ 10.4161/cc.7.3.5295 [DOI] [PubMed] [Google Scholar]
- 38. Lenart P, Petronczki M, Steegmaier M, Di Fiore B, Lipp JJ, Hoffmann M, Rettig WJ, Kraut N, Peters JM. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr Biol 2007; 17:304-15; PMID:17291761; http://dx.doi.org/ 10.1016/j.cub.2006.12.046 [DOI] [PubMed] [Google Scholar]
- 39. Lazo JS, Nemoto K, Pestell KE, Cooley K, Southwick EC, Mitchell DA, Furey W, Gussio R, Zaharevitz DW, Joo B, et al. Identification of a potent and selective pharmacophore for Cdc25 dual specificity phosphatase inhibitors. Mol Pharmacol 2002; 61:720-8; PMID:11901209; http://dx.doi.org/ 10.1124/mol.61.4.720 [DOI] [PubMed] [Google Scholar]
- 40. Coqueret O, Berube G, Nepveu A. The mammalian Cut homeodomain protein functions as a cell-cycle-dependent transcriptional repressor which downmodulates p21WAF1/CIP1/SDI1 in S phase. EMBO J 1998; 17:4680-94; PMID:9707427; http://dx.doi.org/ 10.1093/emboj/17.16.4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xia K, Lee RS, Narsimhan RP, Mukhopadhyay NK, Neel BG, Roberts TM. Tyrosine phosphorylation of the proto-oncoprotein Raf-1 is regulated by Raf-1 itself and the phosphatase Cdc25A. Mol Cell Biol 1999; 19:4819-24; PMID:10373531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Galaktionov K, Jessus C, Beach D. Raf1 interaction with Cdc25 phosphatase ties mitogenic signal transduction to cell cycle activation. Genes Dev 1995; 9:1046-58; PMID:7744247; http://dx.doi.org/ 10.1101/gad.9.9.1046 [DOI] [PubMed] [Google Scholar]
- 43. Wang Z, Wang M, Lazo JS, Carr BI. Identification of epidermal growth factor receptor as a target of Cdc25A protein phosphatase. J Biol Chem 2002; 277:19470-5; PMID:11912208; http://dx.doi.org/ 10.1074/jbc.M201097200 [DOI] [PubMed] [Google Scholar]
- 44. Wang Z, Zhang B, Wang M, Carr BI. Cdc25A and ERK interaction: EGFR-independent ERK activation by a protein phosphatase Cdc25A inhibitor, compound 5. J Cell Physiol 2005; 204:437-44; PMID:15672448; http://dx.doi.org/ 10.1002/jcp.20297 [DOI] [PubMed] [Google Scholar]
- 45. Ma ZQ, Liu Z, Ngan ES, Tsai SY. Cdc25B functions as a novel coactivator for the steroid receptors. Mol Cell Biol 2001; 21:8056-67; PMID:11689696; http://dx.doi.org/ 10.1128/MCB.21.23.8056-8067.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Loffler H, Syljuasen RG, Bartkova J, Worm J, Lukas J, Bartek J. Distinct modes of deregulation of the proto-oncogenic Cdc25A phosphatase in human breast cancer cell lines. Oncogene 2003; 22:8063-71. [DOI] [PubMed] [Google Scholar]
- 47. Theis-Febvre N, Filhol O, Froment C, Cazales M, Cochet C, Monsarrat B, Ducommun B, Baldin V. Protein kinase CK2 regulates CDC25B phosphatase activity. Oncogene 2003; 22:220-32. [DOI] [PubMed] [Google Scholar]
- 48. Baldin V, Theis-Febvre N, Benne C, Froment C, Cazales M, Burlet-Schiltz O, et al. PKB/Akt phosphorylates the CDC25B phosphatase and regulates its intracellular localisation. Biol Cell 2003; 95:547-54; PMID:14630392; http://dx.doi.org/ 10.1016/j.biolcel.2003.08.001 [DOI] [PubMed] [Google Scholar]
- 49. Eot-Houllier G, Venoux M, Vidal-Eychenie S, Hoang MT, Giorgi D, Rouquier S. Plk1 regulates both ASAP localization and its role in spindle pole integrity. J Biol Chem 2010; 285:29556-68; PMID:20615875; http://dx.doi.org/ 10.1074/jbc.M110.144220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. MacKay C, Declais AC, Lundin C, Agostinho A, Deans AJ, MacArtney TJ, Hofmann K, Gartner A, West SC, Helleday T, et al. Identification of KIAA1018/FAN1, a DNA repair nuclease recruited to DNA damage by monoubiquitinated FANCD2. Cell 2010; 142:65-76; PMID:20603015; http://dx.doi.org/ 10.1016/j.cell.2010.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cans C, Ducommun B, Baldin V. Proteasome-dependent degradation of human CDC25B phosphatase. Mol Biol Rep 1999; 26:53-7; PMID:10363647; http://dx.doi.org/ 10.1023/A:1006912105352 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.