

Abstract
Colorectal cancer (CRC) is the third most common cancer in the world and the second leading cause of cancer deaths in the US and Spain. The molecular mechanisms involved in the etiology of CRC are not yet elucidated due in part to the complexity of the human gut microbiota. In this study, we compared the microbiome composition of 90 tumor and matching adjacent tissue (adjacent) from cohorts from the US and Spain by 16S rRNA amplicon sequencing in order to determine the impact of the geographic origin on the CRC microbiome. Data showed a significantly (P < 0.05) higher Phylogenetic Diversity (PD) for the US (PD Adjacent = 26.3 ± 5.3, PD Tumor = 23.3 ± 6.2) compared to the Spanish cohort (PD Adjacent = 18.9 ± 5.9, PD Tumor = 18.7 ± 6.6) while no significant differences in bacterial diversity were observed between tumor and adjacent tissues for individuals from the same country. Adjacent tissues from the Spanish cohort were enriched in Firmicutes (SP = 43.9% and US = 22.2%, P = 0.0001) and Actinobacteria (SP = 1.6% and US = 0.5%, P = 0.0018) compared to US adjacent tissues, while adjacent tissues from the US had significantly higher abundances of Fusobacteria (US = 8.1% and SP = 1.5%, P = 0.0023) and Sinergistetes (US = 0.3% and SP = 0.1%, P = 0.0097). Comparisons between tumor and adjacent tissues in each cohort identified the genus Eikenella significantly over represented in US tumors (T = 0.024% and A = 0%, P = 0.03), and the genera Fusobacterium (T = 10.4% and A = 1.5%, P = <0.0001), Bulleida (T = 0.36% and A = 0.09%, P = 0.02), Gemella (T = 1.46% and A = 0.19%, P = 0.03), Parvimonas (T = 3.14% and A = 0.86%, P = 0.03), Campylobacter (T = 0.15% and A = 0.008%, P = 0.047), and Streptococcus (T = 2.84% and A = 2.19%, P = 0.05) significantly over represented in Spanish tumors. Predicted metagenome functional content from 16S rRNA surveys showed that bacterial motility proteins and proteins involved in flagellar assembly were over represented in adjacent tissues of both cohorts, while pathways involved in fatty acid biosynthesis, the MAPK signaling pathway, and bacterial toxins were over represented in tumors. Our study suggests that microbiome compositional and functional dissimilarities by geographic location should be taken in consideration when approaching CRC therapeutic options.
Introduction
Colorectal cancer (CRC) is the third most common cancer in the world1 with approximately 1.4 million new cases diagnosed in 2012. According to the American Cancer Society, CRC is the third most diagnosed cancer, the most common for both sexes, and the second leading cause of cancer related deaths in the United States, with an incidence of 57.2 for men and 42.5 for women, and a mortality of 21.2 for men and 14.0 for women (per 100,000, age adjusted to the 2000 US standard population).2 In Spain, CRC is the third most diagnosed cancer for men, after lung cancer and prostate cancer, the second after breast cancer for women, and the second leading cause of cancer deaths.3 CRC incidence in Spain was 60.8 for men and 33.8 for women, with an estimated mortality of 28.10 for men and 14.67 for women (per 100,000, age adjusted to the standard European population for the years 2000–2004).4
The human body contains a vast number of microbes essential to its proper functioning. Moreover, the human colon microbiota is composed of 1013 to 1014 microorganisms, primarily bacteria. The three most represented bacterial phyla in the human colon are Firmicutes, Bacteroidetes, and Actinobacteria.5-7 Several metagenomics studies have reported that the microbiota composition is diverse within and between individuals and populations.8,9 In addition, factors including host genetics, diet, and environmental factors have been proposed to have an impact on microbiome composition and on the incidence of certain diseases like diabetes, obesity, and CRC.10-16 Research studies have shown that the composition of the gut microbiome was contingent on dietary habits; with less reported disease incidence directly correlated with diets rich in vegetables, fruits, and olive oil (the “Mediterranean diet”)17,18 compared to diets rich in red/processed meats and low in fiber (the “Western diet”).19 Although the impact of diet in shaping the gut microbiota has not been thoroughly elucidated, a recent human study comparing the gut microbiome of individuals consuming an animal-based diet versus a plant-based diet showed that the food regimen altered the microbiome composition in a very short-term.20 The study showed that the animal-based diet had a profound effect on the gut microbiome significantly increasing the abundance of Alistipes, Bilophila, and Bacteroides and decreasing the abundance of the butyrate producers Roseburia, Eubacterium rectale, and Ruminococcus bromii.20
Research studies indicate that the composition of the gut microbiome is a major factor in CRC risk,15,16,21-23 however mechanisms of modulation are not clearly understood. Early studies showed a direct correlation between increased abundance of Bacteroides sp., Streptococcus gallolyticus (formerly S. bovis), and Clostridium sp in CRC patients.24-26 More recently, microbiome studies using next-generation sequencing approaches have shown that a decrease in bacterial diversity in the gut microbiota was associated with higher CRC risk in stool samples.21 Moreover, CRC patients have a distinct gut bacterial community composition with increased abundance of Fusobacterium, Porphyromonas (which have been related to inflammation21,27,28), Coriobacteridae and Roseburia29 and decreased abundance of Firmicutes, specifically Clostridia (involved in fermentation of dietary fiber30) and Enterobacteriaceae29 in both stool and mucosal samples. Although Fusobacterium has been found over represented in the gut microbiome of colorectal adenoma and carcinoma patients in stool and mucosal samples, more research is needed to elucidate a potential role of this bacterium as a potential CRC etiologic agent.28,31
Published research studies in general have aimed to determine the microbiome composition in CRC or adenoma patients within the same population,32-34 or the composition of the microbiome in response to dietary habits20,35 in different populations. Only a small number of studies have compared the gut microbiome composition of CRC patients from different populations. A study comparing the microbiome of healthy individuals from a high CRC risk population (African-Americans) with a low risk population (Africans)36,37 showed differences in the microbiome composition between the 2 populations, which probably reflect a diet high in fiber and less meat and fat for Africans compared to the African-American diet more comparable to the Western diet. The study found an over abundance of Prevotella, Succinivibrio, and Oscillospira in Africans and a predominance of the genus Bacteroides in African-Americans. In addition, the majority of CRC microbiome studies focused on stools because sample collection protocols are less invasive.
