

Abstract
The tumor suppressive function of PTEN is exerted within 2 different cellular compartments. In the cytosol-membrane, it negatively regulates PI3K-AKT pathway through the de-phosphorylation of phosphatidylinositol (3,4,5)-triphosphate (PIP3), therefore blocking one of the major signaling transduction pathways in tumorigenesis. In the nucleus, PTEN controls genomic stability and cellular proliferation through phosphatase independent mechanisms. Importantly, impairments in PTEN cellular compartmentalization, changes in protein levels and post-transductional modifications affect PTEN tumor suppressive functions. Targeting mechanisms that inactivate PTEN promotes apoptosis induction of cancer cells, without affecting normal cells, with appealing therapeutic implications. Recently, we have shown that BCR-ABL promotes PTEN nuclear exclusion by favoring HAUSP mediated PTEN de-ubiquitination in Chronic Myeloid Leukemia. Here, we show that nuclear exclusion of PTEN is associated with PTEN inactivation in the cytoplasm of CML cells. In particular, BCR-ABL promotes Casein Kinase II-mediated PTEN tail phosphorylation with consequent inhibition of the phosphatase activity toward PIP3. Targeting Casein Kinase II promotes PTEN reactivation with apoptosis induction. We therefore propose a novel BCR-ABL/CKII/PTEN pathway as a potential target to achieve synthetic lethality with tyrosine kinase inhibitors.
Keywords: casein kinase II, Chronic Myeloid Leukemia, PTEN, tumor suppressor
Introduction
The tumor suppressor PTEN is one of the most frequently mutated and deleted tumor suppressors in cancer.1,2 Its tumor suppressive function is exerted within 2 different cellular compartments. In the cytosol-membrane, it negatively regulates PI3K-AKT pathway through the de-phosphorylation of phosphatidylinositol (3,4,5)-triphosphate (PIP3), therefore blocking one of the major signaling transduction pathways in tumorigenesis. In the nucleus, PTEN acts as a tumor suppressor by regulating genomic stability and cellular proliferation through phosphatase independent mechanisms.1,3-5 Recently, PTEN has been proposed as the paradigm for non genomic loss of function of tumor suppressors.6,7 In particular, the tumor suppressive functions of PTEN are tightly controlled by PTEN protein level, cellular compartmentalization and post-trasductional modifications, such as phosphorylation, ubiquitination and sumoylation.1 Importantly, the identification of those cancers, where wild-type PTEN is functionally inhibited, could have dramatic consequences from the therapeutic standpoint. The reactivation of the tumor suppressor could indeed have tremendous and selective consequences in cancer cells without affecting normal cells. Chronic Myeloid Leukemia is a myeloproliferative disorder characterized by the translocation t(9;22), coding for the chimeric protein BCR-ABL.8-11 PTEN plays an essential role in the pathogenesis of Chronic Myeloid Leukemia (CML).12,13 Recently we have shown that BCR-ABL promotes PTEN nuclear exclusion causing loss of PTEN tumor suppressive function in the nucleus.13,14 The shuttling of nuclear PTEN into the cytoplasm could imply that PTEN inhibits PI3K-AKT signaling in CML. On the contrary, it is well demonstrated that BCR-ABL transformation requires PI3K-AKT signaling.15-25 Furthermore, it was also shown that PIP3 levels are dramatically increased in CML.26 Therefore, we hypothesized that PTEN could be functionally inactivated in the cytoplasm of CML cells. Here, we demonstrate that BCR-ABL promotes the inactivation of cytoplasmic PTEN, through the activity of the serine-threonine Casein Kinase II, with potential therapeutic implications.
Results
To test whether cytoplasmic PTEN is able to regulate BCR-ABL-induced PI3K-AKT signaling, we first assessed the phosphorylation status of AKT in CML progenitor cells. We recently showed that Lin-CD34+CD38+ CML progenitor cells and Lin+ CML cells expressed PTEN mostly in the cytoplasm.13 Interestingly, in Lin-CD34+CD38+ CML progenitors cells AKT is mostly phosphorylated, suggesting that PI3K-AKT pathway is active even in the presence of cytosolic PTEN (Fig. 1A; Fig. S1A). Similarly, in BCR-ABL-NIH3T3 cells, where PTEN is mostly cytosolic,13 AKT is phosphorylated, when detected by Western immunoblot (Fig. 1B). As already shown by others,26 we also observed that PIP3 levels are substantially increased in CML CD34 positive cells (Fig. 1C). Furthermore, PI3K-AKT pathway was demonstrated to be necessary for BCR-ABL mediated transformation.15 These data suggest that PTEN activity could be impaired in CML. PTEN protein has been shown to be under-expressed in CML patients.13,24,27 Expression of low levels of PTEN could explain the inability of PTEN to promote PIP3 de-phosphorylation. However, the observation that not all CML samples are characterized by markedly reduction of PTEN levels prompted us to investigate other mechanisms of PTEN activity regulation. In particular, PTEN activity is regulated by the Casein Kinase II-mediated serine/threonine phosphorylation of the tail.1 Therefore, we sought to assess PTEN tail phosphorylation in CML bone marrow samples. As shown in Figure 1D, PTEN is highly phosphorylated. Interestingly, we observed high levels of phosphorylation even in those samples where PTEN is markedly under-expressed (Supplementary Fig. 