

Abstract
ATM is the master regulator of the cellular response to DNA double strand breaks (DSBs). Deficiency of ATM predisposes humans and mice to αβ T lymphoid cancers with clonal translocations between the T cell receptor (TCR) α/δ locus and a 450 kb region of synteny on human chromosome 14 and mouse chromosome 12. While these translocations target and activate the TCL1 oncogene at 14q32 to cause T cell pro-lymphocytic leukemia (T-PLL), the TCRα/δ;14q32 translocations in ATM-deficient T cell acute lymphoblastic leukemia (T-ALL) have not been characterized and their role in cancer pathogenesis remains unknown. The corresponding lesion in Atm-deficient mouse T-ALLs is a chromosome t(12;14) translocation with Tcrδ genes fused to sequences on chromosome 12; although these translocations do not activate Tcl1, they delete the Bcl11b haploinsufficient tumor suppressor gene. To assess whether Tcrδ translocations that inactivate one copy of Bcl11b promote transformation of Atm-deficient cells, we analyzed Atm−/− mice with mono-allelic Bcl11b deletion initiating in thymocytes concomitant with Tcrδ recombination. Inactivation of one Bcl11b copy had no effect on the predisposition of Atm−/− mice to clonal T-ALLs. Yet, none of these T-ALLs had a clonal chromosome t(12;14) translocation that deleted Bcl11b indicating that Tcrδ translocations that inactivate a copy of Bcl11b promote transformation of Atm-deficient thymocytes. Our data demonstrate that antigen receptor locus translocations can cause cancer by deleting a tumor suppressor gene. We discuss the implications of these findings for the etiology and therapy of T-ALLs associated with ATM deficiency and TCRα/δ translocations targeting the 14q32 cytogenetic region.
Keywords: ATM, BCL11B, T-ALL, transformation, translocations, thymocytes
Abbreviations
- ATM
Ataxia Telangiectasia mutated
- DSB
DNA double strand break
- T-ALL
T cell acute lymphoblastic leukemia
- TCR
T cell receptor
- A-T
Ataxia Telangiectasia, Ea, TCRa transcriptional enhancer
- ALL
acute lymphoblastic leukemia
- SKY
spectral karyotyping
Introduction
The cellular response to DSBs coordinates DNA repair with activation of cell cycle checkpoints to maintain genomic stability and suppress malignant transformation1 The Ataxia Telangiectasia mutated (ATM) protein kinase is the master regulator of this response.2 Upon activation by DSBs, ATM induces phosphorylation of numerous proteins to stimulate DSB repair, activate cell cycle checkpoints, and induce apoptosis if DSBs persist. Inherited ATM deficiency in humans causes Ataxia Telangiectasia (A-T), a fatal disease involving progressive neurological degeneration, immunodeficiency, elevated chromosomal instability, and increased predisposition to lymphoid malignancies.3 The average life expectancy of people with A-T is ∼25 years, with chronic lung infection and lymphoid cancers the major causes of death.3 Although childhood acute lymphoblastic leukemia (ALL) is the most common malignancy that occurs in humans with A-T, their cancer genomes have not been characterized beyond the identification of clonal translocations that target a 450 kb region of 14q32 and involve TCRα/δ loci on chromosome 14 or TCRβ loci on chromosome 7.3 In contrast, while adult T-PLLs arise less often in A-T humans, these cancers have been more extensively characterized and found to harbor clonal translocations in which TCRα genes are fused within 160 kb of the TCL1 oncogene at 14q32.1 or the MTCP1 oncogene on the X chromosome.3-7 TCRα;TCL1 and TCRα;MTCP1 translocations are also recurrent clonal lesions in T-PLLs with acquired inactivation of ATM8,9 TCRα;TCL1 and TCRα;MTCP1 translocations likely drive transformation by placing TCL1 or MTCP1 under control of the TCRα transcriptional enhancer (Eα). Demonstrations that enforced expression of TCL1 or MTCP1 causes T cell lymphoma in mice support this notion and provide mouse models of T-PLL.10,11 The absence of similar characterization and models for the recurrent 14q23 translocations in T-ALLs of A-T children provides barriers for elucidating the etiology of ATM-deficient T-ALLs for the development of therapeutics that could cure these fatal lymphoid malignancies.
Germline Atm-deficient (Atm−/−) mice are useful model to assess the role of 14q23 translocations in the pathogenesis of T-ALLs that develop in A-T patients. Like A-T children, Atm−/− mice exhibit immunodeficiency, elevated chromosomal instability, and increased predisposition to T-ALLs arising in the thymus.12-15 These phenotypes result from loss of ATM functions in promoting DSB repair, activating cell cycle checkpoints, and inducing apoptosis during TCR recombination and proliferation of developing αβ T cells.16-20 Atm−/− mice invariably succumb by six months of age to T-ALLs with clonal translocations that typically involve the Tcrα/δ locus on chromosome 14, the Tcrβ locus on chromosome 6, and/or the immunoglobulin heavy chain (Igh) locus on chromosome 12 that recombines D and J segments in thymocytes.19-23 The most frequent clonal translocation in Atm−/− T-ALLs is a t(12;14) chromosome translocation that occurs in ∼60% of these cancers.19-23 The initial cytogenetic characterization of Atm−/− T-ALLs suggested that these clonal t(12;14) translocations occur between Tcrα/δ loci and a region of mouse chromosome 12 that harbors Tcl1 and shares synteny with human chromosome 14q32.22 This led to the hypothesis that these lesions fuse Tcrα genes to chromosome 12 sequences near Tcl1 and drive transormation through the ability of Eα to activate expression of Tcl1 or another oncogene in this region of chromosome 12.22,24 However, our molecular analysis of Atm−/− T-ALLs revealed that these recurrent t(12;14) translocations fuse Tcrδ genes to chromosome 12 sequences, deleting 5-30 Mb of the telomeric end of chromosome 12.21 Notably, these deletions often included Tcl1, and deletions that did not include Tcl1 did not increase Tcl1 expression.21 We also showed that these clonal Tcrδ translocations arise independent of Eα.21 Thus, our data counter the notion that the clonal recurrent t(12;14) translocations of Atm−/− T-ALLs promote transformation by targeting and activating Tcl1 or another oncogene on chromosome 12.