Our study aimed to test the hypothesis that matching adjacent tissue (adjacent) microbiome of individuals from different geographical locations has significant compositional differences potentially reflecting different genetic backgrounds, diet and lifestyle. We also aimed to demonstrate that, regardless of the sample origin, the tumor microenvironment could modulate the microbiome composition leading to an increased abundance of functionally equivalent phylogenetic groups in tumor tissues. We analyzed the microbiome composition of 90 matched pairs of colorectal carcinoma and adjacent tissues specimens from the US and Spain by 16S amplicon pyrosequencing. The purpose of this study was twofold: 1) to identify bacterial taxa over or under represented in tumor vs. adjacent tissues in human samples from the 2 geographical locations, and 2) to assess if differences between tumor and tumor-adjacent tissues from 2 different populations were comparable. Additionally, we used Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt)38 to infer metabolic differences between tumor and adjacent tissues from individuals from the US and Spain.
Results
In this study we analyzed the mucosal-adherent microbiome composition of matched pairs of colorectal carcinoma and adjacent tissues from 22 human subjects from the US and 23 human subjects from Spain. Patient and sample characteristics are summarized in Table 1 and Table S1. A total of 500,946 raw sequences were generated from 90 samples (44 samples from the US and 46 samples from Spain). After quality filtering, the mean length of the remaining sequences was 361.4 ± 10.2 bp, the average quality score was 35.4 ± 3.8, and the average number of reads/sample was 3,467.9 ± 1,967.6. Ninety five percent of the sequences were assigned to a taxonomic group while 5% of the reads were unclassified/unassigned. A total of 4,523 Operational Taxonomic Units (OTUs) and 5,493 OTUs were identified in the Spanish and the US cohorts, respectively after clustering sequences at a 97% similarity threshold (equivalent to the species level classification). The majority of sequences in all samples belonged to the phyla Bacteroidetes, Firmicutes, Fusobacteria, Proteobacteria, and Actinobacteria (Table 2) in agreement with previous reports of the human gut microbiome composition.39
Table 1.
Summary of general characteristics of samples analyzed in this study.
| Cohort |
US |
Spain |
|---|---|---|
| Number of Samples | N = 44 (22 tumor and 22 tumor-adjacent) | N = 46 (23 tumor and 23 tumor-adjacent) |
| Age Range | ||
| 40-49 | 5 | 1 |
| 50-59 | 5 | 2 |
| 60-69 | 5 | 8 |
| 70-79 | 4 | 9 |
| 80-89 | 3 | 3 |
| Sex | ||
| Female | 11 | 8 |
| Male | 11 | 15 |
| Tumor Location | ||
| Cecum | - | 2 |
| Ascending (Right) Colon | 12 | 5 |
| Splenic Flexure | 2 | - |
| Descending (Left) Colon | - | 2 |
| Sigmoid* | 6 | 13 |
| Rectum | 2 | 1 |
Includes recto-sigmoid tumors.
Table 2.
Most represented phyla in tumor and tumor-adjacent samples from the US and Spain. Numbers represent phyla relative abundance (%) ± standard deviation.
| Cohort | Sample Type | Actinobacteria | Bacteroidetes | Firmicutes | Fusobacteria | Proteobacteria |
|---|---|---|---|---|---|---|
| US | Adjacent | 0.5±0.3 | 55.5±22.1 | 22.2±15.5 | 8.1±19.8 | 2.9±4.5 |
| Tumor | 0.5±0.4 | 54.5±19.0 | 27.1±16.4 | 8.4±16.3 | 2.4±2.9 | |
| Spain | Adjacent | 1.6±1.8 | 46.6±19.5 | 43.9±18.3 | 1.6±4.2 | 3.2±8.9 |
| Tumor | 1.7±2.0 | 45.5±16.8 | 36.8±18.4 | 10.5±14.6 | 1.7±2.6 |
The US gut microbiome harbors a higher phylogenetic diversity than Spain
Overall, US samples showed a significantly higher Phylogenetic Diversity (PD) and Species Richness (S) than Spain samples (Fig. 1). Additionally, adjacent samples from both Spain and the US showed higher PD and S values than their corresponding tumor samples, although those differences did not reach statistical significance (Fig. 1).
Figure 1.