1B). These observations prompted us to investigate whether PTEN phosphatase is functionally inactivated through phosphorylation in CML. We performed a PTEN phosphatase assay in CML cells, as described in material and methods, and upon normalization of the PTEN protein amounts (Fig. S1C). Interestingly, in primary CML cells, PTEN activity is decreased when compared to normal bone marrow mononuclear cells (Fig. 1E). PTEN tail phosphorylation on serine/threonine residues by Casein Kinase II is thought to cause a “tail-closed” conformation switch which prevents PTEN from targeting its substrate at the membrane.28-30 Casein Kinase II is a serine/threonine kinase that is involved in tumorigenesis.31 Notably, Casein Kinase II was described as a druggable substrate of BCR-ABL, although its function in CML is still unclear.32-35 Importantly, it was clearly demonstrated that BCR-ABL/CKII complex plays an essential role in the regulation of imatinib resistance.36 To verify whether PTEN tail phosphorylation is regulated by BCR-ABL, the BCR-ABL inhibitor imatinib and/or the casein kinase inhibitor TBB were utilized in BCR-ABL expressing NIH3T3 cells and PTEN phosphorylation levels were assessed by western immunoblot, upon normalization of PTEN levels. As shown in Figure 2A, the association Imatinib and TBB revert PTEN tail phosphorylation induced by BCR-ABL. Although with less extend, similar conclusions were observed in the 32D-BCR-ABL cell lines, as reported in Figure S1D. These observations suggest that BCR-ABL promotes PTEN phosphorylation through Casein Kinase II, that was reported to occur at specific serine/threonine residues located in the tail (Fig. 2B). To better address the role of Casein Kinase II in the phosphorylation of PTEN, we first confirmed that BCR-ABL physically interacts with CKII, as previously reported (Fig. 2C).32,34,36 Next, to determine whether BCR-ABL mediates PTEN phosphorylation by Casein Kinase II, we performed an in vitro kinase assay with purified ABL protein and with PTEN and CKII immunoprecipitated proteins, as described in Material and Methods. As shown in Figure 2D, the presence of ABL increases PTEN phosphorylation induced by CKII. Treatment with imatinib reverts ABL effects on CKII, suggesting that ABL can increase CKII activity toward PTEN. Next, to assess whether PTEN-tail phoshorylation by BCR-ABL/CKII causes PTEN inactivation, we generated PTEN-tail mutants (S370A, TM: S380A/T382A/T383A, S385A) in Myc-tag-pRK5 and pGFP-C2-PTEN vectors. We expressed these vectors in NIH3T3 cells due to the wide cytoplasm/nuclear ratio that allows to better evaluate cellular compartmentalization. These PTEN mutants were already shown to be unstable, able to reach the membrane in un-stimulated conditions and associated with increased PTEN phosphatase activity.37 Expression of myc-tagged-PTEN tail-mutants in BCR-ABL NIH3T3 cells enabled PTEN to reach the membrane (Fig. 2E). Due to the instability of PTEN-S385A and PTEN-S370A, we decided to focus on the more stable PTEN-TM mutant. To test PTEN activity in the presence of BCR-ABL, BCR-ABL-NIH3T3 cells were transfected with GFP-tagged-PTEN-WT and GFP-tagged-PTEN-TM. Interestingly, when we measured GFP-tagged PTEN activity in BCR-ABL-NIH3T3 cells, we observed that PTEN-WT is less active than in parental cells, while expression of PTEN-TM substantially increases PTEN activity, even in the BCR-ABL context (Fig. 3A; Fig. S2A). In line with these considerations, expression of PTEN-TM was associated with increased apoptosis induction (Fig. 3B). Importantly, PTEN-tail de-phosphorylation is therapeutically achievable by treatment with CKII inhibitors. Treatment of BCR-ABL NIH3T3 cells with CKII inhibitor TBB was indeed associated with apoptosis induction (Fig. 3C; Fig. S2B). The utility of Casein Kinase inhibitors has already been tested in CLL and in other hematological disorders.36,38-43 A major therapeutic challenge in the treatment of CML is the therapy of BCR-ABL-T315I and other BCR-ABL mutations that are responsible of mediating tyrosine kinase inhibitors (TKIs) resistance. To test whether CKII promotes apoptosis in imatinib resistant CML, we treated a T315I-BCR-ABL CML bone marrow sample with 60 μM TBB for 10 hours confirming that TBB promotes apoptosis induction in Imatinib-resistant primary cells (Fig. 4A).
Figure 1.

Cytoplasmic PTEN is functionally inhibited by BCR-ABL. (A) Representative Phosho-AKT immunofluorescence staining in primary CML Lin-CD34+CD38+ and Lin+ cells. PI: propidium iodide; P-AKT: phospho-Ser473-AKT. (B) Western immunoblot on Phospho-Ser473-AKT and Phospho-ERK in BCR-ABL-NIH3T3 cells. (C) Representative phosphatidylinositol (3,4,5)-triphosphate (PIP3) staining immunofluorescence in CD34 normal bone marrow and CML sample; PIP3 immunofluorescence staining intensity quantification with ImageJ software in 25 cells per conditions. (D) Upper panel: western immunoblot of primary total bone marrow CML extracts. Phospho-PTEN: Phospho-S380/T382/T383. Lower panel: quantification of Phospho-PTEN/PTEN in CML cases #1,#2,#3 of the western presented in the upper panel and CML cases #4,#5,#6 shown in the Supplementary Figure 1 B compared to the 2 normal bone marrow cases; *P <0.05. (E) Evaluation of PTEN activity in CML cases #1,#2,#3 of panel (d) and 2 normal bone marrow samples. A.U.: arbitrary unity; *P<0.05.
