

Abstract
Abnormal proliferation is one characteristic of cancer-associated fibroblasts (CAFs), which play a key role in tumorigenesis and tumor progression. Oxidative stress (OS) is the root cause of CAFs abnormal proliferation. ATM (ataxia-telangiectasia mutated protein kinase), an important redox sensor, is involved in DNA damage response and cellular homeostasis. Whether and how oxidized ATM regulating CAFs proliferation remains unclear. In this study, we show that there is a high level of oxidized ATM in breast CAFs in the absence of double-strand breaks (DSBs) and that oxidized ATM plays a critical role in CAFs proliferation. The effect of oxidized ATM on CAFs proliferation is mediated by its regulation of cellular redox balance and the activity of the ERK, PI3K-AKT, and Wnt signaling pathways. Treating cells with antioxidant N-acetyl-cysteine (NAC) partially rescues the proliferation defect of the breast CAFs caused by ATM deficiency. Administrating cells with individual or a combination of specific inhibitors of the ERK, PI3K-AKT, and Wnt signaling pathways mimics the effect of ATM deficiency on breast CAF proliferation. This is mainly ascribed to the β-catenin suppression and down-regulation of c-Myc, thus further leading to the decreased cyclinD1, cyclinE, and E2F1 expression and the enhanced p21Cip1 level. Our results reveal an important role of oxidized ATM in the regulation of the abnormal proliferation of breast CAFs. Oxidized ATM could serve as a potential target for treating breast cancer.
Keywords: abnormal proliferation, breast cancer, cancer-associated fibroblasts, oxidized ATM, oxidative stress, proliferation signaling pathways, reactive oxygen species, redox homeostasis
Abbreviations
- CAFs
cancer associated fibroblasts
- ATM
ataxia telangiectasia mutated
- OS
oxidative stress
- ROS
reactive oxygen species
- DSBs
double strand breaks
- NAC
N-acetyl-cysteine
- TM
tumor microenvironment
- NFs
normal fibroblasts
- CDK1
cyclin-dependent kinase 1
- E2F1
E2F transcription factor 1
- CCNA2
cyclin A2
- CCNB2
cyclin B2
- CDKN2B
cyclin-dependent kinase inhibitor 2B
Introduction
Cancer-associated fibroblasts (CAFs) are the most predominant stromal cells residing in the breast tumor microenvironment (TM).1 CAFs play a key role in tumor growth, angiogenesis, invasion, metastasis, inflammation, and metabolism remodeling via cross-talk with cancer cells through paracrine signals.2-4 Our previous work has shown that breast cancer-derived CAFs have a higher proliferation rate compared with normal fibroblasts (NFs).3 Oxidative stress (OS) in TM mediates the conversion of CAFs from various cell types such as NFs and mesenchymal stem cells.2 These work indicates that OS contributes to CAFs abnormal proliferation. Accordingly, a series of cell cycle regulated signaling pathways (e.g., the PI3K pathway) are highly activated in breast cancer-derived CAFs relative to paired NFs.3,5 Recently, OS was shown to be responsible for the activation of proliferation-related signaling pathways in breast CAFs through promoting autophagy-dependent degradation of caveolin1,6 which is one member of the membrane-bound scaffolding protein family and functions as a tumor suppressor by negatively regulating signal transduction, including the WNT and ERK pathways.7,8 However, the molecular mechanisms by which OS accelerates CAFs proliferation are still obscure.
ATM protein kinase plays a critical role in the regulation of DNA damage response and cellular homeostasis. Null mutations of the ATM kinase gene lead to the ataxia-telangiectasia syndrome (A-T).9 Accumulating evidence has shown that ATM associates with cell proliferation. Growth retardation, premature aging, and progressive neurodegeneration are hallmarks of the A-T.9 In addition, various cell types from ATM−/− mouse including neural stem cells, astrocytes, and haematopoietic stem cells have defective proliferation.10-12 These phenotypes of the A-T can't be easily explained by the defective response to physiological double strand breaks (DSBs) in ATM-null cells. This indicates that ATM kinase could be activated in the absence of DSBs and have other functions. Indeed, ATM can be oxidized in cytoplasm independent of DSBs13 and works as a redox sensor.14 Oxidized ATM is required for the full activation of the PI3K-AKT pathway via phosphorylating AKT at Ser473.15 Furthermore, oxidized ATM maintains intracellular redox homeostasis by phosphorylating P53 at Ser 15.16 ATM deficiency results in proliferation defect of astrocytes by increasing cellular ROS levels, which can be partially rescued by N-acetyl-cysteine (NAC),10 suggesting that oxidized ATM may phosphorylate some pathways controlling cell proliferation. Our previous work found that the ATM signaling is activated in breast CAFs.3 But how oxidized ATM regulating the proliferation of CAFs remains unclear.
Here, we show that oxidized ATM promotes breast CAFs proliferation. ATM deficiency leads to increased ROS levels and proliferation defect in breast CAFs. Defective proliferation of breast CAFs is only partially rescued by the antioxidant NAC. Of note, proliferation inhibition of breast CAFs by the ATM kinase specific inhibitor KU60019 or siRNA is associated with the inactivation of ERK, PI3K-AKT and Wnt pathways, 3 major cellular proliferation regulation pathways. ATM deficiency or blockage of ERK, PI3K-AKT and Wnt signaling lead to downregulation of β-catenin and transcriptional inhibition of c-Myc. Accordingly, that further results in down-regulation of the c-Myc dependent cyclin E, cyclin D1 and E2F1 expression, and activation of the p21Cip1. Thus, the role of oxidized ATM in controlling CAFs proliferation is achieved by maintaining cellular redox homeostasis and activating important cellular proliferation associated signaling pathways.
Results
CAFs have a strong proliferation potential
We previously reported that the activated CAFs have a higher proliferation rate than NFs.2,5 Indeed, after analyzing our previous mRNA microarray data of paired breast NFs and CAFs, some cell cycle-related genes such as E2F1, CCNB1, and CCNA2 were up-regulated in CAFs compared with NFs (Table 2), while some other cell cycle-related genes such as CDKN2B and SFN were down-regulated (Table 3). To verify these results, CDK1, E2F1, CCNA2, and CCNB2 were assessed in 3 pair of primary NFs and CAFs derived from breast cancer patients by qRT-PCR and a consistent expression pattern was acquired with that of mRNA microarray data (Fig. 1D).
Table 2.
