

ABSTRACT
Repair of DNA double strand breaks (DSBs) is influenced by the chemical complexity of the lesion. Clustered lesions (complex DSBs) are generally considered more difficult to repair and responsible for early and late cellular effects after exposure to genotoxic agents. Resection is commonly used by the cells as part of the homologous recombination (HR) pathway in S- and G2-phase. In contrast, DNA resection in G1-phase may lead to an error-prone microhomology-mediated end joining. We induced DNA lesions with a wide range of complexity by irradiation of mammalian cells with X-rays or accelerated ions of different velocity and mass. We found replication protein A (RPA) foci indicating DSB resection both in S/G2- and G1-cells, and the fraction of resection-positive cells correlates with the severity of lesion complexity throughout the cell cycle. Besides RPA, Ataxia telangiectasia and Rad3-related (ATR) was recruited to complex DSBs both in S/G2- and G1-cells. Resection of complex DSBs is driven by meiotic recombination 11 homolog A (MRE11), CTBP-interacting protein (CtIP), and exonuclease 1 (EXO1) but seems not controlled by the Ku heterodimer or by phosphorylation of H2AX. Reduced resection capacity by CtIP depletion increased cell killing and the fraction of unrepaired DSBs after exposure to densely ionizing heavy ions, but not to X-rays. We conclude that in mammalian cells resection is essential for repair of complex DSBs in all phases of the cell-cycle and targeting this process sensitizes mammalian cells to cytotoxic agents inducing clustered breaks, such as in heavy-ion cancer therapy.
Keywords: double-strand break repair, complex DNA damage, resection in G1-phase, CtIP, MRE11, EXO1
Abbreviations
- ATM
Ataxia telangiectasia mutated
- ATR
Ataxia telangiectasia and Rad3-related
- BLM
Bloom syndrome protein
- BRCA1
breast cancer 1, early onset
- CENP-F
centromere protein F
- CtIP
CTBP-interacting protein
- DAPI
4',6-diamidino-2-phenylindole
- DSB
double strand break
- EXO1
exonuclease 1
- FCS
fetal calf serum
- HR
homologous recombination
- IR
ionizing radiation
- kd
knockdown
- LET
linear energy transfer
- MEF
mouse embryonic fibroblasts
- MMEJ
microhomology-mediated end joining
- MRE11
meiotic recombination 11 homolog A
- NHEJ
none homologous end joining
- PARP
poly (ADP-ribose) polymerase
- RAD51
DNA repair protein RAD51 homolog 1
- RPA
replication protein A
- siRNA
small interfering RNA
- ssDNA
single stranded DNA
- WRN
Werner syndrome
- wt
wild-type
Introduction
DNA double-strand breaks (DSBs) are generally considered the crucial lesions leading to early and late effects of ionizing radiation (IR). The severity of the damage is related to its chemical and spatial complexity, i. e. to the clustering of different lesions in close proximity (complex DSBs). Radiation track structure simulation analysis suggests that complex DSBs are induced by IR and their frequency increases by increasing the radiation ionization density (linear energy transfer, LET).1 Recent molecular data shows that more severe, clustered DSBs are more difficult to repair and may be responsible for the formation of chromosomal aberrations and other late effects.2
Eukaryotic cells have evolved different mechanisms of DSB repair, and the pathway choice is influenced by cell cycle stage, chromatin structure, and damage complexity.3 The two major pathways to repair DSBs are non homologous end joining (NHEJ) and homologous recombination (HR). While HR is restricted to the late S- and G2-cell cycle phases, when undamaged sister chromatids can be used as a template for faithful repair,3 NHEJ is active throughout the cell cycle and is the predominant repair pathway in mammalian cells.4,5 During NHEJ, DNA break ends are directly ligated, although depending on DSB end processing errors are likely. Mechanistically, classical (c)-NHEJ, which requires binding of the Ku70–80 heterodimer (Ku) to DNA ends and alternative (alt)-NHEJ can be distinguished.6 The latter depends on poly (ADP-ribose) polymerase (PARP) and was identified as a backup NHEJ pathway, whose activity becomes evident in the absence of c-NHEJ factors.6 In addition, alt-NHEJ is often referred to as microhomology-mediated end joining (MMEJ), as it frequently involves CtIP- and MRE11-dependent resection and the presence of microhomologies near the DSB ends.7–12 In contrast to HR, where the generation of ssDNA overhangs via DSB resection is a crucial prerequisite for homology-mediated error-free repair, alternative end joining using short microhomologies is intrinsically error prone and allegedly a major pathway leading to exchange-type chromosomal aberrations and tumorigenesis.11,13,14 Commitment to a repair pathway defines the propensity for errors, and regulating DSB resection is critical to influence the accuracy of repair and thus genomic instability.15
In S- and G2-phase cells, MRE11 and CtIP are responsible for the initiation and short range resection promoting HR.16 The nucleases EXO1 as well as DNA2, the latter together with the helicase Bloom syndrome protein (BLM) or Werner syndrome (WRN), perform long range resection by extending the resected DNA ends. DSB resection in G1-phase has only recently been described and seems to be influenced by the quality of the break.17–19 So far only CtIP has been described to be involved in resection of IR-induced DSBs in G1.18,19 At covalently blocked DSBs, MRE11 and CtIP were shown to be involved in resection of DSBs induced in G1-phase.17 DSB resection is regulated at several levels. CtIP is far more expressed in G2- than G1-phase20 and its phosphorylation pattern and activity are cell-cycle dependent.19 Additionally, CtIP is phosphorylated in response to DNA damage by Ataxia telangiectasia mutated (ATM) and ATR.18,21,22 Upon DNA damage, ATM also phosphorylates EXO1 to a greater extent in G2- than in G1-phase.23 Data on murine lymphocytes suggest that the phosphorylation of the histone H2AX in the vicinity of a DSB represses break resection in G1-cells in the process of antigen receptor gene assembly.24 Furthermore, binding of the c-NHEJ factor Ku to DSB ends protects them from resection.25,26
To assess the role of resection in DNA repair in G1-phase, we have introduced DSBs of increasing complexity using accelerated heavy nuclei. Using specific gene knockdown, we analyzed the key players of resection and how impairment of resection by CtIP depletion affects cell killing by ionizing radiation (IR).