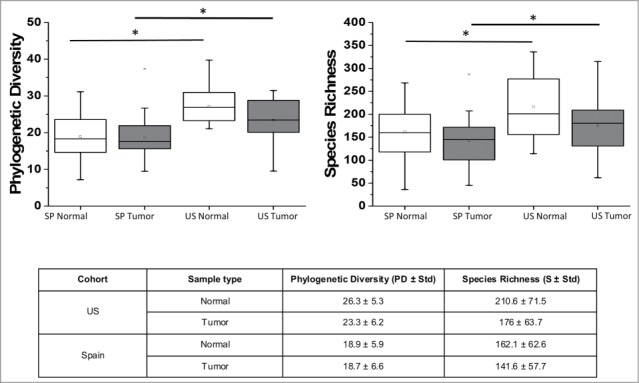
Comparison of phylogenetic diversity (PD) and species richness (S) between adjacent and tumor tissues from the US and Spain. * P < 0.05
Principal Coordinates Analysis (PCoA) of UniFrac distance matrices38 indicated that geographic origin of samples was the primary explanation for the variation in our dataset (Fig. 2). Analysis of Similarities (ANOSIM) and Permutational Multivariate Analysis of Variance (PERMANOVA) analyses showed no statistically significant differences between the microbiome composition of adjacent and tumor tissues for Spain or US cohorts (Figs. 2A and B). However, when we compared adjacent as well as tumor tissues from the different geographic locations, we observed clear clustering by cohort (Figs. 2C and D).
Figure 2.
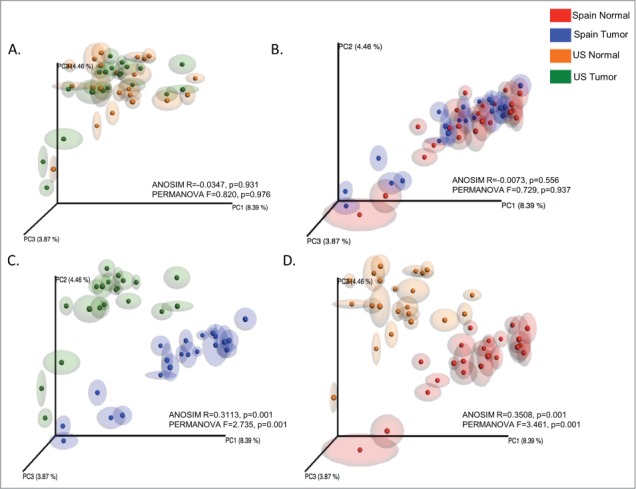
Unweight UniFrac based Principal Coordinates Analysis (PCoA) of microbial communities associated with adjacent tissues and tumors from the US and Spain. ANOSIM and PERMANOVA generated parameters are indicated in each comparative plot.
Due to the low number of participants in each age category we stratified data into 2 groups: ages 40 to 69 and ages 70 to 89 y old. No statistically significant differences between age categories were observed for the US (ANOSIM P = 0.544, R = −0.0129, PERMANOVA P = 0.850) or the Spanish cohorts (ANOSIM P = 0.367, R = 0.0142, PERMANOVA P = 0.710). Likewise, no statistically significant differences were observed in cohorts between adjacent and tumor tissues among females, between adjacent and tumor tissues among males, or in adjacent tissues between males and females. We observed differences in the distribution of bacterial taxa along the different regions of the colon; however, due to the low number of samples originated from each intestinal location, we did not perform statistical analyses to assess significance. Abundance of unassigned bacteria was higher in US tissues in both adjacent and tumor samples compared to Spain samples regardless of tumor location (Fig. 3, Table S2). The phylum Actinobacteria appeared to be predominant in the right colon of the Spanish cohort in both adjacent and tumor samples, although this observation was not replicated in the US cohort. Conversely, Fusobacteria was over represented in the right colon and splenic flexure in the US cohort, enriched in sigmoid and rectal sections only in tumor samples, while this phylum was abundant in left colon only in tumor of the Spanish cohort.
Figure 3.
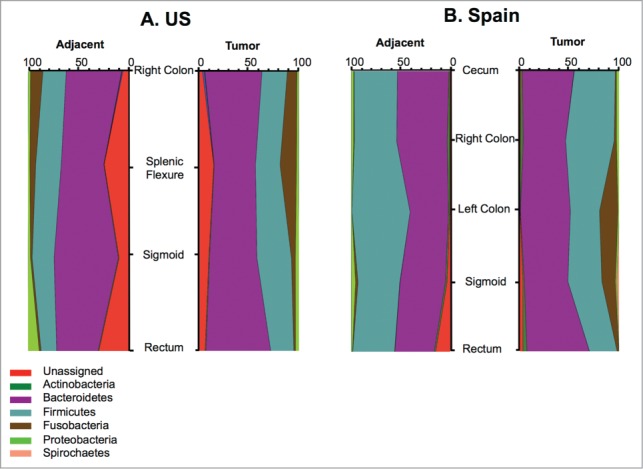
Representation of relative abundances of bacterial phyla in adjacent and tumor according to tumor location in the intestinal tract.
Gut microbiome compositional differences between the US and Spanish cohorts
In this section, we first present an overall comparison of the relative abundance of bacterial taxa between cohorts. Next, we report compositional differences between adjacent tissues to finally list compositional differences between tumor tissues of the different populations.