The recurrent clonal TCRα/δ translocations of T-ALLs that arise in A-T humans and Atm−/− mice are selected from many other translocations formed in ATM-deficient thymocytes, indicating that these TCRα/δ translocations effect genetic changes that promote malignant transformation of immature αβ T cells.3 In this context, t(12;14) translocations are not the most frequent translocations observed in non-malignant αβ T cells of Atm−/− mice,24 despite being the most prevalent clonal translocation in Atm−/− T-ALLs. The recurrent t(12;14) translocations of Atm−/− T-ALLs nearly always delete one copy of Bcl11b, resulting in reduced Bcl11b expression.21 In mice, both copies of Bcl11b are required to inhibit development of T-ALL following γ-radiation,25 demonstrating that Bcl11b is a haploinsufficient tumor suppressor gene. Consistent with this notion, TCRδ translocations or mutations that inactivate a single copy of BCL11B are recurrent clonal lesions in human T-ALLs.26-28 Therefore, we have speculated that TCRδ translocations that delete BCL11B promote development of ATM-deficient T-ALL.21,29 To test this hypothesis, we sought to determine the effect of pervasive mono-allelic Bcl11b inactivation on the spontaneous predisposition of Atm−/− mice to T-ALLs with clonal t(12;14) translocations that delete Bcl11b.
Results
Both αβ and γδ T lymphocytes develop in the thymus from CD4−CD8− (double negative, DN) thymocytes.30 Assembly and expression of TCRγ and TCRδ genes in DN thymoctyes signals differentiation into γδ T cells.30 In contrast, assembly and expression of TCRβ genes in DN cells signals differentiation into CD4+CD8+ (double positive, DP) thymocytes, in which assembly and expression of functional TCRα genes signals differentiation into CD4+ or CD8+ (single positive, SP) thymocytes that become mature CD4+ or CD8+ αβ T cells.30 The Lckcre transgenic mouse provides an experimental approach to pervasively delete and inactivate “floxed” genes in DN thymocytes concomitant with Tcrδ and Tcrβ recombination.31,32 We have shown that Lckcre expression in Atm−/− mice does not alter their predisposition to clonal T-ALLs or the frequency of clonal t(12;14) translocations in these Atm-deficient T-ALLs.23 Consequently, to assess whether Tcrδ translocations that inactivate one allelic copy of Bcl11b promote transformation of Atm-deficient thymocytes, we established and analyzed Lckcre+/−Atm−/−Bcl11bflox/WT (LAb), Lckcre+/−Atm−/− (LA), and Lckcre-Bcl11bflox/WT (Lb) mice. We detected Lckcre-mediated deletion of Bcl11bflox alleles in total thymocytes and splenocytes of LAb and Lb mice, with near complete deletion in thymocytes (data not shown), confirming pervasive mono-allelic inactivation of Bcl11b in developing T cells. Thus, we created and aged parallel cohorts of 26 LAb, 8 LA, and 16 Lb mice to evaluate their predisposition to T-ALL. We analyzed only eight cohort LA mice since we had previously characterized a larger cohort of LA mice.23 We show here that our current cohort LA mice survived cancer-free between 82-138 days with a median age of cancer free-survival of 97 days (Fig. 1A), similar to the published median ages of cancer-free survival of LA and Atm−/− mice.19-23 Our cohort LAb mice survived cancer-free between 74-294 days with a median age of cancer-free survival of 117 days (Fig. 1A), which was not significantly different than the median age of cancer-free survival of cohort LA mice. All cohort LA and LAb mice were euthanized due to thymic cancers that caused respiratory stress, except for one LAb mouse that developed large masses of cancer cells in the spleen and lymph nodes (Table 1; data not shown). All cohort Lb mice survived cancer-free during the one-year study, except for one that succumbed to a thymic malignancy detected during necropsy (Fig. 1A). Our data indicate that pervasive mono-allelic inactivation of Bcl11b starting in DN thymocytes does not accelerate the mortality of Atm−/− mice from thymic malignancies.
Figure 1.

T-ALL Predisposition of Atm−/− Mice with Pervasive Mono-Allelic Deletion of Bcl11b initiating in DN Thymocytes. (A) Kaplan-Meier curve depicting the cancer-free survival of parallel cohorts of 26 LAb, 8 LA, and 16 Lb mice. All cohort mice succumbed to thymic T-ALLs except for LAb mouse #211 that succumbed to a T-ALL in peripheral lymphoid tissues (indicated by asterisk). These mice were of a mixed C57BL6 and 129SvEv background.(B-C) Flow cytometry analysis of LAb T-ALLs no. 321 (B) and no. 277 (C) showing surface expression of TCRβ or CD4 and CD8. This analysis was conducted as described (33). Gates were drawn using normal thymocytes or splenocytes. The percentages of cells in each gate are indicated. (D) Schematic of the Tcrβ locus (top) and Southern blot analyses of Tcrβ rearrangements (bottom). Top, shown are relative locations of the indicated Tcrβ segments, HindIII restriction sites (H3), and 3′Jβ1 and 3′Jβ2 probes. Bottom, Southern blots of HindIII-digested DNA from the indicated LAb thymic cancers (and LAb splenic cancer #211) or from the kidney of a WT mouse using the 3′Jβ1 or 3′Jβ2 probe as previously described.33 Germline (GL) bands for each probe are indicated. The membrane was hybridized with the 3′Jβ1 probe and then stripped and hybridized with the 3′Jβ2 probe, revealing which LAb cancers lack 3′Jβ1-hybridizing band(s) due to Vβ-to-Dβ2-Jβ2 rearrangements on both alleles. The images were cropped from a larger blot. Mouse no. 634 was removed from the cohort for incorrect genotyping.