Figure 2.
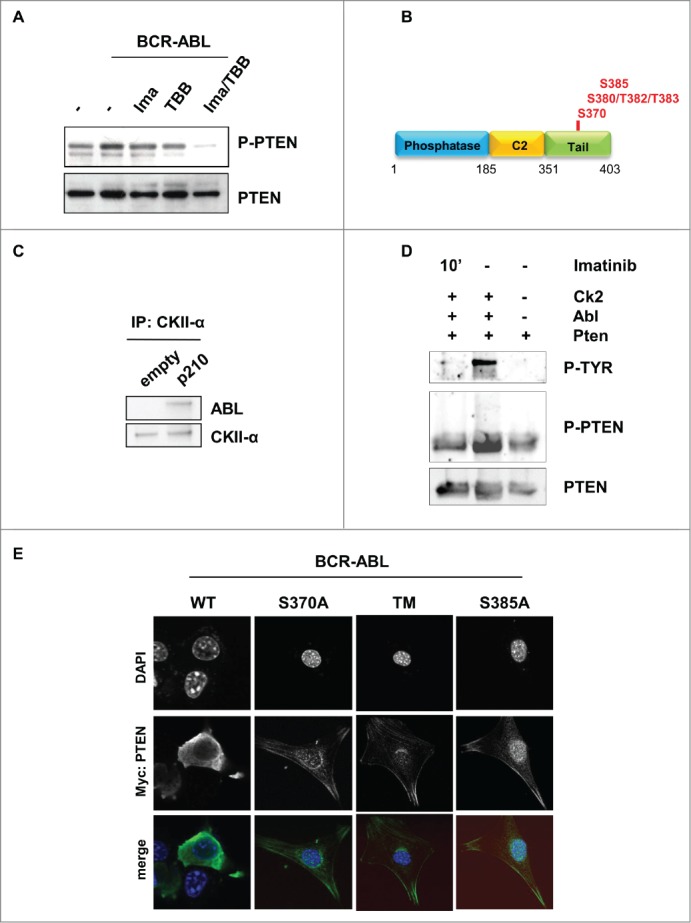
BCR-ABL promotes PTEN inactivation through Casein Kinase II. (A) PTEN-phosphorylation in parental and BCR-ABL-NIH3T3 treated with 1 μM imatinib and 60 μM TBB for 10 hours. To obtain a comparable level of expression of PTEN, the amount of BCR-ABL-NIH3T3 extracts were increased of 20% compared to parental NIH3T3 cells. (B) Schematic representation of PTEN with the indication of PTEN tail phosphorylation sites. (C) Co-immunoprecipitation of CKII-α and p210-BCR-ABL in BCR-ABL-infected NIH3T3 cells. (D) In vitro kinase assay with immunoprecipitated PTEN and CKII-α and purified ABL kinase. One μM Imatinib was added to the in vitro reaction for 10 minutes. Phospho-PTEN: Phospho-S380/T382/T383; P-Tyr: Phospho-Tyrosine: the band corresponds to the BCR-ABL molecular weight. (E) Transient transfection of Myc-PTEN mutants in BCR-ABL-infected NIH3T3 cells. 48 hours after transfection, myc-tag immunofluorescence was performed to verify PTEN compartmentalization. PTEN-TM: PTEN-S380A/T382A/T383A.
Figure 3.
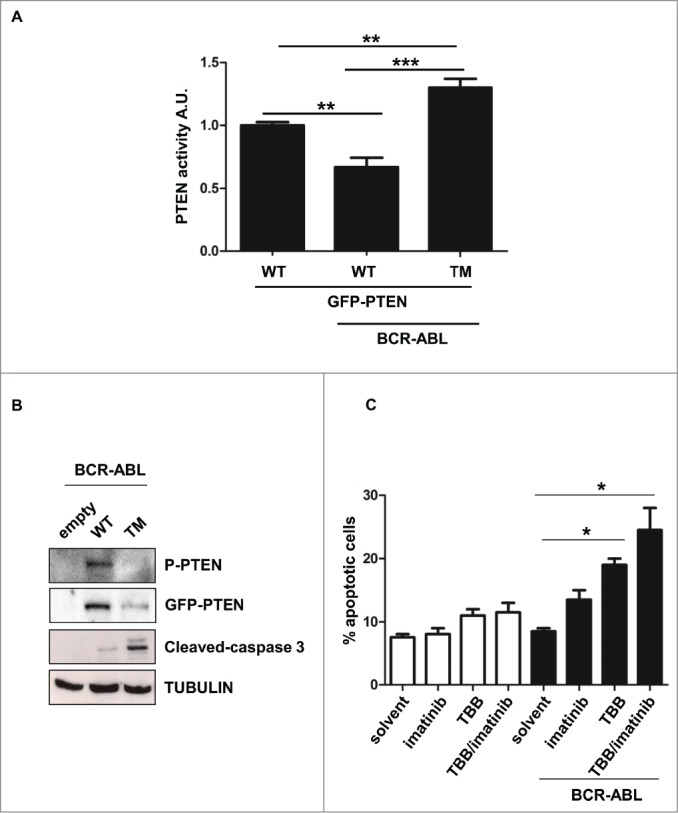
PTEN reactivation promotes apoptosis in BCR-ABL positive cells. (A) Parental and BCR-ABL-NIH3T3 were transfected with the indicated GFP-tagged-PTEN vectors. After 48 hours, PTEN phosphatase assay was performed in immunoprecipitated GFP-PTEN. *P <0.05; **P<0.01. (B) Western immunoblot of BCR-ABL-NIH3T3 cells transfected with the indicated GFP-PTEN mutants. Phospho-PTEN: PTEN-S380/T382/T383. (C) Apoptosis was assessed in NIH3T3 cells treated with 60 μM TBB and 1 μM Imatinib for 10 hours. Data are mean and d.s. of two independent experiments. After short incubation period with inhibitors, in BCR-ABL-NIH3T3 cells, solvent vs. TBB and solvent vs. TBB/imatinib differences are significant with *P<0.05.