The main overall difference between the US and the Spanish cohorts was the significantly higher relative abundance of Actinobacteria, specifically of the class Coriobacteriia, in the Spanish cohort (adjacent and tumor tissues) compared to the US cohort (Fig. 4 and Table 2). The most prevalent genera in both cohorts were Bacteroides and Fusobacterium. Both of them were over represented in the US compared to Spain. Additionally, several taxa within the most represented phyla were also significantly (Steel Dwass All Pairs test P < 0.05) or borderline significantly (P value indicated between brackets) differentially represented in the US cohort compared to the Spanish cohort (Fig. 5, Tables 3 and 4): Actinobacteria (Propionibacterium, Collinsela, and Slackia), Bacteroidetes (Barnesiellaceae, Butyricimonas, Paraprevotella, Prevotella [P = 0.06], and Rikenellaceae [P = 0.052]), Firmicutes (Geobacillus, other Clostridiaceae, Lactobacillus, Coprococcus, Epulopiscium, Oribacterium, Roseburia, Ruminococcaceae, Schwartzia, Selenomonas, and Bulleidia [P = 0.067]), Fusobacteria (Cetobacterium, Leptotrichia, and Fusobacterium), and Proteobacteria (Ralstonia, Bilophila, and Enterobacteriaceae).
Figure 4.
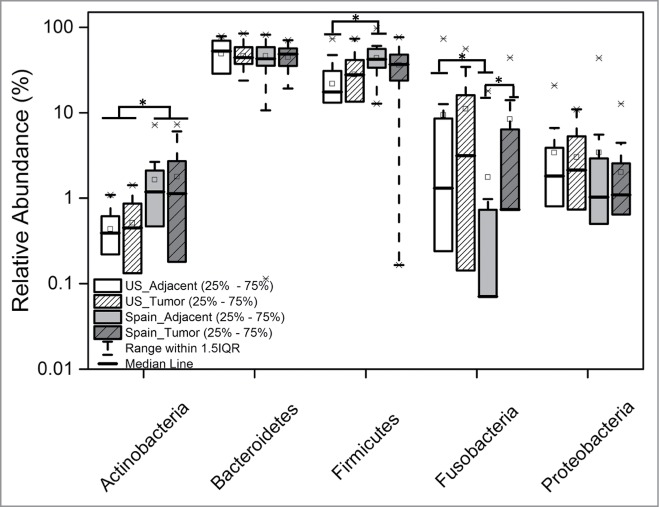
Relative abundances of the most represented bacterial phyla in tumor and adjacent tissues in the US and Spain cohorts. Asterisks represent significant (P < 0.01) differences between groups.
Figure 5.

Relative abundances of genera differentially represented in the tumors or non-tumor tissues of the US and Spanish cohorts. Asterisks represent significant (P < 0.05) differences between groups.
Figure 5.

(Continued)
Table 3.
Comparison of bacterial genera significantly (P<0.05) over or under represented in tumor adjacent tissues from the US and Spain. Numbers represent the relative abundance (%) ± standard deviation. Shaded cells indicate higher relative abundance.
| Phyla | Genera | US | Spain | P-value |
|---|---|---|---|---|
| Actinobacteria | Propionibacterium | 0.1±0.2 | 0.02±0.09 | 0.0452 |
| f_Coriobacteriaceae | 0.02±0.07 | 0.09±0.19 | 0.0096 | |
| Bifidobacterium | 0.0±0.0 | 0.03±0.14 | 0.0059 | |
| Collinsella | 0.1±0.1 | 1.27±1.83 | 0.0001 | |
| Slackia | 0.008±0.02 | 0.05±0.13 | 0.0426 | |
| Bacteroidetes | Bacteroides | 39.9±21.4 | 27.1±17.6 | 0.0469 |
| Butyricimonas | 0.06±0.15 | 0.16±0.25 | 0.0054 | |
| f_[Barnesiellaceae] | 0.38±0.72 | 1.39±2.46 | 0.0075 | |
| Paraprevotella | 0.07±0.31 | 0.17±0.35 | 0.0411 | |
| f_Rikenellaceae | 0.37±0.61 | 1.30±1.96 | 0.0215 | |
| Firmicutes | Selenomonas | 0.6±1.9 | 0.0±0.0 | 0.0037 |
| Geobacillus | 0.3±0.6 | 0.0±0.0 | 0.0008 | |
| Schwartzia | 0.3±1.0 | 0.0±0.0 | 0.0067 | |
| Bulleidia | 0.5±0.8 | 0.1±0.2 | 0.0224 | |
| Lachnospira | 0.002±0.01 | 0.07±0.21 | 0.0123 | |
| Anaerostipes | 0.0±0.0 | 0.01±0.04 | 0.0059 | |
| Dorea | 0.27±0.44 | 1.30±1.61 | 0.0355 | |
| 02d06 | 0.0±0.0 | 0.01±0.04 | 0.0233 | |
| cc_115 | 0.0±0.0 | 0.01±0.02 | 0.0059 | |
| f_Lachnospiraceae_Other | 1.93±2.37 | 4.65±5.44 | 0.0328 | |
| Turicibacter | 0.0±0.0 | 0.02±0.09 | 0.0118 | |
| f_Clostridiaceae_Other | 0.01±0.05 | 0.35±1.43 | 0.0032 | |
| Coprococcus | 0.09±0.12 | 1.41±2.58 | 0.0002 | |
| Lactobacillus | 0.03±0.13 | 0.43±1.00 | 0.0143 | |
| Epulopiscium | 0.0±0.0 | 0.08±0.23 | 0.0454 | |
| f_Christensenellaceae | 0.008±0.02 | 0.31±0.88 | 0.0147 | |
| f_Ruminococcaceae | 1.61±1.63 | 4.93±4.95 | 0.0076 | |
| Fusobacteria | Fusobacterium | 9.4±19.6 | 1.6±3.9 | 0.0017 |
| Leptotrichia | 0.3±0.6 | 0.0±0.0 | 0.0194 | |
| Cetobacterium | 0.0±0.0 | 0.10±0.40 | 0.0233 | |
| Proteobacteria | Campylobacter | 0.5±2.0 | 0.0±0.0 | 0.0122 |
| Ralstonia | 2.1±4.2 | 0.9±4.3 | <.0001 | |
| f_Bradyrhizobiaceae | 0.0±0.0 | 0.16±0.77 | 0.0454 | |
| Acidocella | 0.0±0.0 | 0.08±0.2 | 0.0233 | |
| Stenotrophomonas | 0.0±0.0 | 0.007±0.01 | 0.0454 | |
| Bilophila | 0.0±0.0 | 0.02±0.04 | 0.0014 | |
| Synergistetes | TG5 | 0.3±0.7 | 0.0±0.0 | 0.0011 |
The comparative analysis of adjacent tissues revealed that, in addition to the described overall overrepresentation of Actinobacteria in the Spanish cohort, Firmicutes and Proteobacteria were at a significantly higher relative abundance in the adjacent tissues of the Spanish cohort compared to the US cohort (Table 2, Fig. 4), while Fusobacteria were more prevalent in the US cohort. When we analyzed differences at genus level, we observed a number of groups significantly over or under represented specifically and only in adjacent tissues of the US cohort (Fig. 5, Table 3). Within the Actinobacteria phylum, the genus Bifidobacterium, considered an indicator of a healthy microbial balance, the Firmicutes: Turicibacter, Clostridiaceae 02d06, Anaerostipes, Lachnospira, Erysipelotrichaceae cc_115, and the Alpha Proteobacteria Acidocella were over represented in the Spanish cohort. Conversely, the potential pathogen Campylobacter was enriched in adjacent tissues of the US cohort. We finally compared the taxa relative abundance in tumor tissues between cohorts and observed that, besides Actinobacteria, the Cyanobacteria, a phylum normally represented in proportions below 0.1% in the human gut microbiome,40 were significantly over represented in the Spanish (0.02% ± 0.07) compared to the US cohort (0.002% ± 0.01). Differences in taxa relative abundances exclusively in tumor tissues included one Firmicutes genus, Peptostreptococcus (over represented in tumor in the Spanish cohort), and lineages from the phylum Proteobacteria, Eikenella and Caulobacteraceae (over represented in tumor in the US cohort), and Desulfovibrionaceae and Methylobacteriaceae (over represented in tumor in the Spanish cohort) (Table 4, Fig. 5).
Cohort-specific microbiome compositional differences between adjacent and tumor tissues
Limited differences were detected between adjacent and tumor tissues from each cohort. In the US cohort, Eikenella, a Proteobacteria of the family Neisseriaceae (P = 0.03), and the Actinobacteria family Coriobacteriaceae (P = 0.06) were over represented in tumor tissues while Parabacteroides, a Bacteroidetes of the family Porphyromonadaceae, were over represented in adjacent tissues (P = 0.09) (Fig. 5). The tumor microbiome of the Spanish cohort had a significantly (P < 0.05) higher relative abundance of the phylum Fusobacteria (Fig. 4). At the genus level, the Bacteroidetes Butyricimonas, the Fusobacteria Fusobacterium, and the Proteobacteria Campylobacter, were significantly over represented in Spanish tumor tissues while the Firmicutes Lachnospira and Blautia were more abundant in adjacent tissues. Interestingly, the Bacteroidetes Parabacteroides was the only taxa showing differences between tumor and adjacent tissues in the 2 cohorts (Table 5, Fig. 5).
Predicted functional differences between tumor and adjacent tissues
We next used PICRUSt to identify differences in predicted metabolic functions between tumor and tumor-adjacent adjacent tissues. Since this bioinformatics tool predicts enzymes and pathways based on 16S rRNA data, the obtained data undoubtedly reflects the major compositional differences between cohorts. Our main observation was that metabolic pathways and enzymes differentially represented in tumor tissues were similar in both cohorts. Specifically, bacterial motility proteins and proteins involved in flagellar assembly were over represented in the adjacent tissues of the US (P < 0.1) and the Spanish cohorts (P < 0.05), while pathways involved in fatty acid biosynthesis, the mitogen-activated protein kinases (MAPK) signaling pathway, bacterial toxins, and type II diabetes mellitus pathways were over represented in tumor tissues (Table S3). We identified significantly (Wilcoxon signed-rank test P < 0.1) more pathways and enzymes differentially represented between adjacent and tumor tissues in the Spanish (91 pathways) compared to the US cohort (12 pathways) that could reflect a higher inter individual variation in the US cohort. In addition to proteins involved in cell motility and toxins, the N-glycan biosynthesis pathway over represented in tumors in the US cohort. Other pathways over represented in tumors in the Spanish cohort included genetic and environmental information processing, cellular processes, metabolism, organismal systems, and unclassified proteins (Table S3).
A total of 39 enzymes showed a differential representation in tumor tissues in the Spain cohort, while 10 were over or under represented in US tumors. Three enzymes were under represented in the adjacent tissues in both cohorts: an urea decarboxylase (EC 6.3.4.6), involved in the conversion of urea to urea-1-carboxylate in the arginine and proline metabolism pathway, a transposase from the IS5 family, and a phosphoadenosine phosphosulfate reductase (EC 1.8.4.8), which catalyzes the conversion of 3’-phosphoadenylylsulfate (PAPS) to sulfite in the sulfur metabolism pathway. A cation-transporting P-type ATPase C (EC:3.6.3.-) and a methionine-gamma-lyase (EC:4.4.1.11) were over represented in the tumors of the Spanish cohort while a galactose-6-phosphate isomerase (EC:5.3.1.26), an aminotransferase (EC:2.6.1.-), and 2 transcriptional regulators (LiaR from the NarL family, and the myo-inositol catabolism operon repressor from the DeoR family of transcriptional regulators) were more abundant in US tumors (Table S4).
