

Lipid biosynthesis and storage in tissues must be carefully balanced, as too little or too much lipid storage is detrimental. Lipin-1 is an enzyme that plays a key role in lipid biosynthesis and storage through its conversion of phosphatidic acid to diacylglycerol (DAG), the immediate precursor of the primary fat storage molecule triacylglycerol. Lipin-1 levels are correlated with fat storage in adipose tissue, and lipin-1 also serves as a transcriptional coactivator for fatty acid gene expression in liver.1 It was therefore unexpected that the defining feature of lipin-1 deficiency in humans is severe rhabdomyolysis (muscle breakdown) during childhood, typically triggered by fasting or febrile illness.2,3 Interestingly, lipin-1 haploinsufficient (LPIN1+/–) individuals also develop muscle symptoms (myopathy) after treatment with statin drugs2, which are commonly used to lower plasma cholesterol levels to prevent cardiovascular disease. Myopathy is a side-effect observed in ∼5% of statin drug users, but the mechanism is not fully understood.
Based on the activity of lipin-1 in lipid metabolism, the basis for lipin-1–related rhabdomyolysis has been mysterious. We recently utilized mouse models to identify a role for lipin-1 in muscle homeostasis through effects on autophagy.4 We first established that muscle pathology in lipin-1 mutant mice parallels that observed in humans. When mice were stressed by fasting and refeeding, Lpin1–/– mice had signs of muscle damage (elevated creatine kinase levels, muscle fiber necrosis and myocyte turnover). Lpin1+/– mice appeared normal after fasting stress, but statin drug treatment initiated muscle damage in these mice, and worsened muscle symptoms in Lpin1–/– mice. The restoration of lipin-1 in muscle prevented primary muscle damage and statin myotoxicity in both Lpin1–/– and Lpin1–/+ mice.
An analysis of muscle ultrastructure provided a clue to the mechanism underlying damage in lipin-1–deficient muscle. Lpin1–/– myocytes accumulated enlarged, aberrantly shaped mitochondria, with reduced maximal respiratory activity.4 We also detected autophagosomes in Lpin1–/– muscle, but never in wild-type muscle. These observations suggested that macroautophagy—a critical mechanism for removal of damaged proteins and organelles (including mitochondria) and for cellular survival during starvation—is dysregulated in response to reduced lipin-1 levels. The resulting impaired function of mitochondria, as well as other organelles and proteins, likely contributes to impaired muscle function in individuals with impaired lipin-1 function.
To characterize the role of lipin-1 in autophagy, we systematically evaluated the stages of this process in wild-type and lipin-1–deficient cells. Using a combination of molecular probes and genetic manipulations, we determined that lipin-1 enzymatic activity is required for the maturation of autophagosomes to autolysosomes.4 This was due to a role for the lipin-1 enzymatic product, DAG, in the fusion of autophagosomes with lysosomes (Fig. 1). Earlier work had shown that the substrate for the lipin-1 enzyme activity (phosphatidic acid) is produced by phospholipase D on the surface of autophagosomes.5 The action of lipin-1 on this substrate produces DAG, which activates protein kinase D and leads to phosphorylation and activation of Vps34 lipid kinase. The product, phosphatidylinositol-3-phosphate, has previously been shown to promote lysosome/autophagosome fusion.6 Thus, phospholipase D and lipin-1 may act in sequence to generate the DAG required for membrane fusion in autophagy. It should be noted that Vps34 also has a role in the initiation of autophagy, which is not impaired in lipin1–deficient cells. We suspect that this is due to the presence of Vps34 in distinct protein complexes that act at the initiation and maturation steps of autophagy, with lipin-1 being present only in the latter complex.
Figure 1.