Table 1.
Analysis of LAb tumor cohort
| Mouse | Age at Death | Gross phenotype | Surface expression |
|---|---|---|---|
| 8 | 94 | thymic lymphoma | n.d. |
| 95 | 138 | thymic lymphoma | n.d. |
| 106 | 92 | thymic lymphoma | TCRβ− DP/CD8+ |
| 211 | 98 | disseminated disease | TCRβ− DP |
| 213 | 121 | thymic lymphoma | TCRβ− CD8+ |
| 215 | 77 | thymic lymphoma | TCRβ− DP/CD8+ |
| 232 | 137 | thymic lymphoma | TCRβ− DP |
| 242 | 114 | thymic lymphoma | TCRβ− CD8+ |
| 277 | 115 | thymic lymphoma | TCRβ− CD8+ |
| 282 | 119 | thymic lymphoma | TCRβint DP |
| 292 | 101 | thymic lymphoma | TCRβint DP/CD8+ |
| 321 | 97 | thymic lymphoma | TCRβ− DP |
| 445 | 172 | thymic lymphoma | TCRβ− DP/CD8+ |
| 447 | 130 | thymic lymphoma | TCRβ− CD8+ |
| 538 | 85 | thymic lymphoma | n.d. |
| 557 | 74 | thymic lymphoma | TCRβ− DP |
| 608 | 194 | thymic lymphoma | TCRβ− CD8+ |
| 611 | 294 | thymic lymphoma | TCRβ− DP/CD8+ |
| 612 | 180 | thymic lymphoma | TCRβint DP/CD8+ |
| 646 | 97 | thymic lymphoma | TCRβ− DP/CD8+ |
| 673 | 90 | thymic lymphoma | TCRβ− CD4/8 var |
| 715 | 83 | thymic lymphoma | TCRβ− DP |
| 751 | 276 | thymic lymphoma | n.d. |
| 752 | 152 | thymic lymphoma | TCRβ− DP/CD8+ |
| 771 | 121 | thymic lymphoma | n.d. |
| 938 | 128 | thymic lymphoma | n.d. |
Similar to A-T children,3 Atm−/− and LA mice succumb to mainly clonal T-ALLs that arise from a single thymocyte and do not express surface TCRβ or TCRα but do express CD4 and CD8 or just CD8.14,19-23 To determine the effect of Lckcre-mediated mono-allelic deletion of Bcl11b on the predisposition of Atm−/− mice to clonal T-ALLs from thymocytes of later stages of αβ T cell development, we analyzed TCRβ, CD4, and CD8 expression and TCRβ rearrangements in LAb T-ALLs. First, we used flow cytometry to analyze expression of TCRβ, CD4, and CD8 proteins on cells isolated from the thymuses, spleens, lymph nodes, and bone marrow of euthanized cohort LAb mice. All 19 LAb thymic cancers assayed were TCRβ− and either CD4+CD8+ or CD8+ (Fig. 1B,C; Table 1). Many mice that succumbed to thymic cancers also harbored cells in their spleens, lymph nodes, and bone marrow with identical TCRβ, CD4, and CD8 expression as their malignant thymocytes (Fig. 1C; data not shown), suggesting dissemination of a single T-ALL. The one LAb peripheral lymphoid malignancy was TCRβ− and CD4+CD8+ (Table 1). Next, we conducted Southern blot analysis to identify Tcrβ rearrangements in representative LAb malignancies. Tcrβ rearrangements cause deletion or changes in the size of 3′Jβ1-hybridizing bands and changes in the sizes of 3′Jβ2-hybridizing bands.33 For normal DN thymocytes to survive, proliferate, and differentiate into DP thymocytes, Tcrβ recombination must occur on at least one allele.34 We found that all but three LAb cancers analyzed contained one or two rearranged Tcrβ alleles and therefore arose from a single thymocyte (Fig. 1D). Of the remaining three: one (no. 215) had four Tcrβ rearrangements indicative of two distinct cancers or a single cancer that continued Tcrβ recombination; one (no. 447) had only germline Tcrβ allele(s) consistent with malignant transformation prior to Tcrβ recombination or a cancer with aberrant Tcrβ rearrangement on one allele; and one (no. 292) lacked detectable Tcrβ alleles suggesting aberrant Tcrβ rearrangements on both alleles (Fig. 1D). These flow cytometry and Southern blot analyses of LAb T-ALLs indicate that pervasive mono-allelic inactivation of Bcl11b in DN cells does not effect the predisposition of Atm−/− mice to clonal T-ALLs arising from thymocytes of later stages of αβ T cell development.