Figure 4.
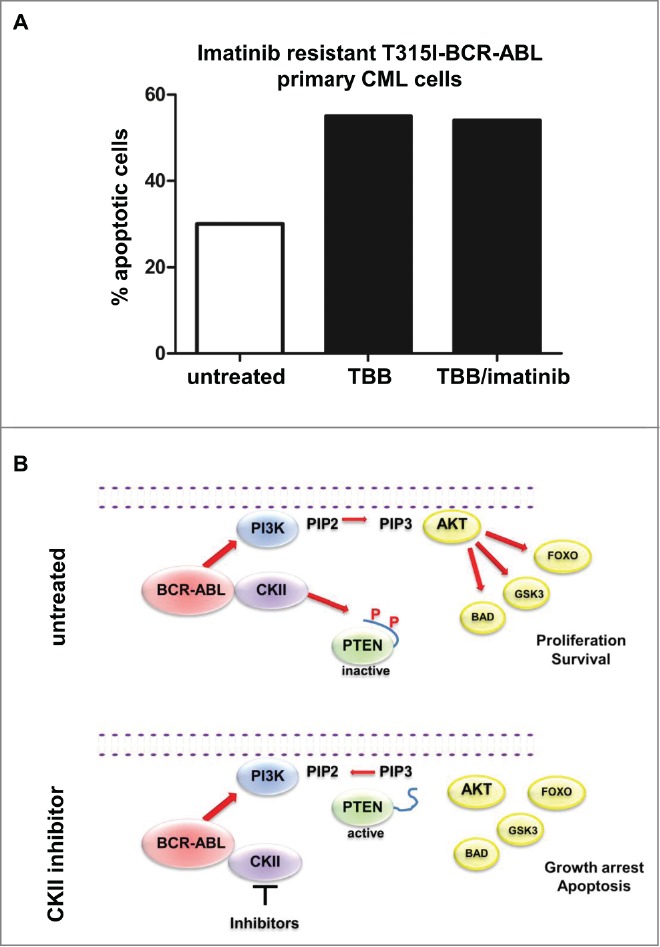
CKII inhibitor promotes apoptosis in T315I-BCR-ABL positive cells. (A) Apoptosis induction of BCR-ABL-T315I bone marrow sample treated with 60 μM TBB and 1 μM Imatinib for 10 hours. (B) Model of the BCR-ABL/CKII/PTEN network in CML.
Discussion
In this work, we have demonstrated that BCR-ABL is able to induce non-genomic loss of function of PTEN by favoring CKII mediated tail phosphorylation, which in turn inhibits PTEN activity (Fig. 4B). Furthermore, we provided evidences that CKII inhibition could promote PTEN reactivation with therapeutic implications especially in those situations of resistance to imatinib, due to BCR-ABL point mutations. The existence of a BCR-ABL/CKII complex has already been solidly demonstr-ated.32-34,36 Moreover, targeting CKII has been described as a powerful therapeutic option in BCR-ABL leukemias.33,36 However, BCR-ABL/CKII mechanisms of activation and targets are still an issue of debate. Previous reports have clearly demonstrated that BCR-ABL promotes CKII activation.32,36 Only one report described that BCR-ABL inhibits CKII.34 The mechanism of CKII activation by BCR-ABL appeared to be highly complex. CKII is indeed referred as a constitutively active kinase, whose regulation depends on protein levels or interactions. However, some tyrosine kinases have also been reported to promote its activation though direct phosphorylation.44 Previous works did confirmed that BCR-ABL induces tyrosine phosphorylation of CKII-α, but this event did not appear to increase its catalytic activity.31,35 Our data are in line with these evidences, although more insights are necessary to investigate how CKII is activated by BCR-ABL. An additional layer of complexity to study BCR-ABL/CKII/PTEN complex in CML is represented by the heterogeneity of CML cells, ranging from the rare leukemia stem cells, to progenitors and differentiate elements. Mechanisms of regulation of the BCR-ABL/CKII/PTEN complex could indeed be differentially affected by protein levels and interactions. Therefore, data regarding BCR-ABL/CKII/PTEN in CML could be affected by the type of samples collected (cell lines versus sorted cells vs. total bone marrow), rendering these studies really challenging. The novelty of our investigation relies on the identification of the target of the BCR-ABL/CKII complex, which is PTEN. We have demonstrated that BCR-ABL/CKII promotes PTEN phosphorylation with impairment of its phosphatase activity. Although PTEN activity could be affected by different mechanisms, in particular protein under-expression/delocalization, it is clear that PTEN inactivation by BCR-ABL/CKII represents a targetable opportunity. The identification of this mechanism could have profound implications from the therapeutic standpoint. The reactivation of PTEN tumor suppressive function in the context of an oncogene addicted cell represents a powerful pro-apoptotic stimulation with potentially few effects on the normal cells.45 Similar observations have been obtained in other hematological cancers, where CKII inhibitors have been associated with dramatic apoptosis induction.36,38–41 From a clinical perspective regarding CML therapy, we suggest that CKII inhibitors could be exploited to target imatinib resistant CML, due to BCR-ABL point mutations, and in particular by BCR-ABL-T315I. In these circumstances, CKII inhibitors promote PTEN reactivation in the presence of a sustained BCR-ABL signaling with a consequent apoptosis induction.