Cell cycle related genes up-regulated in CAFs compared with NFs
| Agilent probe set | Gene symbol | Fold difference (CAFs/NFs) | GenBank no. | Gene name |
|---|---|---|---|---|
| A_23_P259586 | TTK | 8.9005 | NM_003318 | TTK protein kinase |
| A_23_P58321 | CCNA2 | 6.0332 | NM_001237 | cyclin A2 |
| A_23_P118174 | PLK1 | 5.5017 | NM_005030 | polo-like kinase 1 (Drosophila) |
| A_23_P138507 | CDK1 | 5.0987 | NM_001786 | cyclin-dependent kinase 1 |
| A_23_P124417 | BUB1 | 4.7619 | NM_004336 | budding uninhibited by benzimidazoles 1 homolog |
| A_23_P65757 | CCNB2 | 4.3616 | AK023404 | cyclin B2 |
| A_24_P313504 | PLK1 | 4.2402 | NM_005030 | polo-like kinase 1 (Drosophila) |
| A_23_P163481 | BUB1B | 3.7153 | NM_001211 | budding uninhibited by benzimidazoles 1 homolog β |
| A_23_P70249 | CDC25C | 3.6697 | NM_001790 | cell division cycle 25 homolog C (S. pombe) |
| A_23_P122197 | CCNB1 | 3.4454 | NM_031966 | cyclin B1 |
| A_23_P57379 | CDC45L | 3.3416 | NM_003504 | CDC45 cell division cycle 45-like (S. cerevisiae) |
| A_23_P92441 | MAD2L1 | 3.105 | NM_002358 | MAD2 mitotic arrest deficient-like 1 |
| A_23_P80032 | E2F1 | 3.0811 | NM_005225 | E2F transcription factor 1 |
| A_23_P7636 | PTTG1 | 3.0429 | NM_004219 | pituitary tumor-transforming 1 |
| A_23_P18579 | PTTG2 | 2.9614 | NM_006607 | pituitary tumor-transforming 2 |
| A_23_P408955 | E2F2 | 2.9364 | NM_004091 | E2F transcription factor 2 |
| A_23_P149200 | CDC20 | 2.7836 | NM_152623 | cell division cycle 20 homolog (S. cerevisiae) |
| A_24_P105102 | PKMYT1 | 2.6186 | NM_182687 | protein kinase, membrane associated tyrosine/threonine 1 |
| A_23_P148807 | CDC7 | 2.5734 | NM_003503 | cell division cycle 7 homolog (S. cerevisiae) |
| A_23_P254612 | DBF4 | 2.4171 | NM_006716 | DBF4 homolog (S. cerevisiae) |
| A_23_P116123 | CHEK1 | 2.3042 | NM_001274 | CHK1 checkpoint homolog (S. pombe) |
| A_23_P357365 | STAG1 | 2.232 | NM_005862 | stromal antigen 1 |
| A_23_P49972 | CDC6 | 2.22 | NM_001254 | cell division cycle 6 homolog (S. cerevisiae) |
| A_23_P100344 | ORC6L | 2.1264 | NM_014321 | origin recognition complex, subunit 6 like (yeast) |
| A_24_P66125 | STAG2 | 2.0243 | NM_001042749 | stromal antigen 2 |
| A_23_P138631 | SMC3 | 2.0242 | NM_005445 | structural maintenance of chromosomes 3 |
| A_23_P90612 | MCM6 | 1.9995 | NM_005915 | minichromosome maintenance complex component 6 |
| A_23_P132277 | MCM5 | 1.9615 | NM_006739 | minichromosome maintenance complex component 5 |
| A_23_P35916 | ATM | 1.9061 | NM_000051 | ataxia telangiectasia mutated |
| A_23_P93690 | MCM7 | 1.8546 | NM_182776 | minichromosome maintenance complex component 7 |
| A_32_P103633 | MCM2 | 1.8195 | NM_004526 | minichromosome maintenance complex component 2 |
Abbreviations: CAFs, carcinoma-associated fibroblasts; NA, not annotated; NFs, normal fibroblasts.
Table 3.
Cell cycle related genes down-regulated in CAFs compared with NFs
| Agilent probe set | Gene symbol | Fold difference(CAFs/NFs) | GenBank no. | Gene name |
|---|---|---|---|---|
| A_24_P360674 | CDKN2B | 2.4261 | NM_078487 | cyclin-dependent kinase inhibitor 2B (p15, inhibits CDK4) |
| A_23_P63254 | SFN | 2.4041 | NM_006142 | stratifin |
Abbreviations: CAFs, carcinoma-associated fibroblasts; NA, not annotated; NFs, normal fibroblasts.
Figure 1.

A strong proliferation potential of breast CAFs. (A) Growth curves of the paired immortalized NFs and CAFs were performed with MTT assay at designed time points. (*, P < 0.05). (B) Phase contrast photomicrographs of NFs and CAFs after incubation for 3 d (upper panel) and 4 d (lower panel). Scale bars, 50 μm. (C) The cell ratio in S-phase was analyzed using the flow cytometry. (*, P < 0.05). (D) The dysregulated expression of ATM and cell cycle regulating genes was re-proved in 3 pairs of primary NFs and CAFs from breast cancer patients by qRT-PCR. (*, P < 0.05). (E) The levels of p-RB, cyclin D1, and c-Myc were determined by Western blot. (*, P < 0.05).
To expand the observation, the proliferation potential was further tested with breast cancer-derived NFs and CAFs immortalized by the human telomerase reverse transcriptase gene (hTERT). As indicated in Figure 1A and B, CAFs had a faster cell growth rate and higher densities than NFs, and more CAFs cells in S-phase were detected (Fig. 1C). Consistently, the protein levels of p-RB, c-Myc, and cyclin D1 in CAFs were higher than those in NFs (Fig. 1E). These data indcate that CAFs have a stronger proliferation capacity than NFs.
Oxidized ATM kinase is induced by oxidative stress independent of DSBs in CAFs
OS has been shown to mediate the transformation of NFs to CAFs,2,17 suggesting that OS may participate in CAFs proliferation. As a redox sensor, ATM protein kinase could be activated in CAFs. As expected, mRNA expression levels of the ATM gene, which was found to be upregulated in CAFs by microarray analysis, was detected to be increased more than 2.0 folds in primary CAFs compared with NFs by qRT-PCR (Table 2 and Fig. 2A). To exclude its DSBs-dependent ATM activation in CAFs, the protein expression levels of total ATM, p-ATM (s1981), and γH2AX (a biomarker of DSBs) were measured using Western Blot, IF staining, and IHC. Interestingly, p-ATM (s1981) and total ATM protein levels were significantly increased in CAFs, whereas the γH2AX levels were similar between NFs and CAFs (Fig. 2B–C). This was confirmed by IHC staining in tumor tissues without chemo- and radio-therapy (Fig. 2D). In addition, the phosphorylated AKT (s473), a hallmark of ATM activation by oxidation,16 showed a higher level in CAFs than in NFs. The Akt phosphorylation was inhibited by the ATM inhibitor KU60019 in NFs and CAFs (Fig. 2E). These data suggest that the oxidized ATM, independent of DSBs, exists in breast CAFs.
Figure 2.

The activation of oxidized ATM independent of DSBs in CAFs. (A) The mRNA expression of the ATM gene was re-proved by qRT-PCR in CAFs and NFs derived from 3 breast cancer patients, and its relative fold changes in CAFs and NFs were presented. (*, P < 0.05). (B, C) The protein levels of total ATM, p-ATM (s1981), and γH2AX (s139) were analyzed by Western blot (B) and by immunofluorescence staining (C) in the immortalized NFs and CAFs. The relative fold changes of total ATM, p-ATM (s1981), and γH2AX (s139) in NFs and CAFs were displayed. Scale bars, 25 μm (*, P < 0.05). (D) The expression of p-ATM (s1981) and γH2AX (s139) in breast cancer specimens was analyzed by IHC staining. Scale bars, 50 μm. (E) The expression of p-ATM (s1981), ATM, p-AKT (s473), and AKT was determined by Western blot in NFs and CAFs in the presence or absence of KU60019 (5 μM) for 24 h.
To explore whether ATM is oxidized and activated by oxidative stress in a DSBs-independent manner in CAFs, CAFs were treated with antioxidant NAC and total ATM, p-ATM (1981), and γH2AX proteins were examined. Under the NAC treatment, the p-ATM (1981) protein levels were reduced around 80%, with no influence on the protein expression of total ATM and γH2AX in CAFs (Fig. 3A), implying that ATM is oxidized by oxidative stress without DSBs in CAFs. ROS can be produced at many intracellular sites, including mitochondria, NADPH oxidases, xanthine oxidase, and cytochrome P450.18,19 To determine the major sources of ROS in CAFs, we exposed CAFs to a series of ROS production inhibitors, including pyrrolidine dithiocarbamate (PDTC, a scavenger of H2O2 by maintaining cellular stores of reduced glutathione), diphenylene iodonium DPI (an inhibitor of electron transport in flavin-containing systems including NADPH oxidases and mitochondrial complex III), oxypurinol (xanthine oxidase inhibitor), NADPH (cell surface NADPH oxidase inhibitor), chloramphenicol (cytochrome P450 inhibitor), and rotenone (mitochondrial complex I inhibitor). DCF fluorescence staining showed that PDTC, DPI, and rotenone but not the other inhibitors specifically decreased the ROS levels in CAFs (Fig. 3B) and reduced the p-ATM (s1981) protein levels (Fig. 3C), suggesting that mitochondria-derived ROS are responsible for the ATM activation in CAFs. These data indicate that oxidative stress oxidizes and activates ATM kinase in a DSBs-independent manner in CAFs and that oxidized ATM may be involved in the abnormal proliferation of CAFs.
Figure 3.

Mitochondria-derived ROS activate the ATM kinase. (A) CAFs were treated with or without NAC (1 mM) for 24 h. Immunoblotting analyses were done with the indicated antibodies. (B) CAFs were treated with PDTC (20 μM), DPI (10 μM), Oxypurinol (100 μM), NADPH (1 mM), Chloramphenicol (300 μM), and Rotenone (1 μM) for 2 h. The intracellular ROS levels were measured in cultures (*, P < 0.05, CAFs treated with inhibitor vs control CAFs). (C) CAFs were treated as in B. Western blots was done with the indicated antibodies (*, P < 0.05, CAFs treated with inhibitor vs control CAFs).