Results
DSB complexity plays a critical role in the decision for DSB end-resection in G1-cells
For the processing of DSBs the location in the chromatin and the chemical complexity are critical.17,18,27 We induced DNA damage of increasing complexity by irradiation with X-rays (low complexity) or heavy ions with increasing ionization density (specified by the LET) up to very high complexity. As a measure of DSB end resection we examined foci formation of the single strand DNA (ssDNA) binding protein RPA28 in normal human fibroblasts (AG1522D), immortalized human fibroblasts (NFFhTERT), and human osteosarcoma cells (U2-OS). Cell-cycle phases were identified using centromere protein F (CENP-F) co-immunostaining, which generally distinguished around 50% G1-phase cells after irradiation. DSB resection was detected in G1-cells after irradiation with heavy ions only (Fig. 1A). The presence of RPA positive G1-cells was verified in independent experiments with different cell cycle markers (SI Fig. S1). With increasing DSB complexity, an increasing fraction of irradiated cells, both in G1- and S/G2-phase, showed RPA foci at the damage sites (Fig. 1B). S/G2-cells showed a higher fraction of RPA positive cells for all radiation qualities tested. After exposure to X-rays, about 30% of S/G2-cells were RPA positive, whereas hardly any RPA positive G1-cells were detected. Based on Poisson statistics, almost 10% of the cell nuclei are not hit by ions at the particle fluences used in our experiment. We conclude that upon induction of very complex DNA damage, all G2 cells that were irradiated are resection positive. In contrast, in G1-cells after induction of very complex lesions the maximum fraction of resection positive cells did not exceed 80% after 1 h from exposure. After induction of DNA damage of intermediate complexity the fraction of RPA positive cells in G1-phase NFFhTERT cells was found to increase with time post exposure (SI Fig. S2) suggesting that after induction of very complex lesions also all G1-cells will be resection positive.
Figure 1.
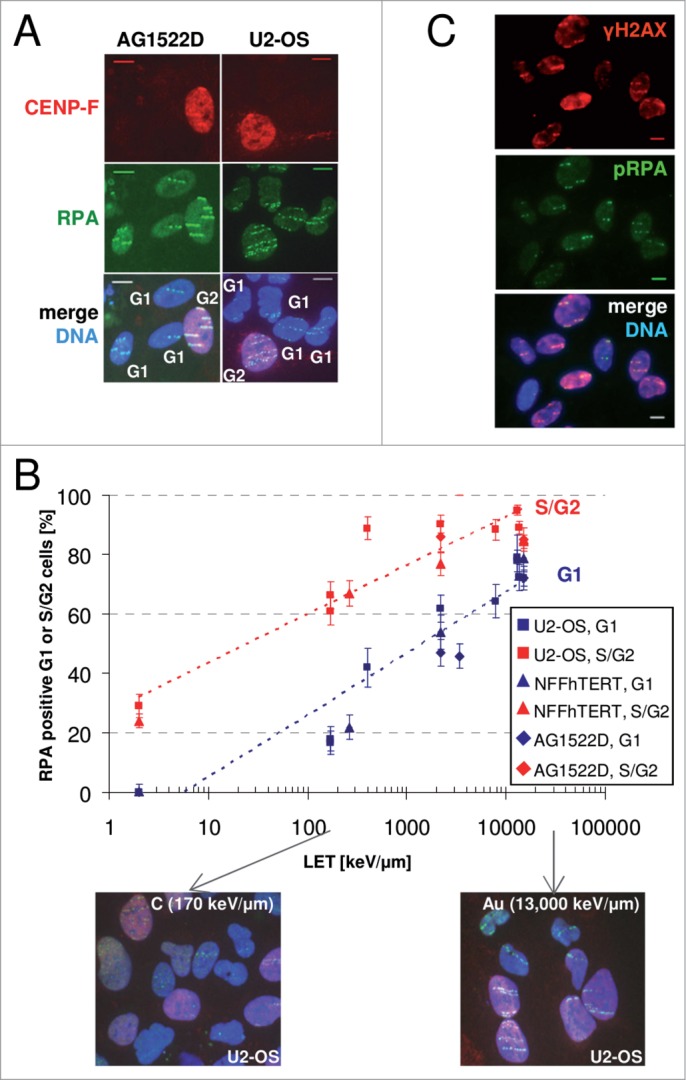
DSB resection occurs in G1- and S/G2-cells upon high-LET irradiation. (A) Human osteosarcoma cells, U2-OS, and normal human fibroblasts, AG1522D, were irradiated with gold ions and fixed 1 h after irradiation. CENP-F immunostaining (red) was used to distinguish between G1- (CENP-F negative) and S/G2-cells (CENP-F positive). RPA immunostaining (green) served as a resection marker. DNA was counter stained with DAPI (blue). Scale bar: 10 μm. (B) Resection of DSBs caused by ionizing radiation is LET dependent. U2-OS, NFFhTERT, and AG1522D cells were irradiated with high-LET (LET ≥ 90 keV/μm; heavy ions) and low-LET irradiation (LET = 2 keV/ μm; X-rays). Cells were fixed 1 h post irradiation and immunostained for RPA (green) and CENP-F (red). DNA was counter stained with DAPI (blue). RPA positive irradiated cells were counted for CENP-F positive (S/G2) cells and CENP-F negative (G1) cells. Each data point represents one experiment, in which at least 50 G1- and S/G2-cells were analyzed. Error bar: binomial error. (C) Confluent normal human fibroblasts AG1522D were irradiated with uranium ions and fixed 1 h after irradiation. Immunostaining was performed against γH2AX (red) and phospho-RPA (green). DNA was counter stained with DAPI (blue). Scale bar: 10 μm. An analogous experiment was performed with lead ions and yielded comparable results.