Discussion
Studies have increasingly addressed the role of the gut microbiome in CRC since a dysbiotic state has been reported in the stools and tissue-adherent microbiome of adenoma and CRC patients (recently reviewed by Keku et al.41). Our study aimed to test the hypothesis that, regardless of the geographic origin, the tumor microenvironment could modulate the microbiota in situ generating compositional and functional similarities in the tumor microbiomes. We determined the microbiome composition of 90 matched pairs of colorectal carcinoma and tumor-adjacent (adjacent) tissues from cohorts from the US and Spain to identify differences between cohorts, and differences between adjacent and tumor tissues in each cohort.
In our study, we observed an overall higher Phylogenetic Diversity (PD) and species richness (S) in the US cohort. Additionally, tumor tissues showed a lower, although non statistically significant, diversity and species richness than adjacent tissues confirming previous studies reporting a decrease of diversity and species richness associated with CRC.42,43 A reduced microbial diversity has been considered an undesirable effect of globalization and its concomitant diet rich in fat, protein, and sugar, and depleted of non-digestible fibers and microorganisms. Few studies have attempted to compare the gut microbiome of humans originating from different geographic locations. A study by Yatsunenko et al.44 determined that diversity was lower in a US cohort compared to Amerindians and Malawians inhabiting rural areas. Likewise, De Filippo et al.45 found a higher microbial richness and biodiversity in samples from a rural African village of Burkina Faso than in European children. To our knowledge, this is the first study comparing tissue-adherent gut microbiomes from 2 populations from urban developed regions.
Differences in adjacent tissues between cohorts were extensive (Fig. 5 and Table 3). Of interest was the enriched abundance, in the Spanish cohort, of Actinobacteria and Bifidobacterium species in particular, which are considered markers of a healthy microbiome due to their ability to generate lactic acid, which aids food digestion, and act as intermediaries in the generation of butyrate by the gut microbiota.46,47 Similarly, adjacent tissues from the Spanish cohort had higher relative abundances of Lactobacillus species. Conversely, Bacteroides, more abundant in adjacent tissue of the US cohort, are generally related to diets rich in protein and animal fat.35 Interestingly, we noted in both the US and the Spanish cohorts, an increased abundance of Bacteroidetes specifically in rectal tumors. The over representation of Bacteroides in countries were CRC is more prevalent was reported in a seminal paper by Hill et al..48 The authors reported that fecal samples from individuals in Britain and the US, countries with high CRC incidence, had higher counts of Bacteroides and lower counts of Enterococci and other aerobic bacteria compared to samples from individuals from Uganda, South India, and Japan, where the incidence of the disease was low. More recently, a study showed that representation of the enterotoxin gene (bft) from Bacteroides fragilis was detected by PCR in 38% of the isolates from CRC patients, but only in 12% of isolates from the control group.49 The Enterotoxigenic Bacteroides Fragilis (ETBF) produces a toxin that can cause acute diarrhea and chronic inflammation by stimulation of T lymphocytes that produce interleukin 17 (IL-17), and can, ultimately, promote CRC in mice.50 In contrast to the extensive differences in relative abundances of bacterial lineages in adjacent tissues, differences in tumors between cohorts were limited to one Firmicutes genus, Peptostreptococcus (over represented in tumor in the Spanish cohort), and lineages from the phylum Proteobacteria: Eikenella and Caulobacteraceae (over represented in tumor in the US cohort), and Desulfovibrionaceae and Methylobacteriaceae (over represented in tumor in the Spanish cohort). These results correlate with our hypothesis that the specific tumor microenvironment could modulate the tumor microbiome resulting in the selection of similar taxa resistant to conditions of hypoxia, occurring due to the insufficient vascularization, low pH and depletion of glucose and other nutrients.51,52
A number of research studies have analyzed the microbiome composition of tissue and fecal samples from CRC patients41 and our study alineates in general with their findings. In our study, however, although we observed a lower phylogenetic diversity and richness in tumor tissues compared to adjacent tissues, Principal Coordinate Analysis of weighted and unweight Unifrac matrices and ANOSIM analysis showed no extensive differences in composition between adjacent and tumor microbial communities in both cohorts, which could be due in part to the fact that adjacent tissues, although non-tumoral, are in close proximity to tumors sharing the same intestinal microenvironment. In the US cohort, the genus Eikenella was significantly over represented in tumors. Eikenella corrodens is the best known species of this genus, a microaerophilic, Gram-negative bacillus, nutritionally fastidious organism that requires 5–10% CO2 and blood agar containing hemin (X factor) for optimal growth.53 E. corrodens is a normal component of the microbiota of the oral cavity54 and the mucosal surface of the gastrointestinal and genitourinary tracts55; however, its potential to act as a pathogen has been well documented.53,56-60 Given its documented history of pathogenicity, further research on the potential role of Eikenella corrodens in CRC etiology is warranted.