Oxidized ATM promotes breast CAFs proliferation
It was recently reported that oxidized ATM can phosphorylate some of downstream substrates in response to various stimuli such as hypoxia and insulin, which are irrelevant to the DSBs repair process.15,20 To validate this speculation, we examined the influence of loss of functional ATM on CAFs proliferation. Indeed, the CAFs proliferation potential was dramatically slowed down as determined by cell growth curve (Fig. 4A), growth densities (Fig. 4B), and the percentage of S-phase cells in the cell cycle (Fig. 4C) after treatment of KU60019, an ATM specific inhibitor. This suggests an important role of ATM in CAFs proliferation. In addition, the effect of KU60019 on CAFs proliferation was confirmed by decreased p-RB and c-Myc protein expression using western blot assays (Fig. 4D). Similarly, the siRNA-mediated knockdown of the ATM gene led to suppressed proliferation of breast CAFs (Fig. 5). These results indicate that oxidized ATM promotes breast CAFs proliferation.
Figure 4.
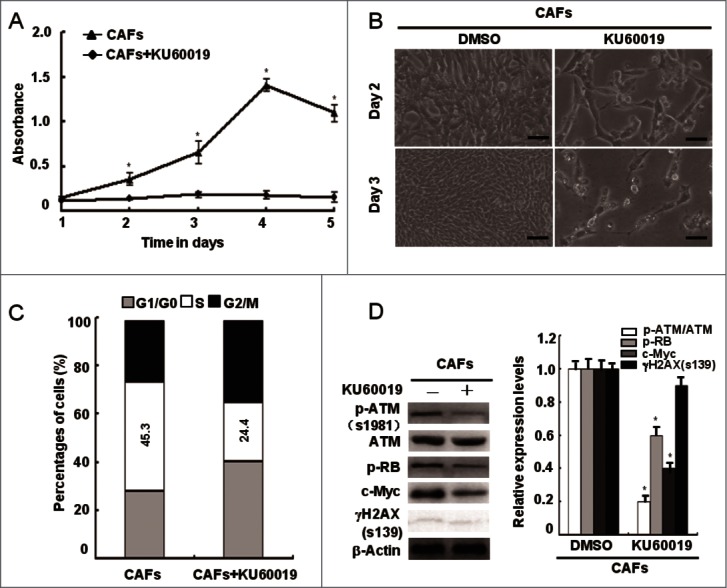
Loss of functional ATM by KU60019 inhibits the breast CAFs proliferation. (A) Growth curves of CAFs under treatment with or without KU60019 (5 μM) were analyzed with MTT assay for 5 d (*, P < 0.05). (B) Photomicrographs of CAFs in the presence or absence of KU60019 (5 μM) for 2 d or 3 d. Scale bars, 50 μm. (C) Flow cytometry analyses of CAFs with or without treatment of KU60019 (5 μM) for 24 h. (D) CAFs were treated with or without KU60019 (5 μM) for 24 h. Immunoblotting analyses were performed with the indicated antibodies. (*, P < 0.05).
Figure 5.

siRNA-mediated knockdown of ATM suppresses the breast CAFs proliferation. (A, B) The knockdown efficiency of the ATM gene by shRNA in CAFs was testified with qRT-PCR and Western Blot (*, P < 0.05, CAF-shGFP vs CAF-shATM). (C) Growth curves of CAF-shGFP and CAF-shATM were analyzed with the MTT assay for 5 d (*, P < 0.05, CAF-shGFP vs CAF-shATM at the designed time points). (D) The cell growth density of CAF-shGFP and CAF-shATM for 2 d or 3 d was displayed by Photomicrographs. Scale bars, 50 μm. (E) Flow cytometry analyses of CAF-shGFP and CAF-shATM. (F) Immunoblotting analyses were performed with the indicated antibodies in both CAF-shGFP and CAF-shATM (*, P < 0.05, CAF-shGFP vs CAF-shATM).
Maintaining of cellular redox homeostasis is not a leading function of oxidized ATM in promoting CAFs proliferation
ATM was reported to maintain intracellular redox homeostasis. OS caused by the deficiency of ATM is involved in defective proliferation.12 Thus, we asked whether ATM could promote CAFs proliferation by regulating intracellular redox homeostasis. To testify this hypothesis, we measured intracellular ROS levels in CAFs with or without treatment of KU60019 using the H2DCFDA probe. CAFs treated with KU60019 had an obvious higher level of intracellular ROS than the cells without KU60019 treatment, and the antioxidant NAC reversed the increased ROS levels in KU60019-treated CAFs to normal levels (Fig. 6A), indicating loss of functional ATM leads to oxidative stress in CAFs. The CAFs proliferation defect under loss of functional ATM, however, was partly rescued by NAC (Fig. 6B7ndash;C). The data are in line with our findings that NAC decreased the proportion of CAFs at G1 phase via partially restoring the proliferation related protein levels of p-RB, cyclin D1, and c-Myc in CAFs in the presence of KU60019 (Fig. 6D and E). These findings suggest that cellular redox homeostasis maintained by ATM is necessary for CAFs survival, but not the major source to promote CAFs proliferation. Other mechanisms associated with oxidized ATM may be involved in regulating the abnormal proliferation of CAFs.
Figure 6.

Maintenance of intracellular redox homeostasis by oxidized ATM plays a minor role in CAFs proliferation. (A) CAFs were pretreated with NAC (1 mM) for 2 h before treatment with KU60019 (5 μM) for 24 h. Fluorescent H2DCFDA levels were measured in these cells (*, P < 0.05 in comparing CAFs treated with or without KU60019); (#, P < 0.05 in comparing CAFs in the presence of KU60019 plus NAC-treated or NAC-untreated). (B) Growth curves of CAFs were measured by MTT assay at the indicated time points. The treatment of CAFs with KU60019 (5 μM) and NAC (1 mM) was as indicated. (*, P < 0.05, CAFs treated with KU60019+NAC vs CAFs with KU60019 only); (#, P < 0.05, CAFs treated with or without KU60019). (C) Photomicrographs of CAFs treated with KU60019 (5 μM) for 2 d in the presence or absence of NAC (1 μM). Scale bars, 50 μm. (D) Flow cytometry analyses of CAFs treated with KU60019 (5 μM) for 24 h in the presence or absence of NAC (1 μM). (E) CAFs was treated with KU60019 (5 μM) for 24 h in the presence of NAC (1 mM). Western blotting analyses were done with the indicated antibodies (*, P < 0.05, and #, P < 0.05 is similar with (B) indicated).
Oxidized ATM enhances CAFs proliferation through controlling the activity of key proliferation-related signaling pathways
There are several critical signaling pathways, including the PI3K-AKT, ERK, and Wnt signaling pathways, which are activated in breast CAFs as described in our previous work.3,21 These pathways respond to extracellular stimuli to regulate cellular proliferation.22,23 To determine whether the oxidized ATM in CAFs promotes cellular proliferation thro-ugh regulating these pathways, their activities were examined in CAFs treated with or without KU60019. As shown in Figure 7A, the PI3K-AKT, ERK, and Wnt signaling pathways were significantly inhibited by KU60019. Consistent with this finding, CAFs proliferation was also decreased as determined by MTT assays (Fig. 7B), cell densities (Fig. 7C), and flow cytometric analysis (Fig. 7D) after the cells were treated with LY294002 (the PI3K inhibitor), U0126 (MEK/ERK inhibitor), or XAV939 (β-catenin inhibitor) alone or in combination (Fig. 7E). Furthermore, the p-RB and c-Myc levels were significantly decreased by theses inhibitors in CAFs (Fig. 7F), which is consistent with the results on KU60019 (see Fig. 4D). These findings indicate that oxidized ATM promotes CAFs proliferation largely by activating the PI3K-AKT, ERK, and Wnt signaling pathways.
Figure 7.