The strong resection signal at complex DSBs observed in G1-cells argues against a major role of H2AX in preventing end processing of IR-induced DSBs in somatic G1-cells, in contrast to RAG-endonuclease-induced DSBs in murine G1-phase lymphocytes.24 This is confirmed by the observation that H2AX deficiency did not cause an increase in resection positive cells after C-ion irradiation: wild-type mouse embryonic fibroblasts (MEFs) showed 18.7 ± 2.9% RPA positive cells and H2AX −/− MEFs 16.7 ± 4.9% (n = 3, ± SEM) 1 h after irradiation. Moreover, we found that in confluent (G1/G0) AG1522D cells irradiated with very high-LET heavy ions a substantial fraction of γH2AX (74 ± 8%, n = 3, ± SEM) co-localizes with phosphorylated RPA (phospho-RPA) in an immunofluorescence analysis (Fig. 1C).
Next, we examined whether in RPA-negative cells Ku was protecting DSB ends from resection, which was described previously.25,26 Evaluating the fraction of RPA and thus resection positive cells upon induction of complex DSBs by C-ion irradiation in Ku proficient and deficient MEFs revealed no change in the fraction of resection positive cells; wild-type cells showed 42.7 ± 4.7% and Ku80 −/− cells 33.7 ± 4.9% (n = 1, ± binomial error) RPA positive cells. Furthermore, the cell cycle distribution of the analyzed Ku proficient and deficient cells was similar (SI Table S1). Therefore, we conclude that Ku does not play a significant role in controlling resection of complex DSBs.
Taken together, increasing DSB complexity leads to a corresponding increase in the detection of RPA at DNA damage sites, indicating that complex lesions need resection for repair both in S/G2- and G1-phase of the cell-cycle.
CtIP, MRE11, and EXO1 are required for the resection of complex DSBs in G1
As CtIP is required for DSB resection in S/G229,30 and in G1-phase cells17–19 we first tested its recruitment to localized, complex DSBs after heavy ion irradiation. CtIP was clearly detected along ion-induced damage tracks both in S/G2-cells and in the majority (about 80%) of the G1-phase cells (Fig. 2A). This CtIP accumulation is readily observed despite the lower level of CtIP expression in this stage of the cell cycle (Fig. 2A: compare G2- with G1-cell).19,20
Figure 2.
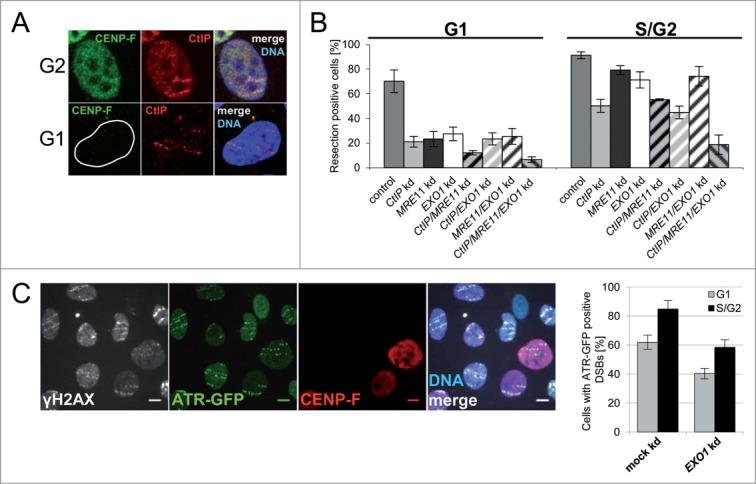
MRE11, CtIP, and EXO1 are important for resection of complex DSBs. (A) CtIP is recruited to DSBs in G1. U2-OS cells were irradiated with uranium ions and fixed 1 h after irradiation. Immunostaining was performed against CENP-F (green; cell cycle marker) and CtIP (red). DNA was counter stained with DAPI (blue). (B) The expression of CtIP, MRE11, and EXO1 was decreased by RNAi. DSB resection positive cells (RPA) were counted 1 h after low angle gold, lead, tin, or uranium-ion irradiation in G1 (CENP-F negative) and S/G2 (CENP-F positive) cells. Each bar represents the average of at least four independent experiments ± standard error of the mean (SEM). All knockdown treated samples have significantly less resection positive cells than mock knockdown samples (Student's t-test, p < 0.05). All single or double knockdown samples but CtIP/MRE11 knockdown in G1 show significantly more resection than the triple knockdown (Student's t-test, p < 0.01). (C) ATR is recruited to complex lesions in G1 in an EXO1 dependent manner. U2-OS ATR-GFP cells were depleted for EXO1 by RNAi, irradiated with gold ions, and fixed 1 h after irradiation. γH2AX served as a DSB marker (white) and CENP-F (red) as cell cycle marker. DNA was counter stained with DAPI (blue). Scale bar: 10 μm. Cells where ATR-GFP is recruited to DSBs were counted in G1- (CENP-F negative) and S/G2-cells (CENP-F positive). At least 60 G1- and S/G2-cells were analyzed. Error bar: binomial error of one experiment.