To determine the effect of Lckcre-mediated mono-allelic deletion of Bcl11b on the frequency of clonal t(12;14) translocations that delete Bcl11b in Atm−/− T-ALLs, we conducted Spectral Karyotyping (SKY) and PCR on representative LAb T-ALLs. SKY is a molecular cytogenetic method of analyzing metaphase spreads that identifies chromosome translocations.35 We and others have previously shown that the clonal t(12;14) translocations that delete the telomeric end of chromosome 12 occur in ∼60% of T-ALLs that arise in Atm−/− and LA mice.21-23 In contrast, we show here that only one of the 10 LAb T-ALLs analyzed by SKY (no. 211) harbored a clonal t(12;14) translocation (Fig. 2A, Table 2). Yet, this T-ALL also contained a clonal t(14;12) translocation and lacked a normal chromosome 12 (Fig. 2A, Table 2), neither of which is observed in Atm−/− or LA T-ALLs with the clonal t(12;14) translocation that deletes the telomeric end of chromosome 12 through Bcl11b.21-23 To assess whether LAb T-ALL no. 211 had a mono-allelic deletion of the Bcl11b locus, we conducted PCR with primers that amplify Bcl11b and distinguish among wild-type (WT), “floxed” (flox), and Lckcre-deleted (Δ) Bcl11b alleles (Fig. 2B). We found equal intensities of Bcl11bWT and Bcl11bΔ products (Fig. 2B), indicating that neither the clonal t(12;14) translocation nor the clonal t(14;12) translocation deleted either copy of Bcl11b. Four other LAb T-ALLs (nos. 8, 277, 292, and 751) also had clonal translocations involving chromosome 12 (Table 2), but each retained a normal chromosome 12 and their Bcl11bWT and Bcl11bΔ loci (Fig. 2B; data not shown). The other five LAb T-ALLs analyzed by SKY and PCR each harbored two normal copies of chromosome 12 (Table 2) and retained their Bcl11bWT and Bcl11bΔ loci (Fig. 2B). In addition, none of the other 12 LAb T-ALLs that we analyzed only by PCR had deletions of either their Bcl11bWT or Bcl11bΔ loci (Fig. 2B). These data indicate that pervasive mono-allelic inactivation of Bcl11b initiating in Atm−/− DN cells concomitant with Tcrδ rearrangements precludes development of T-ALLs with t(12;14) translocations that delete Bcl11b or with independent deletion of Bcl11b.
Figure 2.
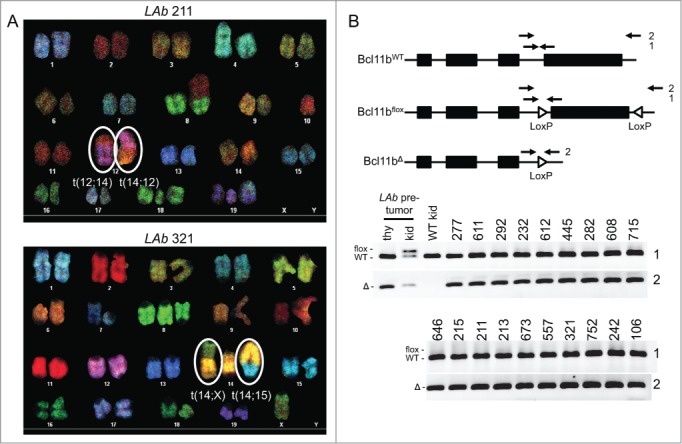
LAb T-ALLs Lack Clonal Chromosome t(12;14) Translocations that Delete Bc111b. (A) SKY images of metaphase spreads prepared from LAb T-ALLs #211 or #321. The clonal chromosome t(12;14) and t(14;12) translocations of LAb T-ALL #211 and the clonal chromosome t(14;X) and t(14;15) translocations of LAb T-ALL #321 are circled. (B) Schematics (top) and PCR analyses (bottom) of Bcl11bWT, Bcl11bflox, and Bcl11bΔ alleles. Top, shown for each allele are locations of the Bcl11b exons, loxP sites, and PCR primers. The no. 1 primer set amplifies distinct bands from Bcl11bWT and Bcl11bflox alleles, while the no. 2 primer set amplifies a band from only Bcl11bΔ alleles. Bottom, Images of PCR no. 1 or no. 2 products from genomic DNA of the indicated LAb T-ALLs, LAb thymocytes, LAb kidney, or WT kidney. The images were cropped from a larger blot.
Table 2.