Fusobacterium has been associated with colorectal tumors and adenomas in several recent studies.27,31,61-65 In the present study, Fusobacterium was significantly over represented in tumor compared to adjacent tissues in the Spanish cohort, while in the US, this phylum was more abundant in both tissues compared to the Spanish cohort, but enriched specifically in tumors in splenic flexure, sigmoid and rectum. More studies are needed to assess the biological significance of these results, which could be related to the ability of these microorganisms to form and maintain biofilms in specific locations of the large intestine as recently shown.66 In addition to Fusobacterium, Campylobacter, Granulicatella, Butyricimonas, and unclassified members of the family Lachnospiraceae were enriched in tumor tissues of the Spanish cohort. However, Blautia and Lachnospira, also of the family Lachnospiraceae, were depleted in tumor tissues. In general there was an overall enrichment of the phylum Firmicutes, specifically the class Clostridia, in the Spanish cohort, which may reflect a higher content of fiber in their diet. Similar observations were previously reported in tumors compared to adjacent tissues,67 and in feces of advanced adenomas68 and CRC patients.69
To investigate if, in addition to a tumor microenvironment compositional impact, there was an impact on bacterial function, we applied PICRUSt to the 16S rRNA amplicon sequencing data to infer bacterial metabolic functions. Interestingly, we found metabolic pathways and enzymes differentially represented in tumor tissues of both cohorts with bacterial motility proteins and proteins involved in flagellar assembly over represented in adjacent tissues, and pathways involved in fatty acid biosynthesis, the MAPK signaling pathway, and bacterial toxins over represented in tumors. The enrichment of virulence genes in colon tumors has been recently reported,70 although in the study by Burns et al.70 over representation of genes encoding bacterial toxins was not statistically significant, likely due to low total gene counts for some categories. The same study found an over representation of bacterial motility proteins in tumors contradicting our findings. In a different study, publically available data was analyzed to test if bacteria in off-tumor sites expressed more toxins.71 The authors focused on particular bacteria, characterized in other studies as “CRC drivers:” Bacteroides, Clostridium, Escherichia, Salmonella and Shigella, and demonstrated that the toxins from E. coli, S. enterica and S. flexneri were the most actively transcribed in tumor tissue and surrounding mucosa from CRC patients compared to toxins from other taxonomic groups.
Finally, in our study, 39 enzymes were differentially represented in the tumors of the Spanish cohort, while 10 were over or under represented in US tumors. Three enzymes were under represented in the adjacent tissues in both cohorts: an urea decarboxylase, involved in the conversion of urea to urea-1-carboxylate in the and arginine and proline metabolism pathway, a transposase from the IS5 family, and a phosphoadenosine phosphosulfate reductase, an oxidoreductase involved in the formation of hydrogen sulfide, a compound that generates free radicals, impairs cytochrome oxidase, suppresses butyrate utilization, and inhibits mucus synthesis and DNA methylation (reviewed in23), which can be generated by sulfure-reducing bacteria from meat, a rich source of dietary sulfur. A cation-transporting P-type ATPase and a methionine-gamma-lyase were over represented in the tumors of the Spanish cohort while a galactose-6-phosphate isomerase, an aminotransferase, and 2 transcriptional regulators (LiaR from the NarL family, and the myo-inositol catabolism operon repressor from the DeoR family of transcriptional regulators) were more abundant in US tumors. The biological significance of these results clearly require further research, although a number of metagenomics studies are beginning to unravel the relationships between CRC and bacterial function.
Although limited by the unavailability of diet information and the lack of samples from non-CRC, healthy controls, our study suggests that geography have a clear impact on the gut microbiome, indicated by the extensive differences between adjacent tissues of the different cohorts. Those differences however were diminished in tumors, characterized by a hypoxic and acidic microenvironment depleted of glucose and other nutrients. Our results may appear paradoxical since more differences in relative abundances could be expected between tumor and adjacent tissues within cohorts, given the extensive differences found in comparisons of adjacent tissues between cohorts; however, this can be explained by the fact that, although, attenuated and modulated by the tumor microenvironment, differences in tumors between cohorts were still significant. Finally, predicted functional information identified potential therapeutic targets in tumors, which bear an over representation of fatty acid biosynthesis, the MAPK signaling, and bacterial toxins pathways. Our work continues the effort of many research groups that intend to understand the correlation between microbiome composition and function, and CRC.
Materials and Methods
Ethics statement
This study was approved by the Institutional Research Board (IRB) of the University of North Carolina (UNC) Tissue Procurement Facility, US and the Principality of Asturias Clinical Research Ethical Committee, Spain (Approval number 67/2014). Patients were provided written informed consent for the collection of samples and subsequent analysis when required.
Samples collection and processing
Twenty-two de-identified tumors and adjacent tissue from the University of North Carolina (UNC) Tissue Procurement Facility. All tissues were snap frozen. Spanish samples were collected from CRC patients who underwent surgery in the Hospital Universitario Central de Asturias (HUCA), Spain. Surgical specimens were sent directly from the operating suites to the pathological anatomy service in sterile plastic boxes. Once there, the specimens were processed in a laminar flow cabinet where tissue's sections of around 3mm each were taken from tumors and adjacent mucosa by the use of sterile scissors and scalpels. Samples were collected in sterile Eppendorf tubes, immediately snap- frozen in isopentane and kept at -80°C until analysis. The mean age and range of US matched tumor and adjacent tissues were 63.6 and 42–88 respectively, and 69.8 and 49–85 for the Spanish cohort (Table S1). Colon tissue specimens were collected from tumor and from adjacent mucosa from the same patient at a resection margin located at a mean distance of 5 cm from the tumor for Spanish patients. As US samples were retrospective, information about distance between tumor and adjacent specimens was not routinely collected. Histologically, all colon tumors were adenocarcinomas with the exception of one in the Spanish cohort that was a carcinoma.