To test whether resection in G1 was performed by CtIP, we measured RPA foci formation at complex DSBs in U2-OS cells after RNAi-mediated knockdown. CtIP depletion strongly decreased but did not completely abolish RPA foci formation in G1-cells (Fig. 2B), indicating that other resection enzymes are also involved. Since CtIP acts in concert with MRE11 in G2-phase29 and the nuclease EXO1 plays a crucial role in DSB repair,23,26 we next tested whether MRE11 and EXO1 also function in the processing of complex DNA lesions. We used RNAi to reduce the expression of CtIP, MRE11, and EXO1 – individually, pairwise, or all together – and analyzed RPA accumulation at ion-induced damage sites in S/G2- and G1-phase U2-OS cells (Fig. 2B). Protein depletion in all combinations tested caused a significant decline of resection, measured as RPA foci positive cells, that was much more pronounced in G1- compared to S/G2-cells. In G1-cells a single or pairwise knockdown of any of the genes, CtIP, MRE11, or EXO1, had a similar effect reducing the fraction of RPA positive cells by 60–70%. This result suggests that these factors are epistatic. The complete suppression of resection in G1-cells after depletion of all three factors implies that they are the only enzymes active in resection of complex lesions in G1-phase. The combined resection data on MRE11/EXO1 and MRE11/EXO1/CtIP depletion suggest despite earlier findings29 that CtIP itself may possess nuclease activity, as it is known for the Saccharomyces cerevisiae homologue Sae2.31
Unlike in G1-phase, a single knockdown of CtIP, MRE11, or EXO1 in S/G2-cells did not show the same effect on the fraction of RPA positive cells. While CtIP depletion caused the strongest effect, with a decrease of 40–50% after induction of complex DSBs, knockdown of MRE11 or EXO1 decreased RPA positive cells only by about 20%. The epistasis of CtIP, MRE11, and EXO1 is also observed in the results of the different combinations of double depletions in S/G2-cells. The depletion of all three resection factors decreases the fraction of RPA positive cells also in S/G2-cells by about 80%, indicating that they are the main players in resection of complex DSBs in all cell-cycle phases. The reduction of RPA foci, observed in irradiated S/G2 double and triple knockdown cells, supports the idea that each resection factor can perform DSB resection on its own, although with different efficiency. The differences in DSB resection activity following depletion of CtIP, MRE11, and EXO1 in G1- and S/G2-phase may be related to cell cycle dependent changes in concentration (Fig. 2A), posttranslational modifications,19,23,32 and/or binding of interaction partners.19,33
In addition to the cell cycle-wide recruitment of ssDNA binding RPA to heavy-ion induced DSBs, we also observed the recruitment of ATR to such damaged sites even in G1-cells (Fig. 2C). The ATR recruitment to complex DSBs was significantly decreased following EXO1 depletion. The described resection of complex DSBs in all analyzed cell cycle phases may thus contribute to the persistent ATR signal transduction induced by complex DNA damage.34
In summary, our data demonstrate that CtIP, MRE11, and EXO1 are key factors involved in the resection of complex DSBs not only in S/G2 but especially in G1. Furthermore, the participation of EXO1 and the DSB recruitment of ATR in G1 indicate extensive resection of complex DSBs also in this cell cycle phase.
Repair of complex DSBs relies on resection independent of the cell cycle stage
To address the relevance of the observed resection of complex DSBs for their repair, we used the DSB marker γH2AX to measure the repair kinetics of carbon ion-induced DSBs in G1 and G2 cells that were impaired for resection by siRNA driven CtIP depletion (Fig. 3A). Initiation of DSB resection in ion-irradiated G1 and G2 cells was verified by RPA staining 2 h after exposure (SI Fig. S4). The obtained repair kinetics clearly revealed impaired DSB repair after carbon irradiation due to the increased lesion density (Fig. 3A) as it has been reported earlier.27,35
Figure 3.
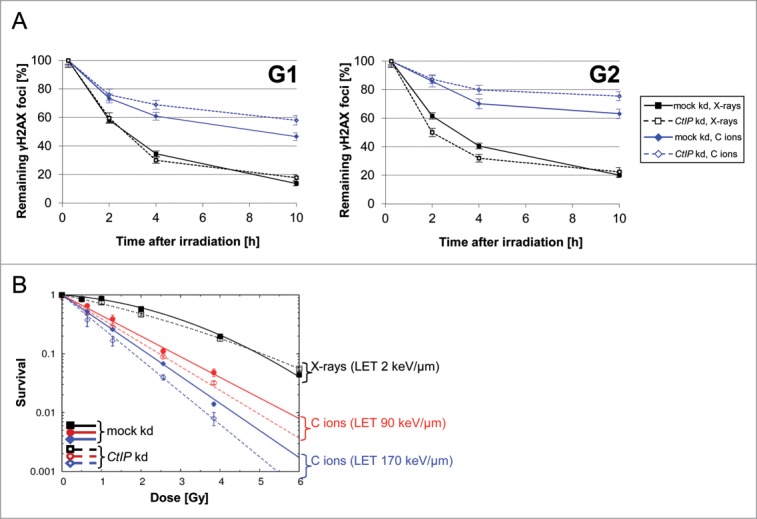
Resection of complex DSBs is important for their repair and cell survival. (A) NFFhTERT cells depleted for CtIP or mock depleted were irradiated with 1.28 Gy X-rays or carbon ions (LET 170 keV/μm) and treated with Aphidicolin right after irradiation to prevent G1-cells from moving on to G2. Aphidicolin treatment was checked not to affect DSB repair (SI Figure S3).50 S phase cells, recognized by their pan-nuclear γH2AX signal, were excluded from the analysis.50 The DSB repair kinetics of G2- (CENP-F positive) and G1-cells (CENP-F negative) was monitored by counting γH2AX foci at several time points after irradiation. The numbers of γH2AX foci were normalized to the γH2AX foci number 15 min after irradiation, which served as the number of DSBs induced. Shown is the mean ± SEM foci number/nucleus of one experiment. At least 50 nuclei per data point were analyzed. (B) Survival upon induction of complex DSBs is disabled in resection impaired cells. Clonogenic survival assay of NFFhTERT cells depleted for CtIP or mock depleted by siRNA and irradiated with 0.5 Gy, 1.0 Gy, 2.0 Gy, or 4.0 Gy of photons or 0.64 Gy, 1.28 Gy, 2.56 Gy, or 3.84 Gy of carbon ions of increasing ionizing density. Shown is the mean ± SEM survival of three replicates.
CtIP depletion further impaired the repair of carbon ion-induced complex DSBs in G2 and G1, but not of X-ray induced DSBs of lesser complexity (Fig. 3A). X-ray induced DSBs in G2-phase cells were repaired even faster when CtIP-dependent resection was impaired, in agreement with previous observations.27 In cells exposed to C-ions the reduced DSB resection after CtIP depletion affected the slow repair kinetics in all analyzed cell cycle stages (Fig. 3A). This suggests that only complex DSBs in the G2- and G1-cell cycle phase depend on resection for repair. DSBs of lower complexity are repaired by a resection-independent pathway in G1-phase, while cells irradiated in G2 seem to switch to a resection-independent pathway only if resection is impaired.