LAb SKY summary
| Tumor | Clonal Translocations | Non-clonal Translocations |
|---|---|---|
| 8 | t(12;1) | none |
| 211 | t(12;14) t(14;12) t(4;4) | t(15;9) t(4;15) t(1;2) t(14;16) t(17;19) t(2;8) t(8;11) t(4;16) |
| 213 | none | t(12;19) t(2;7) t(4;2) t(14;1) t(14;2) t(14;12) t(19;Y) |
| 215 | none | t(7;19) t(12;1) t(6;13) t(12;14) t(14;6) t(2;2) t(11;14) t(10;12) t(19;Y) t(12;19;1) t(6;1) t(12;7) |
| 277 | t(12;16) t(14;16) | t(2;6) t(6;2) t(12;12) |
| 292 | t(12;1) t(11;3) | none |
| 321 | t(14;15) t(14;X) | none |
| 608 | none | t(17;19) t(12;16) t(12;Y) t(15;19;16) t(12;1) t(19;5) t(14;1) |
| 611 | t(14;14) | none |
| 751 | t(6;19) t(19;6) t(12;Y) | none |
Discussion
The selection of clonal t(12;14) translocations that delete the telomeric end of chromosome 12 in Atm−/− T-ALLs indicates that thee recurrent lesions effect genetic changes that promote malignant transformation of Atm-deficient thymocytes. Our finding that pervasive mono-allelic deletion of Bcl11b initiating in Atm−/− DN thymocytes concomitant with Tcrδ rearrangements precludes the development of T-ALLs with t(12;14) translocations that delete Bcl11b demonstrates that inactivation of a single copy of Bcl11b is a major genetic change that drives malignant transformation of Atm−/− thymocytes. However, the inability of such pervasive mono-allelic Bcl11b deletion to cause more rapid onset of T-ALL and/or promote the development of poly-clonal T-ALLs indicates that formation of t(12;14) translocations that delete Bcl11b is not a limiting factor in pathogenesis of Atm−/− T-ALLs. Consistent with this notion, t(12;14) translocations and analogous t(14;14) translocations/inversions arise in ∼1% of ATM-deficient αβ T cells in mice and humans, respectively, however Atm−/− mice and A-T children succumb to T-ALLs that arise from a single immature αβ T cell at ages in their lives when billions of mature αβ T cells have already developed. The Atm−/− T-ALLs with clonal t(12;14) translocations that delete Bcl11b also harbor additional clonal oncogenic lesions, such as Pten deletion or Notch1 activation, that differ among these malignancies.21 Since chromosome t(12;14) translocations in Atm−/− αβ T cells arise concomitant with Tcrβ-mediated proliferation and differentiation of DN thymocytes,36 it is likely that the acquisition and selection of additional oncogenic lesions during DN-to-DP thymocyte expansion is necessary and rate- limiting for transformation of Atm−/− thymocytes lacking one copy of Bcl11b. The t(12;14) translocations of Atm−/− αβ T cells arise from aberrant repair between DSBs induced by the RAG proteins at Tcrδ loci on chromosome 14 and DSBs induced by other factors on the telomeric end of chromosome 12. RAG DSBs induced at Igh loci near the chromosome 12 telomere contribute to generation of t(12;14) translocations.24 However, considering that Atm−/− mice lacking Rag2 and expressing a TCRβ transgene that drives thymocyte proliferation still succumb to T-ALLs with clonal translocation/deletion of the region of chromosome 12 where Bcl11b resides,19 DSBs arising from DNA replication errors also likely contribute to formation of these oncogenic translocations.
Our data reveal that antigen receptor locus translocations can drive malignant transformation of lymphocytes through deletion and inactivation of a tumor suppressor gene. It has long been known that clonal antigen receptor locus translocations found in lymphoid malignancies promote transformation by unleashing the activities of oncogenes. Therefore, therapies for human leukemias and lymphomas are being developed that target and inactivate these oncogenes or their downstream signaling pathways or targets. In addition to their t(12;14) translocations that delete Bcl11b, Atm−/− T-ALLs harbor di-centric chromosome 14 derivatives with amplification of sequences centromeric of Tcrα/δ loci.21 Although we also observed this amplicon in the one human T-ALL analyzed,21 the amplified sequences lack any known oncogenes. In addition, LAb T-ALLs contain neither di-centric chromosome 14 derivatives nor amplification of sequences centromeric of Tcrα/δ loci (data not shown). Accordingly, our data suggest that the clonal TCRα/δ;14q32 translocations in T-ALLs arising in A-T children most likely promote transformation of ATM-deficient thymocytes through deletion and inactivation of one allelic copy of BCL11B. Although strong evidence indicates that BCL11B is a haploinsufficient suppressor of T-ALL, the mechanisms by which bi-allelic BCL11B expression inhibits transformation and by which mono-allelic expression of BCL11B leads to T-ALL are unknown. Expression of Bcl11b in thymocytes silences stem/progenitor cell gene expression programs to prevent TCR-independent self-renewal and cellular proliferation before initiation of TCR recombination and thereby promotes αβ T cell development.37-39 In human and mouse T lineage cells developed beyond the DN thymocyte stage, decreased BCL11B expression appears to compromise the cellular response to DNA replication stress leading to increased apoptosis of cells from DNA damage during S phase.40 Since ATM stimulates DNA repair and induces apoptosis if DNA damage is too severe, mono-allelic BCL11B expression in ATM-deficient thymocytes could lead to increased frequency of additional oncogenic lesions arising from the aberrant repair of DSBs induced during DN-to-DP thymocyte proliferation. LAb mice provide a useful pre-clinical model to elucidate how inherited ATM deficiency and acquired BCL11B haploinsufficiency cooperate to cause T-ALL, and to design and test efficacy of T-ALL therapies that target intrinsic properties of these cancer cells.
Materials and Methods
Mice
Lck-cre31 Atm−/−,41 and Bcl11bflox/flox 42 mice of a mixed 129SvEv and C57BL/6 were bred to create experimental animals. Mice of both sexes and normal weight were studies. Expect for cohort mice, all mice were analyzed between 4-6 weeks of age. Cohort mice were monitored and euthanized on signs of distress. These studies were conducted under the approval and monitoring of the Children's Hospital of Philadelphia IACUC.
Flow cytometry
Cells were stained in PBS with 3% FBS using APC-anti-TCRβ, FITC-anti-CD8, PE-anti-CD4, PE-Cy7-anti-B220, FITC-anti-CD43, and APC-anti-IgM antibodies (BD Pharmingen). Data were collected using a FACSCalibur with CellQuest software (BD Biosciences) and analyzed by FlowJo software (Tree Star). Statistics were performed in Microsoft Excel or Graphpad Prism 5 using a two-tailed unpaired Student's t-test.
Kaplan-meier analysis
Curves were generated in Prism 5 and compared using the log-rank (Mantel-Cox) test.