(See previous page). Oxidized ATM enhances the CAFs proliferation through activating key proliferation-related signaling pathways. (A) CAFs were treated with KU60019 (5 μM) for the indicated time points. Immunoblotting analyses were performed with the indicated antibodies. Total histone 3 functions as a loading control for nuclear proteins. (B) CAFs were treated separately or jointly with LY294002 (20 μM), U0126 (25 μM), and XAV939 (10 μM) for the designed time. Their growth curves were determined by MTT assay (*, P < 0.05, untreated CAFs vs treated CAFs with indicated inhibitor(s)). (C) Photomicrographs of CAFs treated with or without inhibitor(s) as in B indicated for 2 d. Scale bars, 50 μm. (D) Flow cytometry analyses of CAFs treated with indicated inhibitor(s) for 24 h as in B. (E) CAFs were treated separately or jointly with LY294002 (20 μM), U0126 (25 μM), and XAV939 (10 μM) for 24 h. Immunoblotting analysis was done with the indicated antibodies. Total histone 3 functions as a loading control for nuclear proteins. (F) CAFs were treated separately or jointly with LY294002 (20 μM), U0126 (25 μM), and XAV939 (10 μM) for 24 h. The levels of p-RB, c-Myc, p21Cip1, and p16Ink4a were analyzed by western blot. (G) CAFs were treated with KU60019 (5 μM) for the indicated time. Immunoblotting analysis was performed with the indicated antibodies.
We next explore the effects of KU60019 on several key proteins (e.g., p21Cip1 and p16Ink4a) associated with cell proliferation in CAFs. KU60019 treatment stimulated the expression of p21Cip1, but not p16Ink4a in CAFs (Fig. 7G). Furthermore, p21Cip1 expression was upregulated by LY294002, U0126, or XAV939 (Fig. 7F).
Loss of functional ATM inhibits CAFs proliferation partially through suppressing β-catenin-regulated cell cycle genes
As a transcription factor, β-Catenin plays a critical role in promoting cell proliferation by upregulating a series of target genes, such as c-Myc, cyclin D1, cyclin E, and CDC25A.24,25 Glycogen synthase kinase 3β (GSK3β), an upstream negative regulator of β-Catenin, can be inactivated by AKT and ERK.26,27 Thus, we hypothesized ATM may promote CAFs proliferation in a GSK3β/β-Catenin dependent manner. To address this question, protein levels of p-GSK3β (Ser9), GSK3β, p-β-Catenin (Ser37), nuclear β-Catenin and total β-Catenin in CAFs were examined under KU60019 treatment. As shown in Figure 8A, ATM inhibition by KU60019 reduced p-GSK3β (Ser9), nuclear β-Catenin, and total β-Catenin levels and increased p-β-Catenin (Ser37) levels, while did not influence GSK3β levels in CAFs. To further determine the role of ERK and AKT on the GSK3β/β-Catenin axis, LY294002, U0126, and XAV939 were used. As expected, under the treatment with LY294002, U0126, and XAV939 alone or in combination, the decrease of p-GSK3β (Ser9), GSK3β, p-β-Catenin (Ser37), nuclear β-Catenin and total β-Catenin levels in CAFs (Fig. 8B) was similar to that under KU60019 treatment (shown in Fig. 8A). These results indicate that inhibition of ATM decreases CAFs proliferation by directly or indirectly reducing the β-Catenin levels.
Figure 8.

Loss of functional ATM leads to the proliferation defect in CAFs partially through down-regulating β-catenin and cell cycle regulation genes. (A) CAFs were treated with KU60019 (5 μM) for the indicated time. Immunoblotting analyses were performed to detect activity of GSK3β and β-Catenin. Total histone 3 functions as a loading control for nuclear proteins. (B) CAFs were treated separately or jointly with LY294002 (20 μM), U0126 (25 μM), and XAV939 (10 μM) for 24 h. Activated GSK3β and β-Catenin were determined using Immunoblotting analysis. Total histone 3 functions as a loading control for nuclear proteins. (C) CAFs were separately treated with or without inhibitors including LY294002 (20 μM), U0126 (25 μM), and XAV939 (10 μM) for 24 h. ChIP assay was done with an anti-β-catenin antibody for immunoprecipitation followed by PCR with c-MYC promoter-specific primers. (D) CAFs were separately treated with or without inhibitors including LY294002 (20 μM), U0126 (25 μM), and XAV939 (10 μM) for 24 h. Immunoblotting analysis was performed with the indicated antibodies. (E) The c-Myc gene in CAFs was knocked down by the shRNA, and the expression of c-Myc related genes was tested by Immunoblotting analysis with the indicated antibodies. β-Actin worked as loading control. (F) The growth curves of CAFs with or without c-Myc depletion was measured with the MTT assay (*, P < 0.05).
c-Myc plays a core role in promoting cell proliferation through enhancing some positive cell cycle regulators (e.g., cyclin D1, cyclin E, and E2F1) and antagonizing cell cycle negative regulators (e.g., p21Cip1).28 c-Myc expression is transcriptionally activated by nuclear β-Catenin,29 suggesting c-Myc may mediate the proliferation function of ATM in CAFs. After the treatment of CAFs by KU60019, LY294002, U0126, and XAV939, the binding of β-Catenin to the MYC promoter was blocked (Fig. 8C), indicating that oxidized ATM plays a positive role in β-Catenin-mediated c-Myc transcription. As expected, c-Myc knockdown by siRNA decreased the expression of cyclin D1, cyclin E, and E2F1, and increased the expression of p21Cip1 (Fig. 8E), leading to impaired CAFs proliferation (Fig. 8F). Furthermore, the expression levels of c-Myc and c-Myc-dependent critical cell cycle proteins were regulated by the ATM kinase and its downstream key proliferation controlling pathways in breast CAFs (Fig. 9A–D, and Fig. 8D). Herein, these data demonstrate that ATM deficiency indirectly blocks the β-Catenin transactivation, which results in a subsequent dysregulation of c-Myc dependent genes involved in CAFs proliferation.
Figure 9.

c-Myc mediates the functional ATM in promoting the breast CAFs proliferation. (A)The differential expression analysis of the cell cycle core regulators between NFs and CAFs was done by Western blot with the indicated antibodies. (B) The activity of key cellular proliferation regulating pathways between NFs and CAFs was determined using Western blot with the indicated antibodies. (C, D) Western blot analyzes the effect of ATM deficiency caused by KU60019 (5 μM for 24 h) on the protein expression levels of cell cycle-associated proteins (C) and the activity of key cell proliferation regulating pathways (D) in breast CAFs using the indicated antibodies. (E) A cartoon scheme is used to depict the c-Myc mediated function of oxidized ATM in promoting breast CAFs proliferation.
Discussion
Abnormal proliferation is one of CAFs typical features compared with normal fibroblasts.3,30 Oxidative stress (OS) is a critical inducer of CAFs abnormal proliferation.4,5,31 ATM, an important intracellular redox sensor, associates with cellular proliferation.15,32 However, the role of oxidized ATM in the abnormal proliferation of CAFs is not known. In the present study, oxidized ATM was found to be highly expressed in breast CAFs and to promote the proliferation of breast CAFs. Oxidized ATM promoted the proliferation of CAFs not only by maintaining cellular redox balance, but also via activating the activity of the PI3K-AKT, ERK, and Wnt signaling pathways. To our knowledge, this is the first report showing that oxidized ATM promotes the proliferation of CAFs mainly through increasing the activity of key proliferation-regulating signaling pathways.
The activated ATM exists independently of DSBs in breast cancer CAFs. Hitherto, it has been reported that ATM kinase can be activated via 2 different mechanisms and has corresponding downstream substrates to perform distinct biological functions.16 The classic mechanism is that ATM is activated by double strand breaks (DSBs) through interactions with the MRE11-RAD50-NBS1 (MRN) complex and mediates the DSBs repair response through activating signaling cascades via phosphorylating specific substrates such as histone H2AX phosphorylation at Ser139. Another mechanism is that ATM is activated by oxidation of the thiol group of its Cys 2991 independently of DSBs and phosphorylates distinct substrates involved in other cellular processes such as AKT phosphorylation at Ser473 for glucose metabolism. The auto-phosphorylation of ATM at Ser1981 is a hallmark of the activation of the ATM kinase by DSBs and oxidation. Histone H2AX phosphorylation at Ser139 and AKT phosphorylation at Ser473 are the specific biomarkers of the ATM activation by DSBs or oxidation respectively. Our results showed that the expression of p-ATM at Ser1981 was higher in CAFs than in NFs, indicating that the ATM kinase is activated in CAFs. The expression levels of H2AX phosphorylation at Ser139 are not changed in CAFs compared with NFs (Fig. 2B–D), whereas the expression of AKT phosphorylation at Ser473 is higher in CAFs than in NFs and all inhibited by the ATM specific inhibitor KU60019 (Fig. 2E). The above results show that oxidation and activation of ATM is elicited by OS without DSBs in breast CAFs.