To determine the influence of resection on DSB genotoxicity, clonogenic survival was measured in proliferating wild-type and CtIP depleted human fibroblasts (NFFhTERT) after exposure to different radiation qualities. As expected, compared to sparsely IR all cells showed an increasing sensitivity to carbon ions producing increasing lesion densities (Fig. 3B and SI Table S2). Decrease of DSB resection resulted in lower cell survival of ion-irradiated cells, thus mirroring the reduced repair capability of complex lesions.
The observed extensive DSB resection in G1-phase and reports that suggest HR repair in G1-phase of mouse embryonic stem cells36 prompted us to test whether HR might represent a possible pathway for complex DSB repair in G1-phase in our cell system. However, DNA repair protein RAD51 homolog 1 (RAD51) depletion did not influence the repair kinetics of complex DSBs in G1-cells and RAD51 foci were not detected in ion-irradiated G1 fibroblasts (NFFhTERT) upon immunofluorescence analysis revealing that HR is not involved (SI Fig. S5). Taken together, we show that with increasing DNA damage complexity DSB repair becomes increasingly dependent on DSB resection at all cell cycle stages.
Discussion
Repair of DSBs is characterized by biphasic repair kinetics with a fast and slow component independent of the cell cycle stage.37 Complex DSBs are mainly repaired by the slow component,27,35 which in G2-phase represents resection-dependent HR.27,38 Recent data on the repair of covalently blocked DSB ends or IR-induced DSBs in DT40 B cells suggest a function of CtIP and MRE11 also in G1.17,19 This is in line with the observed recruitment of RPA to complex DSBs in G1 (Fig. 1 and Yajima et al.18) Here we show that in human G1-cells the slow component of complex DSB repair requires resection. The extent of resection is such that RPA binding to the produced ssDNA can be clearly visualized as ion irradiation-induced foci by immunofluorescence microscopy (Fig. 1A). The clear correlation of the increase in the number of RPA positive cells with increasing LET indicates a direct connection between DSB complexity and the degree of resection (Fig. 1B). This is in agreement with a low resection activity at DSBs induced by X-rays in G1-phase (SI Fig. S4). The resection taking place at complex DSBs may be the cause of the large deletions induced by high-LET radiation.39 We found the enzymes MRE11, CtIP, and EXO1 to be responsible for the resection of complex DSBs. Our data imply that EXO1 is the sole long range nuclease in G1-phase, while the other long range nuclease DNA2 and the associated helicase BLM are described to be primarily active in G2-phase.40–42 Our data further reveal that the activity of DNA2 is less important for resection of complex DSBs in G2-phase as well. This suggests that the DSB quality may affect the choice of the resection machinery.
We show that CtIP is indispensable for the repair of clustered complex DSBs with slow kinetics both in G1- and G2-phase. Neither in G1- nor in G2-phase can CtIP depleted cells switch to a resection-independent repair pathway, as it seems to be possible in the damage response to simple DNA lesions in G2-phase (Fig. 3A and Shibata et al.27). Thus, DSB resection is crucial for repair of complex DNA damage in all cell-cycle stages. Although G2 cells depleted for CtIP still have a substantial fraction of resected breaks (Fig. 2B), they hardly show any IR-induced DSB repair with slow kinetics (Fig. 3A), where resection-dependent HR is active.27 This implies that CtIP may have additional functions in DSB repair besides DSB resection, which may be connected to its G2 specific complex formation with BRCA1 (breast cancer 1, early onset).33
We conclude that the increased requirement for processing of DNA ends observed on complex DSBs forces the pathway choice in G1-cells towards resection-dependent repair, as already reported for G2-cells.27 Evidence for substantial resection in the G1-phase and lack of HR in this cell cycle stage suggests that MMEJ is a major repair choice of complex DSBs in G1-phase, especially for the subset of lesions repaired with slow kinetics (Fig. 3A). The fact that rejoined DSBs arising from ion-induced clustered DNA damage are often characterized by deletions and flanking microhomologies39 supports this notion.
Charged particles such as protons and carbon ions are nowadays used in several radiotherapy centers for treatment of many solid tumors. In this therapy the normal tissue is exposed to fast ions, similar to X-rays, while in the target volume – the tumor – the slow, densely ionizing ions induce complex DNA damage.43 Our results suggest that these differential effects can be amplified by targeting the resection pathway as reduced resection not only hampers HR in the S/G2-phase but also inhibits resection dependent repair in the G1-phase in cancer cells (exposed to high-LET), but not by normal tissue cells (exposed to low-LET; see Fig. 1B). As a result, targeting the resection pathway can enhance the therapeutic window between normal tissue effects and tumor cell killing in particle therapy.
Materials and Methods
Cells, cell culture, siRNA transfection, and survival assay
The human osteosarcoma cell line U2-OS (ATCC) and the murine cell lines MEF wild-type, MEF H2AX −/−44, and MEF Ku80 −/−45 were grown in DMEM with 10% fetal calf serum (FCS). U2-OS ATR-GFP cells46 were cultured in DMEM with 10% FCS and 1 μg/ml Puromycin. U2-OS cells expressing the GFP-tagged phosphorylation dependent subcellular localization control domain of helicase B (GE Healthcare) were grown in DMEM with 10% FCS and 500 μg/ml G418. NFFhTERT cells, immortalized human fibroblasts, were cultivated in DMEM with 15% FCS, while normal human fibroblasts AG1522D (passage 10–16) were grown in EMEM with 10% FCS, 1% Glutamine. All cell lines were kept in a humidified incubator at 37°C/5% CO2. For clonogenic survival experiments the cells were seeded in 25 cm2 culture flasks and methylene blue stained after 10 days of incubation. For gene knockdowns in U2-OS and U2-OS ATR-GFP the cells were transfected with siRNA (Eurofins MWG/Operon) using Interferin (Polyplus-transfection) according to the manufacturer's protocol. siRNA transfection in NFFhTERT was performed with HiPerfect (Qiagen) according to the manufacturer's instruction. For siRNA mediated gene knockdowns the end-concentration of siRNA was 10 nM. The incubation times with siRNA were 48 h (CtIP and EXO1 siRNA) and 72 h (MRE11, EXO1, and CtIP siRNA) before irradiation. CtIP siRNA transfection in cells seeded for survival assays was performed 48 h and 6 h before irradiation and 96 h after irradiation. The sequence of EXO1 siRNA was CCACCUAGGACGAGAAAUAdTdT, The CtIP and MRE11 siRNA sequence were described earlier.29,47 Successful gene knockdown was ensured by Western blot analyses (SI Fig. S6).