Materials and Methods
Cells and reagents
The following antibodies were used: ABL (#131; Santa Cruz Biotechnology), Phospho-ERK (#9101; Cell Signaling), Phospho-AKT S473 (#3787; Cell Signaling), Tubilin (#2146; Cell Signaling), phospho-PTEN-S385 (#54434; Ana Spec), phospho-PTEN-Ser370 (#11062; Signalway antibody), phospho-PTEN-Ser380/Thr382/383 (#9549; Cell Signaling), PTEN (#9552; Cell Signaling), PtdIns(3,4,5)P3 (#A21328; Molecular Probes), Casein Kinase II-α (#12738; Santa Cruz Biotechnologies), Phospho-Tyrosine (#7020; Santa Cruz Biotechnologies). Secondary antibodies were: goat anti-mouse IgG-HRP (Santa Cruz, # 2005) and goat anti-rabbit IgG-HRP (Santa Cruz, #2004). 32D cells line and Wehi-3B cells were purchased from DSMZ vendor. NIH3T3 cells were from ATCC. Parental 32D cells were growth in the presence of 10% Wehi-3B conditioned medium. TBB was purchased from Sigma-Aldrich. TBB and imatinib were used at a concentration of 60 μM and 1 μM, respectively, for 10 hours, unless specifically indicated.
Plasmid, mutagenesis and transfection
pBABE-BCR-ABL, pRK5-myc-tag-PTEN and pEGFP-C2-PTEN expressing vectors have been described elsewhere.13 PTEN mutants were generated using the Quick-change II-XL (Stratagene, #200523) site directed mutagenesis kit. Transfection was performed using Lipofectamine 2000 (Invitrogen) or X-treme GeneHP (Roche). NIH3T3 infections with pBABE-BCR-ABL vector and empty-pBABE vector were performed as previously reported.13 BCR-ABL-32D cell lines were generated by electroporation of pBABE-BCR-ABL followed by puromycin selection.
Western immunoblot, immunoprecipitation and Immunofluorescence
Western immunoblot and immunofluorescence was performed as previously described.13 Each western immunoblot and immunoprecipitation experiments were repeated at least 2 times. PIP3 immunofluorescence intensity staining was measured with ImageJ software.
PTEN activity assay.
PTEN activity was measured accordingly to manufacturer's protocol (Echelon, #K4700). In particular, equal amounts of PTEN proteins were immunoprecipitated as the source for the enzyme reaction. Standard curve and dose response (variable slope) correlation was analyzed using GraphPad software.
Human CML samples and ethics statement
Primary CML bone marrow samples were collected with informed consent at the time of diagnosis from untreated patients. CD34 positive cells were isolated from bone marrow aspirate accordingly to the Miltenyi Biotec protocol for CD34 purification (Miltenyi Biotec, #130–094–531). This project was reviewed and approved by the San Luigi Hospital Institutional Ethical Committee (Code # 10/2013).
Flow cytometry
Sorting of Lin-CD34+CD38+ cells were performed as previously described.13,46 Apoptosis quantification was performed with PE-annexin V/7-AAD (BD Biosciences).
Kinase assay
PTEN Kinase assay has been performed as previously described.13 In particular, PTEN and CKII were immunoprecipitated from untreated cells and incubate with purified full-length ABL protein (Invitrogene, #P3049) and 10 μM ATP. When indicated, 1 μM imatinib was added to the in vitro reaction for 10 minutes.
Statistical analyses
Two-side Student's t test and one-way ANOVA were calculated using GraphPad Prism software. P values < 0,05 were considered statistically significant. Data represent the average ± SE. *P<0.05; **P<0.01.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thanks all the member of Prof. Saglio's and Pandolfi's laboratories for help and discussions.