Mitochondria-derived ROS oxidize and activate ATM protein kinase independent of DSB in breast CAFs. ATM protein kinase can be activated by DSBs in nucleus, but also directly by OS in cytoplasm.13,19 OS in CAFs may oxidize and activate ATM kinase in the cytoplasm of breast CAFs. In this current work, we demonstrated that both total ATM and p-ATM (s1981) mainly located in the cytoplasm of breast NFs and CAFs. It is well known that intracellular redox balance depends on the cellular ROS levels and antioxidant capacity and that ROS levels is strictly regulated in cells to achieve cellular redox homeostasis.18 Overproduction of ROS at different subcellular sites results in disruption of cellular redox status and subsequently OS.19 It is unknown that which source(s) of ROS oxidize the ATM kinase in breast CAFs. In the present study, we identified that ROS mainly derive from mitochondria and lead to OS-dependent oxidization and activation of the ATM kinase in breast CAFs.
Oxidized ATM promotes the proliferation of breast CAFs. Recently ATM has been found to can also be oxidized and activated by oxidative stress in cytoplasm in a DSBs-independent manner.13 These evidence suggests that ATM has functions out of nucleus. Oxidized ATM kinase associates with cell proliferation.10,12 For example, in hypoxia state, oxidized ATM suppresses the activity of mTORC1 through activating the HIF1 pathway by directly phosphorylating HIF1α at Ser696.20 In addition, after insulin treatment, oxidized ATM-dependent phosphorylation of AKT at Ser473 is required for the full activation of the PI3K/AKT signaling pathway,32 which positively regulates the activity of the mTORC1, acting as a chief controller for cell proliferation in response to extracellular cues such as hypoxia.16,23 These studies strongly suggest that oxidized ATM probably regulates the proliferation of CAFs. In this study, we demonstrated that loss of functional ATM by KU60019 treatment or by the siRNA technology led to severe proliferation defect in breast CAFs. To our knowledge, it is first to report that oxidized ATM promotes the proliferation of breast CAFs.
The antioxidant capacity of oxidized ATM plays a limited role in breast CAFs abnormal proliferation. Low levels of ROS promote cell proliferation, but high doses of ROS impair cell proliferation by triggering cell cycle arrest, senescence, and apoptosis.14,33 One important function of oxidized ATM is to maintaining intracellular redox balance by reducing the cellular ROS levels and enhancing the cellular antioxidative capacity.16,19 For example, oxidized ATM decreases the mitochondrial ROS production by suppressing oxidative phosphorylation in a LKB1/AMPK/TSC2/mTORC1 dependent manner after H2O2 treatment.34 However, the role of the cellular redox status maintaining function of oxidized ATM in the proliferation of CAFs is unknown. Unexpectedly, we observed that OS caused by ATM deficiency played a minor role in the abnormal proliferation rate of breast CAFs, indicating that oxidized ATM may regulate breast CAFs proliferation via other underlying mechanisms. This finding is supported by the Kim' study that OS is not the leading cause of proliferation defect in astrocytes with ATM deficiency.10 However, the antioxidant capacity of oxidized ATM plays a key role in the proliferation of haematopoietic stem cells.12 The reason for this discrepancy may indicate the cellular specificity of the antioxidant function of oxidized ATM in regulating cell proliferation.
Redox state regulates the proliferation of breast CAFs through regulating positive and negative key cell cycle regulators by balancing the activity of positive and negative proliferation regulation pathways. A moderate level promotes the cell proliferation by activating the positive proliferation controlling pathways such as the ATM and PI3K-AKT pathways, which promote cell proliferation via indirectly controlling positive key cell cycle regulators such as c-Myc.14 A high level of ROS can lead to adverse cellular events, for instance, proliferation arrest and autophagy, via the p53/p21Cip1 axis.14,18 The p53/p21Cip1 axis can arrest cell cycle by indirectly inactivating pRB and c-Myc.35,36 The degree of ROS in CAFs probably approach the high levels because the OS-activated breast CAFs have abnormal proliferation and harmful cellular events such as autophagy simultaneously.3,4 OS may activate the proliferation controlling signaling pathways such as the PI3K/AKT pathway and the p53 pathway in CAFs at the same time. Indeed, our previous publications support this speculation.3,21,37 This is also consistent with our results in this study (Fig. 6E). These evidence suggests that NAC treatment not only reduced the activity of the ATM signaling, which is necessary for the full activation of proliferation regulating pathways such as the PI3K/AKT pathway, but also decreased the activation of the p53 pathway (Fig. 6E). As a result, the net effect of NAC treatment may be that the activity of positive proliferation regulation pathways exceeds that of negative proliferation control pathways (Fig. 6E). KU60019 treatment-mediated ATM deficiency in CAFs not only decreased the activity of the positive proliferation regulation pathways such as the PI3K/AKT pathway, but also increased the activity of the p53 signaling (Fig. 6E) via disturbing the intracellular redox balance (Fig. 6A). Likewise, NAC treatment did not influence the activity of the ATM signaling in the presence of KU60019, but reduced the activation of the p53 pathway by alleviating ROS levels (Fig. 6A and E ). So, this may reasonably explain the contradictory phenomenon which is that NAC rescued the decreased expression of p-RB, cyclin D1 and c-Myc, in response to KU60019 treatment. Our results and other studies are not completely consistent. The p21Cip1 and p16INK4a CDK inhibitor are key negative cell cycle regulators. The p21Cip1 CDK inhibitor may be partially responsible for the proliferation arrest of neural stem cells induced by ATM deficiency.11 Whereas, the p16INK4a CDK inhibitor may in part contribute to the proliferation defect of ATM-deficient astrocytes and haematopoietic stem cells.10,12 This may result from different cell types and the residual activity of ATM in the presence of NAC and KU60019.
C-Myc mediates the role of ATM in promoting the proliferation of breast CAFs. C-Myc is a well-known transcription factor which promotes cell cycle progression through controlling the expression key cell cycle regulators.28 As far as we know, it is not reported that whether ATM regulates the expression of c-Myc. However, accumulative evidence implys that ATM may indirectly regulate the expression of c-Myc. First, c-Myc is located downstream of the ERK-MAPK, PI3K-AKT, and Wnt signaling pathways. These pathways are the chief mechanisms in response to extracellular stimuli to control cell proliferation.22,25 β-catenin, which is a core component of the Wnt pathway and functions as a transcription factor, plays an important role in regulating cell proliferation by trans-activating up-regulation of cell cycle genes, such as cyclin D, c-Myc, and cyclin E.22,25 Both the ERK and PI3K-AKT pathways can activate the β-catenin through inactivating the downstream GSK3β kinase by phosphorylating it at Ser9,23,38 which can phosphorylate β-Catenin at Ser33, Ser37, and Thr41 in cytoplasm, leading to its degradation via the way of ubiquitylation-mediated proteasome.27 Second, these pathways are active in breast CAFs.26,39,40 Finally, oxidized ATM can regulate the activity of many signaling pathways through phosphorylating specific substrates.16 However, whether oxidized ATM promotes CAFs abnormal proliferation in a c-Myc dependent manner by enhancing the activity of these pathways needs to be determined. In this study, we found that oxidized ATM promotes the proliferation of breast CAFs by increasing the expression of c-Myc through activating the ERK-MAPK, PI3K-AKT, and Wnt signaling pathways (Fig. 8 and Fig. 9A–D). Based on our results in the study, a model was proposed to depicting the role of c-Myc in mediating the oxidized ATM-regulated proliferation of breast CAFs (Fig. 9E).
Protein phosphorylation is a key post-translational modification (PTM) in signal transduction by modulating substrate activity, stability, interaction with other proteins, subcellular localization, and its additional PTMs. Phosphorylation and dephosphorylation can fine-tune the function of a kinase substrate. Oxidized ATM kinase may control the activity of the ERK, PI3K-AKT, and Wnt signaling pathways through phosphorylating the core components on these pathways. Indeed, oxidized ATM fully activates the PI3K-AKT signaling pathway via directly phosphorylating AKT at Ser473 after insulin treatment.32 The underlying mechanisms need to be investigated in our next study.