Irradiation
Cells were exposed to X-rays (X-ray tube IV320-13, Seifert, Germany; 250 keV, 16 mA; LET 2 keV/μm) or heavy ions at the GSI Helmholtz Center for Heavy Ion Research (Darmstadt, Germany). Irradiations at the UNILAC linear accelerator were performed with ions at a primary energy of 11.4 MeV/nucleon and a fluence of 3 × 106 particles/cm2: 238U ions (LET 15,000 keV/μm), 207Pb ions (LET 13,500 keV/μm), 197Au ions (LET 13,000 keV/μm), 119Sn ions (LET 7,880 keV/μm), 59Ni (LET 3,430 keV/μm), 48Ti (LET 2,180 keV/μm), 14N ions (LET 400 keV/μm), and 12C ions (LET 170 keV/μm). Further experiments with 12C (100 MeV/nucleon, 90 keV/μm), 40Ca (186 MeV/nucleon, 200 keV/μm), or 59Ni ions (265 MeV/nucleon, 350 keV/μm) were carried out at the SIS beam line. Low angle irradiation at the UNILAC was as described before.48 The selection of ions available is limited; hence some ion data are from single experiments (as indicated).
Immunostaining
Cells were fixed in 2% formaldehyde and permeabilized as described earlier.49 For immunofluorescence staining primary antibodies were diluted in 1× PBS, 0.4% BSA: αCENP-F (rabbit, Novus, NB500-101, 1:750), αCtIP (rabbit, Bethyl Laboratories, A300–488A, 1:100), αRPA/p34 (mouse, clone 9H8, Thermo Scientific, MS-691-P1, 1:300), αphospho-RPA32 (S4/S8) (rabbit, Bethyl Laboratories, A300–345A, 1:200), αγH2AX (Ser139) clone JBW301 (mouse, Millipore, 05–636, 1:500), αRAD51 (rabbit, Oncogene, PC130, 1:500). Secondary Alexa 488- and Alexa 568-conjugated goat αmouse and αrabbit antibodies (Invitrogen, A11017, A11070, A11019, A21069) were used (1:400). DNA was counterstained with DAPI (Oncor, S1335-4, 1 μg/ml).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank O. Becherel (Radiation Biology and Oncology Laboratory, Queensland Institute of Medical Research, Brisbane, Australia) for NFFhTERT cells, J. Bartek and C. Lukas for U2-OS ATR-GFP cells, and A. Nussenzweig for MEF H2AX −/− and Ku80 −/− cells, A. L. Leifke and G. Becker for excellent technical assistance, and the biophysics accelerator team for the great support during beam times. This work was partially funded by the German Federal Ministry of Education and Research [02NUK001A] and the Beilstein Institute, Frankfurt/Main, Germany (NanoBiC collaboration).
Supplemental Materials
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Goodhead DT, Fifth Warren K. Sinclair keynote address: issues in quantifying the effects of low-level radiation. HealthPhys 2009; 97:394–406; PMID: 19820449; http://dx.doi.org/10.1097/HP.0b013e3181ae8acf [DOI] [PubMed] [Google Scholar]
- 2. Asaithamby A, Hu B, Chen DJ. Unrepaired clustered DNA lesions induce chromosome breakage in human cells. Proc Natl Acad SciUSA 2011; 108:8293–8; PMID: 21527720; http://dx.doi.org/10.1073/pnas.1016045108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Goodarzi AA, Jeggo PA. The repair and signaling responses to DNA double-strand breaks. AdvGenet 2013; 82:1–45; PMID: 23721719; http://dx.doi.org/10.1016/B978-0-12-407676-1.00001-9 [DOI] [PubMed] [Google Scholar]
- 4. Mao Z, Bozzella M, Seluanov A, Gorbunova V. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. CellCycle 2008; 7:2902–6; PMID: 18769152; http://dx.doi.org/10.4161/cc.7.18.6679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beucher A, Birraux J, Tchouandong L, Barton O, Shibata A, Conrad S, Goodarzi AA, Krempler A, Jeggo PA, Lobrich M.. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EmboJ 2009; 28:3413–27; PMID: 19779458; http://dx.doi.org/10.1038/emboj.2009.276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mladenov E, Iliakis G. Induction and repair of DNA double strand breaks: the increasing spectrum of non-homologous end joining pathways. MutatRes 2011; 711:61–72; PMID: 21329706; http://dx.doi.org/10.1016/j.mrfmmm.2011.02.005 [DOI] [PubMed] [Google Scholar]
- 7. Rahal EA, Henricksen LA, Li Y, Williams RS, Tainer JA, Dixon K. ATM regulates Mre11-dependent DNA end-degradation and microhomology-mediated end joining. CellCycle 2010; 9:2866–77; PMID: 20647759; http://dx.doi.org/10.4161/cc.9.14.12363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rass E, Grabarz A, Plo I, Gautier J, Bertrand P, Lopez BS. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat Struct MolBiol 2009; 16:819–24; PMID: 19633668; http://dx.doi.org/10.1038/nsmb.1641 [DOI] [PubMed] [Google Scholar]
- 9. Xie A, Kwok A, Scully R. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nat Struct MolBiol 2009; 16:814–8; PMID: 19633669; http://dx.doi.org/10.1038/nsmb.1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bennardo N, Cheng A, Huang N, Stark JM. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoSGenet 2008; 4:e1000110; PMID: 18584027; http://dx.doi.org/10.1371/journal.pgen.1000110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang Y, Jasin M. An essential role for CtIP in chromosomal translocation formation through an alternative end-joining pathway. Nat Struct MolBiol 2011; 18:80–4; PMID: 21131978; http://dx.doi.org/10.1038/nsmb.1940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Truong LN, Li Y, Shi LZ, Hwang PY, He J, Wang H, Razavian N, Berns MW, Wu X.. Microhomology-mediated end joining and homologous recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc Natl Acad SciUSA 2013; 110:7720–5; PMID: 23610439; http://dx.doi.org/10.1073/pnas.1213431110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mladenov E, Magin S, Soni A, Iliakis G. DNA double-strand break repair as determinant of cellular radiosensitivity to killing and target in radiation therapy. FrontOncol 2013; 3:113; PMID: 23675572; http://dx.doi.org/10.3389/fonc.2013.00113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Durante M, Bedford JS, Chen DJ, Conrad S, Cornforth MN, Natarajan AT, Gent DC, Obe G.. From DNA damage to chromosome aberrations: joining the break. MutatRes 2013; 756:5–13; PMID: 23707699; http://dx.doi.org/10.1016/j.mrgentox.2013.05.014 [DOI] [PubMed] [Google Scholar]