Southern blotting
DNA (20 μg) was digested with 100 units of indicated enzymes (New England Biolabs), separated on 0.8% TAE gels, transferred onto Zeta-probe (BioRad), and hybridized with32 P-labeled 3′Jβ1, 3′Jβ2, 5′Jα, 5′Tp53 or 3′Tp53 probes as described.33
Spectral karyotyping
Metaphases were prepared as described.23 Spectral karyotyping was performed per instructions (Applied Spectral Imaging, ASI). Slides were examined with a BX61 microscope (600× magnification) from Olympus controlled by a LAMBDA 10-B Smart Shutter (Sutter Instrument). Images were captured using a LAMBDA LS light source (Sutter) and a COOL-1300QS camera (ASI), and analyzed by Case Data Manager Version 5.5 (ASI).
PCR. Reactions were performed as describe.42 The no. 1 PCR primer set used the following two oliogs: 5′-ACTGCACACGTGACTCCAAG-3′ and 5′-AAGCCATGTGTGTTCTGTGC-3′ primers. The no. 2 PCR primer set used the following two oligos: 5′-CGTGTTCCCTTGCCGTCGGGGGAGG-3′ and 5′-GCTTCCCTCTACGTCACTTGCGAGT-3′.
Funding Statement
This research was supported by training grant 5T32CA009615 of the Cancer Center Research Training Program at the University of Pennsylvania (L.A.E.); the Department of Pathology and Laboratory Medicine and the Center for Childhood Cancer Research of the Children's Hospital of Philadelphia Research Institute, the Abramson Family Cancer Research Institute of the Perelman School of Medicine of the University of Pennsylvania, a Cookies for Cancer foundation grant, a Leukemia and Lymphoma Society Scholar Award, and the National Institutes of Health R01 Grants CA125195 and CA136470 (C.H.B).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1. Bassing CH, Alt FW. The cellular response to general and programmed DNA double strand breaks. DNA repair 2004; 3:781-96; PMID:15279764; http://dx.doi.org/ 10.1016/j.dnarep.2004.06.001 [DOI] [PubMed] [Google Scholar]
- 2. Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Bio 2013; 14:197-210; http://dx.doi.org/ 10.1038/nrm3546 [DOI] [PubMed] [Google Scholar]
- 3. Taylor AM, Metcalfe JA, Thick J, Mak YF. Leukemia and lymphoma in ataxia telangiectasia. Blood 1996; 87:423-38; http://dx.doi.org/ [PubMed] [Google Scholar]
- 4. Russo G, Isobe M, Gatti R, Finan J, Batuman O, Huebner K, Nowell PC, Croce CM. Molecular analysis of a t(14;14) translocation in leukemic T-cells of an ataxia telangiectasia patient. Proc Natl Acad Sci U S A 1989; 86:602-6; PMID:2783489; http://dx.doi.org/ 10.1073/pnas.86.2.602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pekarsky Y, Hallas C, Isobe M, Russo G, Croce CM. Abnormalities at 14q32.1 in T cell malignancies involve two oncogenes. Proc Natl Acad Sci U S A 1999; 96:2949-51; http://dx.doi.org/ 10.1073/pnas.96.6.2949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Baer R, Heppell A, Taylor AM, Rabbitts PH, Boullier B, Rabbitts TH. The breakpoint of an inversion of chromosome 14 in a T-cell leukemia: sequences downstream of the immunoglobulin heavy chain locus are implicated in tumorigenesis. Proc Natl Acad Sci U S A 1987; 84:9069-73; http://dx.doi.org/ 10.1073/pnas.84.24.9069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Davey MP, Bertness V, Nakahara K, Johnson JP, McBride OW, Waldmann TA, Kirsch IR. Juxtaposition of the T-cell receptor alpha-chain locus (14q11) and a region (14q32) of potential importance in leukemogenesis by a 14;14 translocation in a patient with T-cell chronic lymphocytic leukemia and ataxia-telangiectasia. Proc Natl Acad Sci U S A 1988; 85:9287-91; PMID: 3194425; http://dx.doi.org/ 10.1073/pnas.85.23.9287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dearden CE. T-cell prolymphocytic leukemia. Medical Oncology 2006; 23:17-22; http://dx.doi.org/ 10.1385/MO:23:1:17 [DOI] [PubMed] [Google Scholar]
- 9. Stoppa-Lyonnet D, Soulier J, Lauge A, Dastot H, Garand R, Sigaux F, Stern MH. Inactivation of the ATM gene in T-cell prolymphocytic leukemias. Blood 1998; 91:3920-6; PMID:9573030 [PubMed] [Google Scholar]
- 10. Virgilio L, Lazzeri C, Bichi R, Nibu K, Narducci MG, Russo G, Rothstein JL, Croce CM. Deregulated expression of TCL1 causes T cell leukemia in mice. Proc Natl Acad Sci U S A 1998; 95:3885-9; PMID:9520462; http://dx.doi.org/ 10.1073/pnas.95.7.3885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gritti C, Dastot H, Soulier J, Janin A, Daniel MT, Madani A, Grimber G, Briand P, Sigaux F, Stern MH. Transgenic mice for MTCP1 develop T-cell prolymphocytic leukemia. Blood 1998; 92:368-73; PMID:9657733 [PubMed] [Google Scholar]