In summary, our findings demonstrate that oxidized ATM plays a key role in promoting the proliferation of breast CAFs through keeping intracellular redox homeostasis and activating the ERK, PI3K-AKT, and Wnt pathways. These findings provide important insights into the mechanisms by which oxidized ATM promotes the abnormal proliferation of CAFs and new targets for treating breast cancer.
Materials and Methods
Cell lines and cell culture
Paired immortalized NFs and CAFs cell lines derived from breast cancer have been established as described previously.37 These cells were routinely maintained in full DMEM (41965–062, Gibco) supplemented with 10% fetal bovine serum (100099–133, Gibco) at 37°C in humidified atmosphere containing 5% CO2.
MTT assay and flow cytometric analysis
The cell proliferation rate was determined by the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay. Cells were plated at 3 × 103 cells per well in 96-well plates. When reaching 60% confluence, cells were treated with KU60019 (S1570, Selleck Chemicals), LY294002 (S1105, Selleck Chemicals), U0126 (A1901, Beyotime), and XAV939 (S1180, Selleck Chemicals). Following the treatment, 10 μl MTT solution (C0009, Beyotime) (5 mg/ml in PBS) was added into each well and continually cultured for 4 h. Then the medium was carefully discarded, 200 μl DMSO was added into each well and shaked gently on a rotator for 10 min in dark at room temperature. The absorbance was readed on an ultraviolet spectrophotometric reader at a wave-length of 492 nm (BioteK). The experiments were repeated 3 times.
The cell cycle S-phase was analyzed by flow cytometry. Cells were processed by standard methods using propidium iodide staining of DNA as previously described.41 Briefly, after cells were treated under specific conditions when reaching 60% confluence, the cells were trypsinized, washed twice with cold PBS, fixed by 75% ethanol, incubated with RNAase and stained by propidium iodide (P4170, Sigma Aldrich). Tritiated thymidine uptake of cells was determined using a cytomics FC device (Beckman coulter). The experiments were performed in triplicate.
Cell growth density and cellular morphology
Cells were plated at 5 × 105 cells in 2 ml full DMEM per well in 6-well plates and cultured to reach 60% confluence. After treatments by specific reagents according to experimental design, the density and morphology of cells were observed by using phase contrast microscopy (Nikon, TE 2000-V) at designed time points.
Western blot analysis
Western blot assays were done as described previously.41 Briefly, total cell proteins were harvested in RIPA lysis buffer (P0013B, Beyotime), quantified using the BCA protein assay kit (P0012, Beyotime), separated in 10% SDS-PAGE gel, and subjected to protein gel blotting. The primary antibodies included monoclonal mouse p21Cip1 antibody (AP021, Beyotime, 1:500 dilution), polyclonal rabbit p16INK4a antibody (BS6431, Bioworld, 1:500 dilution), polyclonal rabbit p-RB (s795) antibody (BS6414, Bioworld, 1:500 dilution), monoclonal rabbit total histone 3 antibody (AH433, Beyotime, 1:500 dilution), monoclonal mouse β-Actin antibody (AA128, Beyotime, 1:1000 dilution), monoclonal rabbit p-ERK1/2 (T202/Y204) antibody (14474, Cell Signaling Technology Co (CST), 1:1000 dilution), monoclonal rabbit ERK1/2 antibody (4348, CST, 1:1000 dilution), monoclonal rabbit c-Myc antibody (5605, CST, 1:1000 dilution), monoclonal rabbit cyclin D1 antibody (2978, CST, 1:1000 dilution), monoclonal mouse cyclin E (4129, CST, 1:1000 dilution), polyclonal rabbit E2F1 antibody (3742, CST, 1:1000 dilution), monoclonal rabbit ATM antibody (2873, CST, 1:1000 dilution), monoclonal rabbit p-ATM antibody (s1981) (5883, CST, 1:1000 dilution), monoclonal rabbit γH2AX (s139) antibody (9718, CST, 1:1000 dilution), monoclonal mouse p-AKT (s473) antibody (12694, CST, 1:1000 dilution), monoclonal rabbit AKT antibody (4685, CST, 1:1000 dilution), polyclonal rabbit β-Catenin antibody (9562, CST, 1:1000 dilution), polyclonal rabbit p-β-Catenin (s37) antibody (sc101651, Santa Cruz, 1:1000 dilution), polyclonal rabbit p-GSK3β antibody (sc-11757, Santa Cruz, 1:1000 dilution), and polyclonal rabbit GSK3β antibody (sc-9166, Santa Cruz, 1:1000 dilution). After incubating with primary antibodies for overnight at 4°C, the appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies (Beyotime) were subsequently applied and immunodetection was done using the enhanced chemiluminescence system (Cool-Imager). All experiments were repeated at least 3 times.
Quantitative real-time PCR (qRT-PCR)
The qRT-PCR primers for each gene were listed in the Table 1. Total RNA was isolated from NFs and CAFs using Trizol (Invitrogen) according to the manufacturer's protocol. RNA quantity was determined by agarose gel electrophoresis and by spectrophotometry. qRT-PCR was performed using the Primescript RT reagent kit (Takara) and SYBR Premix Ex TaqTM (Takara) according to the protocol of the manufacturer. β-Actin was used as internal reference gene. All experiments were done at least 3 times.
Table 1.
Primers sequences for qRT-PCR
| Gene name | Forward 5′-3′ | Reverse 5′-3′ | Size (bp) |
|---|---|---|---|
| ATM | ACTATCTCAGCTTCTACCCC | TGCTCAGAACTTATACCACG | 183 |
| CDK1 | GGATCTACCATACCCATTGAC | TGGCTACCACTTGACCTGT | 120 |
| E2F1 | ACTCTGCCACCATAGTGTCACCA | GACAACAGCGGTTCTTGCTCC | 103 |
| CCNA2 | AACAGTATGAGAGCTATCCTCGT | ATGTAGTTCACAGCCAAATGCAG | 95 |
| CCNB2 | TACCAGTTCCCAAATCCGAGA | AGCTCCATCAAATACTTGGCTA | 156 |
| CDKN2B | AATCCCCTTATGACTTGCTAC | CAATCTAGGCGTTTGCACT | 118 |
| β-Actin | TGACGTGGACATCCGCAAAG | CTGGAAGGTGGACAGCGAGG | 205 |
Abbreviations: ATM, ataxia telangiectasia mutated; CDK1, cyclin-dependent kinase 1; E2F1, E2F transcription factor 1; CCNA2, cyclin A2; CCNB2, cyclin B2; CDKN2B, cyclin-dependent kinase inhibitor 2B (p15, inhibits CDK4).
Immunohistochemistry (IHC) staining
IHC was done as described previously.42 Briefly, tumor and its adjacent normal tissues were fixed with formalin. Paraffin-embedded specimens were then sectioned into 4-μm sections and stained with H&E staining according to standard histopathological techniques. After treated with 3% hydrogen peroxide in methanol for 30 min to exhaust endogenous peroxidase activity, the sections were incubated with monoclonal rabbit anti-p-ATM antibody (2873, CST, 1:50 dilution) and monoclonal rabbit γH2AX antibody (5883, CST, 1:50 dilution) for overnight at 4°C. Then, the sections were sequentially incubated with polymer helper solution (ZSBiO) for 20 min, polyperoxidase-anti-mouse/rabbit IgG (PV-9000, ZSBiO, 1:100 dilution) for 30 min at 37°C, and stained with diaminobenizidine. Specimens used in this study were approved by the Ethics committees of Chongqing Medical University.
Immunofluorescence assay was done as described previously.37 Briefly, NFs and CAFs were grown on pre-prepared coverlips and fixed within 4% paraformaldehyde for 15 min at room temperature when reaching 40% confluency. Following washing 3 times with PBS, cells were incubated with 1% triton-100 for 20 min at room temperature and then blocked with 10% goat serum solution for 30 min at 37°C. Subsequently, cells were separately incubated with antibodies specifically against p-ATM (2783, CST, 1:250 dilution) and γH2AX (5883, CST, 1:250 dilution) for overnight at 4°C. After washing, cells were then incubated with an appropriate FITC-labeled secondary antibody (ZSbio) for 1 h at 37°C in a humidified incubator. Sections were immersed in aqueous medium containing 10% DAPI as a nuclear counterstain (Beyotime). A negative control was done to ensure the specificity of immunofluorescence assay by using a normal mouse or rabbit IgG as the primary antibodies.