Funding
This work has been supported by Giovani Ricercatori – Ricerca Finalizzata 2010 funding from Italian Ministero Salute to A.M. and AIRC to G.S.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol 2012; 13(5):283-96; PMID:22473468 [DOI] [PubMed] [Google Scholar]
- 2. Shi Y, Paluch BE, Wang X, Jiang X. PTEN at a glance. J Cell Sci 2012; 125(Pt 20):4687-92; PMID:23223894; http://dx.doi.org/ 10.1242/jcs.093765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, Yin Y. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell 2007; 128(1):157-70; PMID:17218262; http://dx.doi.org/ 10.1016/j.cell.2006.11.042 [DOI] [PubMed] [Google Scholar]
- 4. Bassi C, Ho J, Srikumar T, Dowling RJO, Gorrini C, Miller SJ, Mak TW, Neel BG, Raught B, Stambolic V. Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Science 2013; 341(6144):395-9; PMID:23888040; http://dx.doi.org/ 10.1126/science.1236188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Song MS, Carracedo A, Salmena L, Song SJ, Egia A, Malumbres M, Pandolfi PP. Nuclear PTEN regulates the APC-CDH1 tumor-suppressive complex in a phosphatase-independent manner. Cell 2011; 144(2):187-99; PMID:21241890; http://dx.doi.org/ 10.1016/j.cell.2010.12.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Leslie NR, Foti M. Non-genomic loss of PTEN function in cancer: not in my genes. Trends Pharmacol Sci 2011; 32(3):131-40. [DOI] [PubMed] [Google Scholar]
- 7. Correia NC, Gírio A, Antunes I, Martins LR, Barata JT. The multiple layers of non-genetic regulation of PTEN tumour suppressor activity. Eur J Cancer Oxf Engl 1990 2014; 50(1):216-25 [DOI] [PubMed] [Google Scholar]
- 8. Saglio G, Morotti A, Mattioli G, Messa E, Giugliano E, Volpe G, Rege-Cambrin G, Cilloni D. Rational approaches to the design of therapeutics targeting molecular markers: the case of chronic myelogenous leukemia. Ann N Y Acad Sci. 2004; 1028: 423-31; PMID:15650267 [DOI] [PubMed] [Google Scholar]
- 9. Melo JV, Barnes DJ. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat Rev Cancer 2007; 7(6):441-53; PMID:17522713; http://dx.doi.org/ 10.1038/nrc2147 [DOI] [PubMed] [Google Scholar]
- 10. Morotti A, Panuzzo C, Fava C, Saglio G. Kinase-inhibitor-insensitive cancer stem cells in chronic myeloid leukemia. Expert Opin Biol Ther 2014; 14(3):287-99; PMID:24387320; http://dx.doi.org/ 10.1517/14712598.2014.867323 [DOI] [PubMed] [Google Scholar]
- 11. Jabbour E, Kantarjian H. Chronic myeloid leukemia: 2014 update on diagnosis, monitoring, and management. Am J Hematol 2014; 89(5):547-56; http://dx.doi.org/ 10.1002/ajh.23691 [DOI] [PubMed] [Google Scholar]
- 12. Peng C, Chen Y, Yang Z, Zhang H, Osterby L, Rosmarin AG, Li S. PTEN is a tumor suppressor in CML stem cells and BCR-ABL-induced leukemias in mice. Blood 2010; 115(3):626-35; PMID:19965668; http://dx.doi.org/ 10.1182/blood-2009-06-228130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Morotti A, Panuzzo C, Crivellaro S, Pergolizzi B, Familiari U, Berger AH, Saglio G, Pandolfi PP. BCR-ABL disrupts PTEN nuclear-cytoplasmic shuttling through phosphorylation-dependent activation of HAUSP. Leukemia 2014; 28(6):1326-33; PMID:24317448; http://dx.doi.org/ 10.1038/leu.2013.370 [DOI] [PubMed] [Google Scholar]
- 14. Morotti A, Panuzzo C, Crivellaro S, Carrà G, Guerrasio A, Saglio G. HAUSP compartmentalization in Chronic Myeloid Leukemia. Eur J Haematol 2014; PMID:25082234 [DOI] [PubMed] [Google Scholar]
- 15. Skorski T, Kanakaraj P, Nieborowska-Skorska M, Ratajczak MZ, Wen SC, Zon G, Gewirtz AM, Perussia B, Calabretta B. Phosphatidylinositol-3 kinase activity is regulated by BCR/ABL and is required for the growth of Philadelphia chromosome-positive cells. Blood 1995; 86(2):726-36; PMID:7606002 [PubMed] [Google Scholar]
- 16. Calabretta B, Skorski T. BCR/ABL regulation of PI-3 kinase activity. Leuk Lymphoma 1996; 23(5-6): 473-6; PMID:9031078; http://dx.doi.org/ 10.3109/10428199609054856 [DOI] [PubMed] [Google Scholar]
- 17. Kharas MG, Janes MR, Scarfone VM, Lilly MB, Knight ZA, Shokat KM, Fruman DA. Ablation of PI3K blocks BCR-ABL leukemogenesis in mice, and a dual PI3K/mTOR inhibitor prevents expansion of human BCR-ABL+ leukemia cells. J Clin Invest 2008; 118(9):3038-50; PMID:18704194; http://dx.doi.org/ 10.1172/JCI33337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kharas MG, Fruman DA. ABL oncogenes and phosphoinositide 3-kinase: mechanism of activation and downstream effectors. Cancer Res. 2005; 65(6):2047-53; http://dx.doi.org/ 10.1158/0008-5472.CAN-04-3888 [DOI] [PubMed] [Google Scholar]