- 12. Borghesani PR, Alt FW, Bottaro A, Davidson L, Aksoy S, Rathbun GA, Roberts TM, Swat W, Segal RA, Gu Y. Abnormal development of Purkinje cells and lymphocytes in Atm mutant mice. Proc Natl Acad Sci U S A 2000; 97:3336-41; PMID:10716718; http://dx.doi.org/ 10.1073/pnas.97.7.3336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Elson A, Wang Y, Daugherty CJ, Morton CC, Zhou F, Campos-Torres J, Leder P. Pleiotropic defects in ataxia-telangiectasia protein-deficient mice. Proc Natl Acad Sci U S A 1996; 93:13084-9; PMID:8917548; http://dx.doi.org/ 10.1073/pnas.93.23.13084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xu Y, Ashley T, Brainerd EE, Bronson RT, Meyn MS, Baltimore D. Targeted disruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis, immune defects, and thymic lymphoma. Genes Dev 1996; 10:2411-22; http://dx.doi.org/ 10.1101/gad.10.19.2411 [DOI] [PubMed] [Google Scholar]
- 15. Barlow C, Brown KD, Deng CX, Tagle DA, Wynshaw-Boris A. Atm selectively regulates distinct p53-dependent cell-cycle checkpoint and apoptotic pathways. Nat Gen 1997; 17:453-6; http://dx.doi.org/ 10.1038/ng1297-453 [DOI] [PubMed] [Google Scholar]
- 16. Callen E, Jankovic M, Difilippantonio S, Daniel JA, Chen HT, Celeste A, Pellegrini M, McBride K, Wangsa D, Bredemeyer AL, et al. ATM prevents the persistence and propagation of chromosome breaks in lymphocytes. Cell 2007; 130:63-75; PMID:17599403; http://dx.doi.org/ 10.1016/j.cell.2007.06.016 [DOI] [PubMed] [Google Scholar]
- 17. Bredemeyer AL, Sharma GG, Huang CY, Helmink BA, Walker LM, Khor KC, Nuskey B, Sullivan KE, Pandita TK, Bassing CH, et al. ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature 2006; 442:466-70; PMID:16799570; http://dx.doi.org/ 10.1038/nature04866 [DOI] [PubMed] [Google Scholar]
- 18. Dujka ME, Puebla-Osorio N, Tavana O, Sang M, Zhu C. ATM and p53 are essential in the cell-cycle containment of DNA breaks during V(D)J recombination in vivo. Oncogene 2010; 29:957-65; http://dx.doi.org/ 10.1038/onc.2009.394 [DOI] [PubMed] [Google Scholar]
- 19. Petiniot LK, Weaver Z, Barlow C, Shen R, Eckhaus M, Steinberg SM, Ried T, Wynshaw-Boris A, Hodes RJ. Recombinase-activating gene (RAG) 2-mediated V(D)J recombination is not essential for tumorigenesis in Atm-deficient mice. Proc Natl Acad Sci U S A 2000; 97:6664-9; PMID:10841564; http://dx.doi.org/ 10.1073/pnas.97.12.6664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Petiniot LK, Weaver Z, Vacchio M, Shen R, Wangsa D, Barlow C, Eckhaus M, Steinberg SM, Wynshaw-Boris A, Ried T, et al. RAG-mediated V(D)J recombination is not essential for tumorigenesis in Atm-deficient mice. Molec Cell Bio 2002; 22:3174-7; PMID:11940674; http://dx.doi.org/ 10.1128/MCB.22.9.3174-3177.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zha S, Bassing C.H., Sanda T., Brush J.W., Patel H., Goff P.H., Gatti R., Look A.T., Alt F.W. ATM-deficient thymic lymphoma is associated with aberrant TCRd rearrangement and gene amplification. J Exp Med 2010; 207:1369-80; http://dx.doi.org/ 10.1084/jem.20100285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liyanage M, Weaver Z, Barlow C, Coleman A, Pankratz DG, Anderson S, Wynshaw-Boris A, Ried T. Abnormal rearrangement within the alpha/delta T-cell receptor locus in lymphomas from Atm-deficient mice. Blood 2000; 96:1940-6; PMID:10961898 [PubMed] [Google Scholar]
- 23. Yin B, Lee BS, Yang-Iott KS, Sleckman BP, Bassing CH. Redundant and nonredundant functions of ATM and H2AX in alphabeta T-lineage lymphocytes. J Immunology 2012; 189:1372-9; http://dx.doi.org/ 10.4049/jimmunol.1200829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Callen E, Bunting S, Huang CY, Difilippantonio MJ, Wong N, Khor B, Mahowald G, Kruhlak MJ, Ried T, Sleckman BP, et al. Chimeric IgH-TCRalpha/delta translocations in T lymphocytes mediated by RAG. Cell cycle 2009; 8:2408-12; PMID:19556863; http://dx.doi.org/ 10.4161/cc.8.15.9085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kamimura K, Ohi H, Kubota T, Okazuka K, Yoshikai Y, Wakabayashi Y, Aoyagi Y, Mishima Y, Kominami R. Haploinsufficiency of Bcl11b for suppression of lymphomagenesis and thymocyte development. Bio Bio Res Com 2007; 355:538-42; PMID:17306224; http://dx.doi.org/ 10.1016/j.bbrc.2007.02.003 [DOI] [PubMed] [Google Scholar]
- 26. Przybylski GK, Dik WA, Wanzeck J, Grabarczyk P, Majunke S, Martin-Subero JI, Siebert R, Dölken G, Ludwig WD, Verhaaf B, et al. Disruption of the BCL11B gene through inv(14)(q11.2q32.31) results in the expression of BCL11B-TRDC fusion transcripts and is associated with the absence of wild-type BCL11B transcripts in T-ALL. Leukemia 2005; 19:201-08; PMID:15668700; http://dx.doi.org/ 10.1038/sj.leu.2403619 [DOI] [PubMed] [Google Scholar]