Analysis of intracellular ROS
Intracellular ROS levels were measured using the Reactive Oxygen Species Assay Kit (S0033,Beyotime, 1:200 dilution) according to the instruction of the manufacture. Briefly, 10 μM 2′,7′-Dichlorofluorescene diacetate (H2DCFHDA), which can be oxidized by ROS and forms a fluorescent compound, dichlorofluorescein,43 was added to cells and continually incubated for 30 min. Then cells were lysed with RIPA buffer (Beyotime), and fluorescence was determined by scanning the total cell lysate using Cary Eclipse (Agilent) at excitation and emission wavelengths of 485 and 535 nm.
ChIP assay
ChIP was performed as previously described using a Thermo Fisher Biotechnology kit (26156).22 Chromatin prepared from cells in a 30-cm2 bottle was used to determine total DNA input and for overnight incubation with the primary antibody against β-Catenin (9562, CST, 1:100 dilution) or with normal rabbit IgG (A7016, Beyotime, 1:100 dilution). The human MYC promoter-specific primers used in PCR were 5′-CAGCCCGAGACTGTTGC-3′ (forward) and 5′-CAGAGCGTGGGATGTTAG-3′ (reverse).
siRNA interference
The lentiviral plasmids pLKO.1-ATM and control plasmid pLKO.1-GFP were kindly provided by Dr. Wen Chen (Sun Yat-sen University, Guangzhou). The lentiviral vectors containing hairpin oligoes against ATM and GFP (negative control) were transfected into 293T cells respectively. The CAFs were infected with Supernatant virus and screened with 1 μg/ml puromycin, generating cell lines named as CAF-shATM and CAF-shGFP respectively. For the c-Myc siRNA plasmid, synthetic DNA inserts for expressing the specific c-Myc shRNA were cloned into pLVX-shRNA1 (Clontech). The c-Myc shRNA was stably transferred into CAFs. The target sequence for c-Myc shRNA is 5′-GATCCGAACGGAGGGAGGGATCGCGCTTTTTCAAGAGAAGCGCGATCCCTCCCTCCGTTCTTA-3′. Negative co-ntrol shRNA sequence, which does not target any known human cDNAs, is 5′-AAGGTGTCAGAAACTGACGAT-3′.
Statistical analysis
Statistical analysis was performed using the SPSS standard version 13.0 software. The data were presented as the means ± SD. Each experiment was repeated at least 3 times. For comparisons between multiple groups, ANOVA followed by the Student-Newman-Keuls multiple comparisons test was used; and for single comparisons between 2 groups, the Student t test was used. A value of P < 0 .05 were considered significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Prof. Xiaojiang Cui from Samuel Oschin Comprehensive Cancer Institute, Cedars Sinai Medical Center, Los Angeles, for constructive suggestions and critical reading of the manuscript.
Funding
This work was supported in part by National Natural Science Foundation of China (NSFC8147247, NSFC81402180, and NSFC 31171336); the Doctoral Fund of Ministry of Education, China (20125503110001); the outstanding Doctor Fund of Chongqing Medical University (2013) to Shifu Tang.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646–74; PMID: 21376230; http://dx.doi.org/ 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- 2.Costa A, Scholer-Dahirel A, Mechta-Grigoriou F. The role of reactive oxygen species and metabolism on cancer cells and their microenvironment. Semin Cancer Biol 2014; 25:23-32; PMID:24406211; http://dx.doi.org/ 10.1016/j.semcancer.2013.12.007 [DOI] [PubMed] [Google Scholar]
- 3.Peng Q, Zhao L, Hou Y, Sun Y, Wang L, Luo H, Peng H, Liu M. Biological characteristics and genetic heterogeneity between carcinoma-associated fibroblasts and their paired normal fibroblasts in human breast cancer. PLoS One 2013; 8:e60321; PMID:23577100; http://dx.doi.org/ 10.1371/journal.pone.0060321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pavlides S, Vera I, Gandara R, Sneddon S, Pestell RG, Mercier I, Martinez-Outschoorn UE, Whitaker-Menezes D, Howell A, Sotgia F, et al.. Warburg meets autophagy: cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid Redox Signal 2012; 16:1264-84; PMID:21883043; http://dx.doi.org/ 10.1089/ars.2011.4243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jezierska-Drutel A, Rosenzweig SA, Neumann CA. Role of oxidative stress and the microenvironment in breast cancer development and progression. Adv Cancer Res 2013; 119:107-25; PMID:23870510; http://dx.doi.org/ 10.1016/B978-0-12-407190-2.00003-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sotgia F, Martinez-Outschoorn UE, Howell A, Pestell RG, Pavlides S, Lisanti MP. Caveolin-1 and cancer metabolism in the tumor microenvironment: markers, models, and mechanisms. Annu Rev Pathol 2012; 7: 423-67; PMID:22077552; http://dx.doi.org/ 10.1146/annurev-pathol-011811-120856 [DOI] [PubMed] [Google Scholar]
- 7.Sotgia F, Williams TM, Cohen AW, Minetti C, Pestell RG, Lisanti MP. Caveolin-1-deficient mice have an increased mammary stem cell population with upregulation of Wnt/beta-catenin signaling. Cell Cycle 2005; 4:1808-16; PMID:16294019; http://dx.doi.org/ 10.4161/cc.4.12.2198 [DOI] [PubMed] [Google Scholar]
- 8.Park DS, Lee H, Frank PG, Razani B, Nguyen AV, Parlow AF, Russell RG, Hulit J, Pestell RG, Lisanti MP. Caveolin-1-deficient mice show accelerated mammary gland development during pregnancy, premature lactation, and hyperactivation of the Jak-2/STAT5a signaling cascade. Mol Biol Cell 2002; 13:3416-30; PMID:12388746; http://dx.doi.org/ 10.1091/mbc.02-05-0071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol 2013; 14:197-210; PMID:23847781; http://dx.doi.org/ 10.1038/nrm3546 [DOI] [PubMed] [Google Scholar]
- 10.Kim J, Wong PK. Oxidative stress is linked to ERK1/2-p16 signaling-mediated growth defect in ATM-deficient astrocytes. J Biol Chem 2009; 284:14396-404; PMID:19321450; http://dx.doi.org/ 10.1074/jbc.M808116200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim J, Hwangbo J, Wong PK. p38 MAPK-Mediated Bmi-1 down-regulation and defective proliferation in ATM-deficient neural stem cells can be restored by Akt activation. PLoS One 2011; 6:e16615; PMID:21305053; http://dx.doi.org/ 10.1371/journal.pone.0016615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ito K, Hirao A, Arai F, Matsuoka S, Takubo K, Hamaguchi I, Nomiyama K, Hosokawa K, Sakurada K, Nakagata N, et al.. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 2004; 431:997-1002; PMID:15496926; http://dx.doi.org/ 10.1038/nature02989 [DOI] [PubMed] [Google Scholar]
- 13.Guo Z, Kozlov S, Lavin MF, Person MD, Paull TT. ATM activation by oxidative stress. Science 2010; 330: 517-21; PMID:20966255; http://dx.doi.org/ 10.1126/science.1192912 [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Yang J, Yi J. Redox sensing by proteins: oxidative modifications on cysteines and the consequent events. Antioxid Redox Signal 2012; 16:649-57; PMID:21967570; http://dx.doi.org/ 10.1089/ars.2011.4313 [DOI] [PubMed] [Google Scholar]
- 15.Yang DQ, Kastan MB. Participation of ATM in insulin signalling through phosphorylation of eIF-4E-binding protein 1. Nat Cell Biol 2000; 2:893-8; PMID:11146653; http://dx.doi.org/ 10.1038/35046542 [DOI] [PubMed] [Google Scholar]