- 19. Keeshan K, Cotter TG, McKenna SL. Bcr-Abl upregulates cytosolic p21WAF-1/CIP-1 by a phosphoinositide-3-kinase (PI3K)-independent pathway. Br J Haematol 2003; 123(1):34-44; PMID:14510940; http://dx.doi.org/ 10.1046/j.1365-2141.2003.04538.x [DOI] [PubMed] [Google Scholar]
- 20. Chu S, Li L, Singh H, Bhatia R. BCR-tyrosine 177 plays an essential role in Ras and Akt activation and in human hematopoietic progenitor transformation in chronic myelogenous leukemia. Cancer Res 2007; 67(14):7045-53; PMID:17638918; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-4312 [DOI] [PubMed] [Google Scholar]
- 21. Wöhrle FU, Halbach S, Aumann K, Schwemmers S, Braun S, Auberger P, Schramek D, Penninger JM, Laßmann S, Werner M, et al. Gab2 signaling in chronic myeloid leukemia cells confers resistance to multiple Bcr-Abl inhibitors. Leukemia 2013; 27(1):118-29; PMID:22858987; http://dx.doi.org/ 10.1038/leu.2012.222 [DOI] [PubMed] [Google Scholar]
- 22. Ding J, Romani J, Zaborski M, MacLeod RAF, Nagel S, Drexler HG, Quentmeier H. Inhibition of PI3K/mTOR overcomes nilotinib resistance in BCR-ABL1 positive leukemia cells through translational down-regulation of MDM2. PloS One 2013; 8(12):e83510; PMID:24349524; http://dx.doi.org/ 10.1371/journal.pone.0083510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kharas MG, Deane JA, Wong S, O’Bosky KR, Rosenberg N, Witte ON, Fruman DA. Phosphoinositide 3-kinase signaling is essential for ABL oncogene-mediated transformation of B-lineage cells. Blood 2004; 103(11):4268-75; PMID:14976048; http://dx.doi.org/ 10.1182/blood-2003-07-2193 [DOI] [PubMed] [Google Scholar]
- 24. Huang F-F, Zhang L, Wu D-S, Yuan X-Y, Chen F-P, Zeng H, Yu YH, Zhao XL. PTEN regulates BCRP/ABCG2 and the side population through the PI3K/Akt pathway in chronic myeloid leukemia. PloS One 2014; 9(3):e88298; PMID:24603487; http://dx.doi.org/ 10.1371/journal.pone.0088298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pellicano F, Scott MT, Helgason GV, Hopcroft LEM, Allan EK, Aspinall-O’Dea M, Copland M, Pierce A, Huntly BJ, Whetton AD, et al. The Antiproliferative Activity of Kinase Inhibitors in Chronic Myeloid Leukemia Cells Is Mediated by FOXO Transcription Factors. Stem Cells Dayt Ohio 2014; 32(9):2324-37; http://dx.doi.org/ 10.1002/stem.1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hamzah HG, Pierce A, Stewart WA, Peter Downes C, Gray A, Irvine A, Spooncer E, Whetton AD. Chronic myeloid leukemia CD34+ cells have elevated levels of phosphatidylinositol 3,4,5 trisphosphate (PtdIns(3,4,5)P3) and lack a PtdIns(3,4,5)P3 response to cytokines and chemotactic factors; effects reversed by imatinib. Leukemia 2005; 19(10):1851-3; PMID:16107888; http://dx.doi.org/ 10.1038/sj.leu.2403919 [DOI] [PubMed] [Google Scholar]
- 27. Panuzzo C, Crivellaro S, Carrà G, Guerrasio A, Saglio G, Morotti A. BCR-ABL promotes PTEN downregulation in chronic myeloid leukemia. PloS One 2014; 9(10):e110682; PMID:25343485; http://dx.doi.org/ 10.1371/journal.pone.0110682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Torres J, Pulido R. The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J Biol Chem 2001; 276(2):993-8; PMID:11035045; http://dx.doi.org/ 10.1074/jbc.M009134200 [DOI] [PubMed] [Google Scholar]
- 29. Miller SJ, Lou DY, Seldin DC, Lane WS, Neel BG. Direct identification of PTEN phosphorylation sites. FEBS Lett 2002; 528(1-3):145-53; PMID:12297295; http://dx.doi.org/ 10.1016/S0014-5793(02)03274-X [DOI] [PubMed] [Google Scholar]
- 30. Vazquez F, Grossman SR, Takahashi Y, Rokas MV, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail acts as an inhibitory switch by preventing its recruitment into a protein complex. J Biol Chem 2001; 276(52):48627-30; PMID:11707428; http://dx.doi.org/ 10.1074/jbc.C100556200 [DOI] [PubMed] [Google Scholar]
- 31. Battistutta R. Protein kinase CK2 in health and disease: Structural bases of protein kinase CK2 inhibition. Cell Mol Life Sci CMLS 2009; 66(11-12):1868-89; http://dx.doi.org/ 10.1007/s00018-009-9155-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mishra S, Reichert A, Cunnick J, Senadheera D, Hemmeryckx B, Heisterkamp N, Groffen J. Protein kinase CKIIalpha interacts with the Bcr moiety of Bcr/Abl and mediates proliferation of Bcr/Abl-expressing cells. Oncogene 2003; 22(51):8255-62; PMID:14614449; http://dx.doi.org/ 10.1038/sj.onc.1207156 [DOI] [PubMed] [Google Scholar]