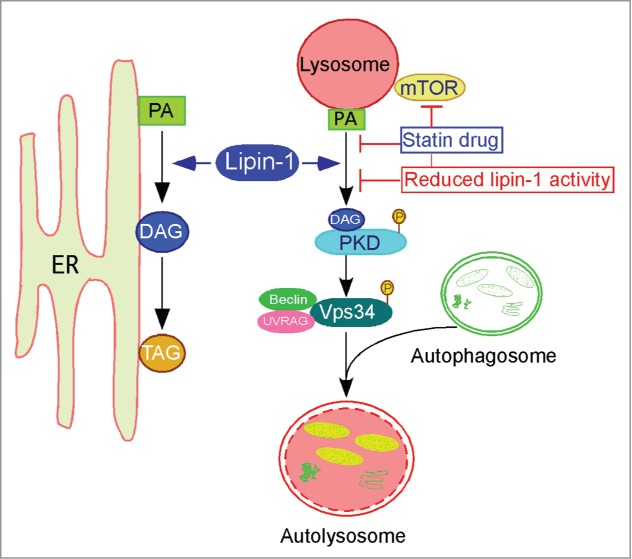
Lipin-1 has roles in lipid synthesis and autophagy clearance. Lipin-1 resides in the cytosol and translocates to the ER membrane (and other specific intracellular membranes) to interact with its substrate, phosphatidic acid (PA), to produce diacylglycerol (DAG). Lipin-1 generated DAG may serve as a precursor of triacylglycerol (TAG), and may also activate protein kinase D (PKD) at the lysosomal surface, with subsequent phosphorylation and activation of Vps34, promoting maturation of autolysosomes. Reduced lipin-1 levels and statin treatment converge at the level of PKD activation, leading to impaired autophagy clearance and muscle damage.
Paradoxically, lipin-1–deficient muscles in humans and mice accumulate neutral lipids.3,7 It has been hypothesized that triglycerides accumulate due to loss of lipin-1 coactivator activity for induction of fatty acid oxidation genes.7 However, analysis of muscle from Lpin1–/– mouse showed normal expression of fatty acid oxidation genes, and revealed that the lipid droplets are comprised of cholesteryl esters rather than triglycerides.4 To our knowledge, this is a unique feature of lipin-1 deficiency compared to other muscle lipidoses. We surmise that muscle accumulates fatty acids and cholesteryl esters to adapt to an inadequate capacity for triglyceride synthesis; the persistence of these lipid droplets may be related to impaired degradation by autophagy. Thus, impaired autophagy in lipin-1–deficient muscle likely leads to impaired turnover of both lipid and protein components.
How does statin action intersect with lipin-1 activity in autophagy? We noticed that muscle from wild-type mice treated with statin had attenuated protein kinase D activation similar to that resulting from lipin-1 deficiency.4 We also determined that statin treatment of wild-type cells causes lipin-1 to translocate to lysosomes/autolysosomes, suggesting that statins may induce the autophagy pathway. Statins would therefore introduce stress in cells with reduced lipin-1 activity by stimulating autophagy in an environment in which autophagic flux is reduced. The intersection between statin and lipin-1 haploinsufficiency in the autophagy pathway may constitute ‘2 hits’ that together impair autophagic flux and contribute to muscle dysfunction in Lpin1+/– mice and LPIN1+/– humans.
Do these findings have therapeutic implications? Based on data from the 1000 Genomes project (http://www.1000genomes.org/data), LPIN1 missense and nonsense mutations are present at significant levels in the “healthy” population. Lipin-1 haploinsufficiency may represent a genetic risk factor for statin induced myopathy, and further evaluation of this possibility in relevant patient populations is warranted. Additionally, the studies of Zhang et al.4 suggest that any type of metabolic stress that pushes myocytes to rely upon autophagy for cellular homeostasis may trigger rhabdomyolitic episodes in LPIN1–/– individuals; this may help to demystify the basis for the sometimes random-seeming occurrence of episodes. Treatment of LPIN1–/– individuals with compounds that selectively promote autophagy clearance might be useful to prevent rhabdomyolytic episodes.
References
- 1. Csaki LS, et al. Prog Lipid Res 2013; 52:305-16; PMID:23603613; http://dx.doi.org/ 10.1016/j.plipres.2013.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zeharia A, et al. Am J Hum Genet 2008; 83:489-94; PMID:18817903; http://dx.doi.org/ 10.1016/j.ajhg.2008.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Michot C, et al. Hum Mutat 2010; 31:E1564-73; PMID:20583302; http://dx.doi.org/ 10.1002/humu.21282 [DOI] [PubMed] [Google Scholar]
- 4. Zhang P, et al. Cell Metab 2014; 20:267-79; PMID:24930972; http://dx.doi.org/ 10.1016/j.cmet.2014.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dall’Armi C, et al. Curr Biol 2013; 23:R33-45; PMID:23305670; http://dx.doi.org/ 10.1016/j.cub.2012.10.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Eisenberg-Lerner A, Kimchi A. Cell Death Differ 2012; 19:7788-97; PMID:22095288; http://dx.doi.org/ 10.1038/cdd.2011.149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Michot C, et al. Biochim Biophys acta 2013; 1832:2103-14; PMID:23928362; http://dx.doi.org/ 10.1016/j.bbadis.2013.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]