- 16.Ditch S, Paull TT. The ATM protein kinase and cellular redox signaling: beyond the DNA damage response. Trends Biochem Sci 2012; 37:15-22; PMID:22079189; http://dx.doi.org/ 10.1016/j.tibs.2011.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martinez-Outschoorn UE, Lin Z, Trimmer C, Flomenberg N, Wang C, Pavlides S, Pestell RG, Howell A, Sotgia F, Lisanti MP. Cancer cells metabolically “fertilize” the tumor microenvironment with hydrogen peroxide, driving the Warburg effect: implications for PET imaging of human tumors. Cell Cycle 2011; 10: 2504-20; PMID:21778829; http://dx.doi.org/ 10.4161/cc.10.15.16585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 2012; 24: 981-90; PMID:22286106; http://dx.doi.org/ 10.1016/j.cellsig.2012.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang S, Yang L, Tang X, Liu M. The role of oxidized ATM in the regulation of oxidative stress-induced energy metabolism reprogramming of CAFs. Cancer Lett 2014; 353:133-44; PMID:25069040; http://dx.doi.org/ 10.1016/j.canlet.2014.07.028 [DOI] [PubMed] [Google Scholar]
- 20.Cam H, Easton JB, High A, Houghton PJ. mTORC1 signaling under hypoxic conditions is controlled by ATM-dependent phosphorylation of HIF-1alpha. Mol Cell 2010; 40:509-20; PMID:21095582; http://dx.doi.org/ 10.1016/j.molcel.2010.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao L, Sun Y, Hou Y, Peng Q, Wang L, Luo H, Tang X, Zeng Z, Liu M. MiRNA expression analysis of cancer-associated fibroblasts and normal fibroblasts in breast cancer. Int J Biochem Cell Biol 2012; 44: 2051-9; PMID:22964023; http://dx.doi.org/ 10.1016/j.biocel.2012.08.005 [DOI] [PubMed] [Google Scholar]
- 22.Yang W, Zheng Y, Xia Y, Ji H, Chen X, Guo F, Lyssiotis CA, Aldape K, Cantley LC, Lu Z. ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat Cell Biol 2012; 14:1295-304; PMID:23178880; http://dx.doi.org/ 10.1038/ncb2629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci 2011; 36:320-8; PMID:21531565; http://dx.doi.org/ 10.1016/j.tibs.2011.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vijayakumar S, Liu G, Rus IA, Yao S, Chen Y, Akiri G, Grumolato L, Aaronson SA. High-frequency canonical Wnt activation in multiple sarcoma subtypes drives proliferation through a TCF/beta-catenin target gene, CDC25A. Cancer Cell 2011; 19: 601-12; PMID:21575861; http://dx.doi.org/ 10.1016/j.ccr.2011.03.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duronio RJ, Xiong Y. Signaling pathways that control cell proliferation. Cold Spring Harb Perspect Biol 2013; 5:a008904; PMID:23457258; http://dx.doi.org/ 10.1101/cshperspect.a008904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prasad CP, Rath G, Mathur S, Bhatnagar D, Parshad R, Ralhan R. Expression analysis of E-cadherin, Slug and GSK3beta in invasive ductal carcinoma of breast. BMC Cancer 2009; 9:325; PMID:19751508; http://dx.doi.org/ 10.1186/1471-2407-9-325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu D, Pan W. GSK3: a multifaceted kinase in Wnt signaling. Trends Biochem Sci 2010; 35:161-8; PMID:19884009; http://dx.doi.org/ 10.1016/j.tibs.2009.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bretones G, Delgado MD, Leon J. Myc and cell cycle control. Biochim Biophys Acta 2015; 1849:506-516; PMID:24704206; http://dx.doi.org/ 10.1016/j.bbagrm.2014.03.013 [DOI] [PubMed] [Google Scholar]
- 29.Yang W, Xia Y, Hawke D, Li X, Liang J, Xing D, Aldape K, Hunter T, Alfred Yung WK, Lu Z. PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 2012; 150:685-96; PMID:22901803; http://dx.doi.org/ 10.1016/j.cell.2012.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Commandeur S, Ho SH, de Gruijl FR, Willemze R, Tensen CP, El Ghalbzouri A. Functional characterization of cancer-associated fibroblasts of human cutaneous squamous cell carcinoma. Exp Dermatol 2011; 20:737-42; PMID:21615509; http://dx.doi.org/ 10.1111/j.1600-0625.2011.01305.x [DOI] [PubMed] [Google Scholar]
- 31.Lisanti MP, Martinez-Outschoorn UE, Sotgia F. Oncogenes induce the cancer-associated fibroblast phenotype: metabolic symbiosis and “fibroblast addiction” are new therapeutic targets for drug discovery. Cell Cycle 2013; 12:2723-32; PMID:23860382; http://dx.doi.org/ 10.4161/cc.25695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Halaby MJ, Hibma JC, He J, Yang DQ. ATM protein kinase mediates full activation of Akt and regulates glucose transporter 4 translocation by insulin in muscle cells. Cell Signal 2008; 20:1555-63; PMID:18534819; http://dx.doi.org/ 10.1016/j.cellsig.2008.04.011 [DOI] [PubMed] [Google Scholar]
- 33.Benhar M, Engelberg D, Levitzki A. ROS, stress-activated kinases and stress signaling in cancer. EMBO Rep 2002; 3:420-5; PMID:11991946; http://dx.doi.org/ 10.1093/embo-reports/kvf094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alexander A, Cai SL, Kim J, Nanez A, Sahin M, MacLean KH, Inoki K, Guan KL, Shen J, Person MD, et al.. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci U S A 2010; 107:4153-8; PMID:20160076; http://dx.doi.org/ 10.1073/pnas.0913860107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Drayton S, Brookes S, Rowe J, Peters G. The significance of p16INK4a in cell defenses against transformation. Cell Cycle 2004; 3:611-5; PMID:15044857; http://dx.doi.org/ 10.4161/cc.3.5.837 [DOI] [PubMed] [Google Scholar]
- 36.Bartek J, Lukas J. Pathways governing G1/S transition and their response to DNA damage. FEBS Lett 2001; 490:117-22; PMID:11223026; http://dx.doi.org/ 10.1016/S0014-5793(01)02114-7 [DOI] [PubMed] [Google Scholar]
- 37.Wang L, Hou Y, Sun Y, Zhao L, Tang X, Hu P, Yang J, Zeng Z, Yang G, Cui X, et al.. c-Ski activates cancer-associated fibroblasts to regulate breast cancer cell invasion. Mol Oncol 2013; 7:1116-28; PMID:24011664; http://dx.doi.org/ 10.1016/j.molonc.2013.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yost C, Torres M, Miller JR, Huang E, Kimelman D, Moon RT. The axis-inducing activity, stability, and subcellular distribution of beta-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes Dev 1996; 10:1443-54; PMID:8666229; http://dx.doi.org/ 10.1101/gad.10.12.1443 [DOI] [PubMed] [Google Scholar]
- 39.Yan D, Avtanski D, Saxena NK, Sharma D. Leptin-induced epithelial-mesenchymal transition in breast cancer cells requires beta-catenin activation via Akt/GSK3- and MTA1/Wnt1 protein-dependent pathways. J Biol Chem 2012; 287:8598-612; PMID:22270359; http://dx.doi.org/ 10.1074/jbc.M111.322800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mylona E, Vamvakaris I, Giannopoulou I, Theohari I, Papadimitriou C, Keramopoulos A, Nakopoulou L. An immunohistochemical evaluation of the proteins Wnt1 and glycogen synthase kinase (GSK)-3beta in invasive breast carcinomas. Histopathology 2013; 62:899-907; PMID:23551536; http://dx.doi.org/ 10.1111/his.12095 [DOI] [PubMed] [Google Scholar]
- 41.Liu M, Sakamaki T, Casimiro MC, Willmarth NE, Quong AA, Ju X, Ojeifo J, Jiao X, Yeow WS, Katiyar S, et al.. The canonical NF-kappaB pathway governs mammary tumorigenesis in transgenic mice and tumor stem cell expansion. Cancer Res 2010; 70:10464-73; PMID:21159656; http://dx.doi.org/ 10.1158/0008-5472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu W, Wang Z, Zhang W, Qian K, Li H, Kong D, Li Y, Tang Y. Mutated K-ras activates CDK8 to stimulate the epithelial-to-mesenchymal transition in pancreatic cancer in part via the Wnt/beta-catenin signaling pathway. Cancer Lett 2014; 356:613-27; PMID:25305448; http://dx.doi.org/ 10.1016/j.canlet.2014.10.008 [DOI] [PubMed] [Google Scholar]
- 43.Woolley JF, Stanicka J, Cotter TG. Recent advances in reactive oxygen species measurement in biological systems. Trends Biochem Sci 2014; 38:556-65; PMID:24120034; http://dx.doi.org/ 10.1016/j.tibs.2013.08.009 [DOI] [PubMed] [Google Scholar]