- 15. You Z, Bailis JM. DNA damage and decisions: CtIP coordinates DNA repair and cell cycle checkpoints. Trends CellBiol 2010; 20:402–9; PMID: 20444606; http://dx.doi.org/10.1016/j.tcb.2010.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Niu H, Raynard S, Sung P. Multiplicity of DNA end resection machineries in chromosome break repair. GenesDev 2009; 23:1481–6; PMID: 19571177; http://dx.doi.org/10.1101/gad.1824209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Quennet V, Beucher A, Barton O, Takeda S, Lobrich M. CtIP and MRN promote non-homologous end-joining of etoposide-induced DNA double-strand breaks in G1. Nucleic AcidsRes 2011; 39:2144–52; PMID: 21087997; http://dx.doi.org/10.1093/nar/gkq1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yajima H, Fujisawa H, Nakajima NI, Hirakawa H, Jeggo PA, Okayasu R, Fujimori A.. The complexity of DNA double strand breaks is a critical factor enhancing end-resection. DNA Repair (Amst) 2013 [DOI] [PubMed] [Google Scholar]
- 19. Yun MH, Hiom K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature 2009; 459:460–3; PMID: 19357644; http://dx.doi.org/10.1038/nature07955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yu X, Chen J. DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol CellBiol 2004; 24:9478–86; PMID: 15485915; http://dx.doi.org/10.1128/MCB.24.21.9478-9486.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li S, Ting NS, Zheng L, Chen PL, Ziv Y, Shiloh Y, Lee EY, Lee WH.. Functional link of BRCA1 and ataxia telangiectasia gene product in DNA damage response. Nature 2000; 406:210–5; PMID: 10910365; http://dx.doi.org/10.1038/35018134 [DOI] [PubMed] [Google Scholar]
- 22. Peterson SE, Li Y, Wu-Baer F, Chait BT, Baer R, Yan H, Gottesman ME, Gautier J.. Activation of DSB processing requires phosphorylation of CtIP by ATR. MolCell 2013; 49:657–67; PMID: 23273981; http://dx.doi.org/10.1016/j.molcel.2012.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bolderson E, Tomimatsu N, Richard DJ, Boucher D, Kumar R, Pandita TK, Burma S, Khanna KK.. Phosphorylation of exo1 modulates homologous recombination repair of DNA double-strand breaks. Nucleic AcidsRes 2010; 38:1821–31; PMID: 20019063; http://dx.doi.org/10.1093/nar/gkp1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Helmink BA, Tubbs AT, Dorsett Y, Bednarski JJ, Walker LM, Feng Z, Sharma GG, McKinnon PJ, Zhang J, Bassing CH, et al.. H2AX prevents CtIP-mediated DNA end resection and aberrant repair in G1-phase lymphocytes. Nature 2011; 469:245–9; PMID: 21160476; http://dx.doi.org/10.1038/nature09585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shao Z, Davis AJ, Fattah KR, So S, Sun J, Lee KJ, Harrison L, Yang J, Chen DJ.. Persistently bound Ku at DNA ends attenuates DNA end resection and homologous recombination. DNA Repair (Amst) 2012; 11:310–6; PMID: 22265216; http://dx.doi.org/10.1016/j.dnarep.2011.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tomimatsu N, Mukherjee B, Deland K, Kurimasa A, Bolderson E, Khanna KK, Burma S.. Exo1 plays a major role in DNA end resection in humans and influences double-strand break repair and damage signaling decisions. DNA Repair (Amst) 2012; 11:441–8; PMID: 22326273; http://dx.doi.org/10.1016/j.dnarep.2012.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shibata A, Conrad S, Birraux J, Geuting V, Barton O, Ismail A, Kakarougkas A, Meek K, Taucher-Scholz G, Lobrich M, et al. Factors determining DNA double-strand break repair pathway choice in G2 phase. EmboJ 2011; 30:1079–92; PMID: 21317870; http://dx.doi.org/10.1038/emboj.2011.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Raderschall E, Golub EI, Haaf T. Nuclear foci of mammalian recombination proteins are located at single-stranded DNA regions formed after DNA damage. Proc Natl Acad SciUSA 1999; 96:1921–6; PMID: 10051570; http://dx.doi.org/10.1073/pnas.96.5.1921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP.. Human CtIP promotes DNA end resection. Nature 2007; 450:509–14; PMID: 17965729; http://dx.doi.org/10.1038/nature06337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. You Z, Shi LZ, Zhu Q, Wu P, Zhang YW, Basilio A, Tonnu N, Verma IM, Berns MW, Hunter T.. CtIP links DNA double-strand break sensing to resection. MolCell 2009; 36:954–69; PMID: 20064462; http://dx.doi.org/10.1016/j.molcel.2009.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lengsfeld BM, Rattray AJ, Bhaskara V, Ghirlando R, Paull TT. Sae2 is an endonuclease that processes hairpin DNA cooperatively with the Mre11/Rad50/Xrs2 complex. MolCell 2007; 28:638–51; PMID: 18042458; http://dx.doi.org/10.1016/j.molcel.2007.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang H, Shi LZ, Wong CC, Han X, Hwang PY, Truong LN, Zhu Q, Shao Z, Chen DJ, Berns MW, et al.. The interaction of CtIP and Nbs1 connects CDK and ATM to regulate HR-mediated double-strand break repair. PLoSGenet 2013; 9:e1003277; PMID: 23468639; http://dx.doi.org/10.1371/journal.pgen.1003277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen L, Nievera CJ, Lee AY, Wu X. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J BiolChem 2008; 283:7713–20; PMID: 18171670; http://dx.doi.org/10.1074/jbc.M710245200 [DOI] [PubMed] [Google Scholar]