- 27. Gutierrez A, Kentsis A, Sanda T, Holmfeldt L, Chen SC, Zhang J, Protopopov A, Chin L, Dahlberg SE, Neuberg DS, et al. The BCL11B tumor suppressor is mutated across the major molecular subtypes of T-cell acute lymphoblastic leukemia. Blood 2011; 118:4169-73; PMID:21878675; http://dx.doi.org/ 10.1182/blood-2010-11-318873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. De Keersmaecker K, Real PJ, Gatta GD, Palomero T, Sulis ML, Tosello V, Van Vlierberghe P, Barnes K, Castillo M, Sole X, et al. The TLX1 oncogene drives aneuploidy in T cell transformation. Nat Med 2010; 16:1321-27; PMID:20972433; http://dx.doi.org/ 10.1038/nm.2246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yin B, Bassing CH. V(D)J recombination causes dangerous chromosome liaisons in developing thymocytes. Cell Cycle 2009; 8:2486-7; http://dx.doi.org/ 10.4161/cc.8.16.9349 [DOI] [PubMed] [Google Scholar]
- 30. Bhandoola A, von Boehmer H, Petrie HT, Zuniga-Pflucker JC. Commitment and developmental potential of extrathymic and intrathymic T cell precursors: plenty to choose from. Immunity 2007; 26:678-89; http://dx.doi.org/ 10.1016/j.immuni.2007.05.009 [DOI] [PubMed] [Google Scholar]
- 31. Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S, Makar KW, Pérez-Melgosa M, Sweetser MT, Schlissel MS, Nguyen S, et al. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity 2001; 15:763-74; PMID:11728338; http://dx.doi.org/ 10.1016/S1074-7613(01)00227-8 [DOI] [PubMed] [Google Scholar]
- 32. Yin B, Yang-Iott KS, Chao LH, Bassing CH. Cellular context-dependent effects of H2ax and p53 deletion on the development of thymic lymphoma. Blood. 2011; 117:175-85; http://dx.doi.org/ 10.1182/blood-2010-03-273045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brady BL, Oropallo MA, Yang-Iott KS, Serwold T, Hochedlinger K, Jaenisch R, Weissman IL, Bassing CH. Position-dependent silencing of germline Vb segments on TCRb alleles containing preassembled VbDJbCb1 genes. J Immunol. 2010; 185:3564-73; PMID:20709953; http://dx.doi.org/ 10.4049/jimmunol.0903098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brady BL, Steinel NC, Bassing CH. Antigen receptor allelic exclusion: an update and reappraisal. J Immunol. 2010; 185: 3801-8; http://dx.doi.org/ 10.4049/jimmunol.1001158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liyanage M, Coleman A, du Manoir S, Veldman T, McCormack S, Dickson RB, Barlow C, Wynshaw-Boris A, Janz S, Wienberg J, et al. Multicolour spectral karyotyping of mouse chromosomes. Nat Gen. 1996; 14:312-5; PMID:8896561; http://dx.doi.org/ 10.1038/ng1196-312 [DOI] [PubMed] [Google Scholar]
- 36. Isoda T, Takagi M, Piao J, Nakagama S, Sato M, Masuda K, Ikawa T, Azuma M, Morio T, Kawamoto H, et al. Process for immune defect and chromosomal translocation during early thymocyte development lacking ATM. Blood. 2012; 120:789-99; PMID:22709691; http://dx.doi.org/ 10.1182/blood-2012-02-413195 [DOI] [PubMed] [Google Scholar]
- 37. Wakabayashi Y, Watanabe H, Inoue J, Takeda N, Sakata J, Mishima Y, Hitomi J, Yamamoto T, Utsuyama M, Niwa O, et al. Bcl11b is required for differentiation and survival of alphabeta T lymphocytes. Nat immunol. 2003; 4:533-9; PMID:12717433; http://dx.doi.org/ 10.1038/ni927 [DOI] [PubMed] [Google Scholar]
- 38. Li L, Leid M, Rothenberg EV. An early T cell lineage commitment checkpoint dependent on the transcription factor Bcl11b. Science. 2010; 329:89-3; http://dx.doi.org/ 10.1126/science.1188989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ikawa T, Hirose S, Masuda K, Kakugawa K, Satoh R, Shibano-Satoh A, Kominami R, Katsura Y, Kawamoto H, et al. An essential developmental checkpoint for production of the T cell lineage. Science. 2010; 329:93-6; PMID:20595615; http://dx.doi.org/ 10.1126/science.1188995 [DOI] [PubMed] [Google Scholar]
- 40. Kamimura K, Mishima Y, Obata M, Endo T, Aoyagi Y, Kominami R. Lack of Bcl11b tumor suppressor results in vulnerability to DNA replication stress and damages. Oncogene. 2007; 26:5840-50; http://dx.doi.org/ 10.1038/sj.onc.1210388 [DOI] [PubMed] [Google Scholar]
- 41. Barlow C, Liyanage M, Moens PB, Tarsounas M, Nagashima K, Brown K, Rottinghaus S, Jackson SP, Tagle D, Ried T, et al. Atm deficiency results in severe meiotic disruption a early as leptonema of prophase I. Development. 1998; 125:4007-17; PMID:9735362 [DOI] [PubMed] [Google Scholar]
- 42. Kastner P, Chan S, Vogel WK, Zhang LJ, Topark-Ngarm A, Golonzhka O, Jost B, Le Gras S, Gross MK, Leid M, et al. Bcl11b represses a mature T-cell gene expression program in immature CD4(+)CD8(+) thymocytes. Eur J Immunol. 2010; 40:2143-54; PMID: 20544728; http://dx.doi.org/ 10.1002/eji.200940258 [DOI] [PMC free article] [PubMed] [Google Scholar]