- 33. Mishra S, Pertz V, Zhang B, Kaur P, Shimada H, Groffen J, Kazimierczuk Z, Pinna LA, Heisterkamp N. Treatment of P190 Bcr/Abl lymphoblastic leukemia cells with inhibitors of the serine/threonine kinase CK2. Leukemia 2007; 21(1):178-80; PMID:17082777; http://dx.doi.org/ 10.1038/sj.leu.2404460 [DOI] [PubMed] [Google Scholar]
- 34. Hériché JK, Chambaz EM. Protein kinase CK2alpha is a target for the Abl and Bcr-Abl tyrosine kinases. Oncogene 1998; 17(1):13-8; http://dx.doi.org/ 10.1038/sj.onc.1201900 [DOI] [PubMed] [Google Scholar]
- 35. Trembley JH, Wang G, Unger G, Slaton J, Ahmed K. Protein kinase CK2 in health and disease: CK2: a key player in cancer biology. Cell Mol Life Sci CMLS 2009; 66(11-12):1858-67; http://dx.doi.org/ 10.1007/s00018-009-9154-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Borgo C, Cesaro L, Salizzato V, Ruzzene M, Massimino ML, Pinna LA, Donella-Deana A. Aberrant signalling by protein kinase CK2 in imatinib-resistant chronic myeloid leukaemia cells: biochemical evidence and therapeutic perspectives. Mol Oncol 2013; 7(6):1103-15; PMID:24012109; http://dx.doi.org/ 10.1016/j.molonc.2013.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Odriozola L, Singh G, Hoang T, Chan AM. Regulation of PTEN activity by its carboxyl-terminal autoinhibitory domain. J Biol Chem 2007; 282(32):23306-15; PMID:17565999; http://dx.doi.org/ 10.1074/jbc.M611240200 [DOI] [PubMed] [Google Scholar]
- 38. Prins RC, Burke RT, Tyner JW, Druker BJ, Loriaux MM, Spurgeon SE. CX-4945, a selective inhibitor of casein kinase-2 (CK2), exhibits anti-tumor activity in hematologic malignancies including enhanced activity in chronic lymphocytic leukemia when combined with fludarabine and inhibitors of the B-cell receptor pathway. Leukemia 2013; 27(10):2094-6; PMID:23900138; http://dx.doi.org/ 10.1038/leu.2013.228 [DOI] [PubMed] [Google Scholar]
- 39. Buontempo F, Orsini E, Martins LR, Antunes I, Lonetti A, Chiarini F, Tabellini G, Evangelisti C, Evangelisti C, Melchionda F, et al. Cytotoxic activity of the casein kinase 2 inhibitor CX-4945 against T-cell acute lymphoblastic leukemia: targeting the unfolded protein response signaling. Leukemia 2014; 28(3):543-53; PMID:24253024; http://dx.doi.org/ 10.1038/leu.2013.349 [DOI] [PubMed] [Google Scholar]
- 40. Martins LR, Perera Y, Lúcio P, Silva MG, Perea SE, Barata JT. Targeting chronic lymphocytic leukemia using CIGB-300, a clinical-stage CK2-specific cell-permeable peptide inhibitor. Oncotarget 2014; 5(1):258-63; PMID:24473900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Martins LR, Lúcio P, Silva MC, Anderes KL, Gameiro P, Silva MG, Barata JT. Targeting CK2 overexpression and hyperactivation as a novel therapeutic tool in chronic lymphocytic leukemia. Blood 2010; 116(15):2724-31; PMID:20660292; http://dx.doi.org/ 10.1182/blood-2010-04-277947 [DOI] [PubMed] [Google Scholar]
- 42. Silva A, Yunes JA, Cardoso BA, Martins LR, Jotta PY, Abecasis M, Nowill AE, Leslie NR, Cardoso AA, Barata JT. PTEN posttranslational inactivation and hyperactivation of the PI3K/Akt pathway sustain primary T cell leukemia viability. J Clin Invest 2008; 118(11):3762-74; PMID:18830414; http://dx.doi.org/ 10.1172/JCI34616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kim JS, Eom JI, Cheong J-W, Choi AJ, Lee JK, Yang WI, Min YH. Protein kinase CK2alpha as an unfavorable prognostic marker and novel therapeutic target in acute myeloid leukemia. Clin Cancer Res Off J Am Assoc Cancer Res 2007; 13(3):1019-28; http://dx.doi.org/ 10.1158/1078-0432.CCR-06-1602 [DOI] [PubMed] [Google Scholar]
- 44. Sommercorn J, Mulligan JA, Lozeman FJ, Krebs EG. Activation of casein kinase II in response to insulin and to epidermal growth factor. Proc Natl Acad Sci U S A 1987; 84(24):8834-8; PMID:3321056; http://dx.doi.org/ 10.1073/pnas.84.24.8834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Epstein RJ. The unpluggable in pursuit of the undruggable: tackling the dark matter of the cancer therapeutics universe. Front Oncol 2013; 3:304.; PMID:24377088; http://dx.doi.org/ 10.3389/fonc.2013.00304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ito K, Bernardi R, Morotti A, Matsuoka S, Saglio G, Ikeda Y, Rosenblatt J, Avigan DE, Teruya-Feldstein J, Pandolfi PP. PML targeting eradicates quiescent leukaemia-initiating cells. Nature 2008; 453(7198):1072-8; PMID:18469801; http://dx.doi.org/ 10.1038/nature07016 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.