- 34. Saha J, Wang M, Cucinotta FA. Investigation of switch from ATM to ATR signaling at the sites of DNA damage induced by low and high LET radiation. DNA Repair (Amst) 2013; 12:1143-51. [DOI] [PubMed] [Google Scholar]
- 35. Tommasino F, Friedrich T, Scholz U, Taucher-Scholz G, Durante M, Scholz M. A DNA double- strand break kinetic rejoining model based on the local effect model. RadiatRes 2013; 180:524-38. [DOI] [PubMed] [Google Scholar]
- 36. Serrano L, Liang L, Chang Y, Deng L, Maulion C, Nguyen S, Tischfield JA.. Homologous recombination conserves DNA sequence integrity throughout the cell cycle in embryonic stem cells. Stem CellsDev 2011; 20:363–74; PMID: 20491544; http://dx.doi.org/10.1089/scd.2010.0159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jeggo PA, Geuting V, Lobrich M. The role of homologous recombination in radiation-induced double-strand break repair. RadiotherOncol 2011; 101:7–12; PMID: 21737170; http://dx.doi.org/10.1016/j.radonc.2011.06.019 [DOI] [PubMed] [Google Scholar]
- 38. Zafar F, Seidler SB, Kronenberg A, Schild D, Wiese C. Homologous recombination contributes to the repair of DNA double-strand breaks induced by high-energy iron ions. RadiatRes 2010; 173:27–39; PMID: 20041757; http://dx.doi.org/10.1667/RR1910.1 [DOI] [PubMed] [Google Scholar]
- 39. Singleton BK, Griffin CS, Thacker J. Clustered DNA damage leads to complex genetic changes in irradiated human cells. CancerRes 2002; 62:6263–9; PMID: 12414656 [PubMed] [Google Scholar]
- 40. Sanz MM, Proytcheva M, Ellis NA, Holloman WK, German J. BLM, the Bloom’s syndrome protein, varies during the cell cycle in its amount, distribution, and co-localization with other nuclear proteins. Cytogenet CellGenet 2000; 91:217–23; PMID: 11173860; http://dx.doi.org/10.1159/000056848 [DOI] [PubMed] [Google Scholar]
- 41. Dutertre S, Ababou M, Onclercq R, Delic J, Chatton B, Jaulin C, Amor-Gueret M.. Cell cycle regulation of the endogenous wild type Bloom’s syndrome DNA helicase. Oncogene 2000; 19:2731–8; PMID: 10851073; http://dx.doi.org/10.1038/sj.onc.1203595 [DOI] [PubMed] [Google Scholar]
- 42. Chen X, Niu H, Chung WH, Zhu Z, Papusha A, Shim EY, Lee SE, Sung P, Ira G.. Cell cycle regulation of DNA double-strand break end resection by Cdk1-dependent Dna2 phosphorylation. Nat Struct MolBiol 2011; 18:1015–9; PMID: 21841787; http://dx.doi.org/10.1038/nsmb.2105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Loeffler JS, Durante M. Charged particle therapy–optimization, challenges and future directions. Nat Rev ClinOncol 2013; 10:411–24; PMID: 23689752; http://dx.doi.org/10.1038/nrclinonc.2013.79 [DOI] [PubMed] [Google Scholar]
- 44. Petersen S, Casellas R, Reina-San-Martin B, Chen HT, Difilippantonio MJ, Wilson PC, Hanitsch L, Celeste A, Muramatsu M, Pilch DR,. et al. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature 2001; 414:660–5; PMID: 11740565; http://dx.doi.org/10.1038/414660a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nussenzweig A, Chen C, da Costa Soares V, Sanchez M, Sokol K, Nussenzweig MC, Li GC.. Requirement for Ku80 in growth and immunoglobulin V(D)J recombination. Nature 1996; 382:551–5; PMID: 8700231; http://dx.doi.org/10.1038/382551a0 [DOI] [PubMed] [Google Scholar]
- 46. Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, Jackson SP.. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat CellBiol 2006; 8:37–45; PMID: 16327781; http://dx.doi.org/10.1038/ncb1337 [DOI] [PubMed] [Google Scholar]
- 47. Yuan J, Chen J. MRE11-RAD50-NBS1 complex dictates DNA repair independent of H2AX. J BiolChem 2010; 285:1097–104; PMID: 19910469; http://dx.doi.org/10.1074/jbc.M109.078436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jakob B, Splinter J, Taucher-Scholz G. Positional stability of damaged chromatin domains along radiation tracks in mammalian cells. RadiatRes 2009; 171:405–18; PMID: 19397441; http://dx.doi.org/10.1667/RR1520.1 [DOI] [PubMed] [Google Scholar]
- 49. Jakob B, Splinter J, Durante M, Taucher-Scholz G. Live cell microscopy analysis of radiation-induced DNA double-strand break motion. Proc Natl Acad SciUSA 2009; 106:3172–7; PMID: 19221031; http://dx.doi.org/10.1073/pnas.0810987106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lobrich M, Shibata A, Beucher A, Fisher A, Ensminger M, Goodarzi AA, Barton O, Jeggo PA.. gammaH2AX foci analysis for monitoring DNA double-strand break repair: strengths, limitations and optimization. CellCycle 2010; 9:662–9; PMID: 20139725; http://dx.doi.org/10.4161/cc.9.4.10764 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.