DNA isolation
Total DNA was isolated from frozen sections of 90 matched pairs of colorectal carcinoma and adjacent tissues specimens from the US and Spain. DNA isolation was carried out using the DNeasy Qiagen Blood and Tissue protocol (Qiagen, Germantown, MD). The Qiagen protocol was modified to ensure an optimal isolation of DNA from Gram-positive bacteria by adding 180 μl of lysozyme (20mg/ml) to samples already containing 200 μl of buffer ATL. Subsequent steps were performed as recommended by the manufacturer's protocol. Briefly, 20 μl of Proteinase K were added to samples, vortexed, and incubated overnight at 56°C. The supernatants were transferred to a new 1.5 ml microcentrifuge tube, 200 μl of buffer AL were added to the tube, and the mix was incubated at 70°C for 10 minutes. Then, 200 μl of 100% ethanol were added to the mixture and they were transferred into a column. Following this, 500 μl of buffer AW1 and AW2 were added to the column separately and the flow-through was discarded in each step. Finally, 200 μl of Buffer AE were added to the column to elute the DNA.
16S amplicon sequencing
Amplification of the V1-V2 hypervariable region of the bacterial 16S rRNA was performed on total DNA from collected samples as previously described.72 The reaction mix contained the Qiagen Hotstar Hi-Fidelity Polymerase Kit (Qiagen, Valencia CA) with a forward primer composed of the Roche Titanium Fusion Primer A (5′-CCATCTCATCCCTGCGTGTCTCCGACTCAG-3′), a 10 bp Multiplex Identifier (MID) sequence (Roche, Indianapolis, IN), and the universal bacterial primer 8F (5′-AGAGTTTGATCCTGGCTCAG-3′).73 The reverse primer was composed of the Roche Titanium Primer B (5′-CCTATCCCCTGTGTGCCTTGGCAGTCTCAG-3′), the identical 10 bp MID sequence, and the reverse bacterial primer 338R (5′-GCTGCCTCCCGTAGGAGT-3′).74 The 16S rDNA amplicons from the pooled sample were sequenced on a 454 Genome Sequencer FLX Titanium instrument (Roche, Indianapolis, IN) in the Microbiome Core Facility (University of North Carolina, Chapel Hill, NC) using the GS FLX Titanium XLR70 sequencing reagents and corresponding protocols. Initial data analysis, base pair calling, and trimming of each sequence to yield high quality reads, were performed by Research Computing at the University of North Carolina at Chapel Hill.
Sequencing data analysis
Bioinformatics analysis of bacterial 16S amplicon pyrosequencing data was done using the Quantitative Insights Into Microbial Ecology (QIIME v.1.8.0) software pipeline.75 The combined raw sequencing data were demultiplexed and filtered; all reads with a length below 200 bp and with a quality score below 25 were removed. Sequencing data was denoised using Denoiser.76 Then, sequences were clustered into operational taxonomic units (OTU) at 97% similarity threshold using UCLUST77 from QIIME, and they were aligned in order to build a phylogenetic tree.78 A random selection of 719 sequences from each sample was used for rarefaction analysis to measure α diversity using observed species (S) and Phylogenetic Diversity (PD) metrics on rarefied OTU tables. Beta diversity and principal coordinates analysis (PCoA) were also calculated within QIIME using weighted and unweight Unifrac distances79 between samples at a depth of 719 sequences per sample to evaluate dissimilarities between the samples.
Bioinformatics analysis of gene functionality
We used the Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt)38 (version 1.0.0) algorithm to predict metagenome functional content from 16S sequencing input data. The Closed Reference OTU picking within QIIME using UCLUST77 was used against the GG GreenGenes database to create the OTU table adequate for input into PICRUSt. The OTU table was normalized by dividing abundance of each OTU by the predicted 16S abundance before creating the metagenome functional predictions table. The PICRUSt output was used in HMP Unified Metabolic Analysis Network (HUMAnN)80 pipeline to identify and group metagenomic data according to the presence or absence of microbial KEGG pathways and their relative abundance.
Statistical analyses
We computed the distance matrix between OTUs using Analysis of Similarities (ANOSIM) and Permutational Multivariate Analysis of Variance (PERMANOVA) tests within QIIME to evaluate similarities or dissimilarities between groups. Paired and unpaired t-tests were performed to identify significant differences in phylogenetic diversity (PD) and species richness (S) between the different samples (adjacent versus tumor tissues). The Wilcoxon Signed test was used to identify significant differences in relative abundances of bacterial taxa between paired samples while Steel-Dwass All Pairs tests were used for comparisons between cohorts with an α level set at 0.05. The Wilcoxon Signed test was also used for the identification of metabolic pathways and enzymes signficantly over or under represented within cohorts while Steel-Dwass All Pairs tests were used for comparisons between cohorts, we set an α level of 0.1 for both tests. All statistical analysis was performed in JMP genomics (SAS, JMP Genomics 10.0).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to the Consejeria de Sanidad del Principado de Asturias for making this collaboration possible.
Funding
IA is a Fulbright Scholar. The Microbiome Core Facility is supported in part by the NIH/NIDDK grant P30 DK34987. The Instituto Universitario de Oncología del Principado de Asturias receives financial support from the Fundación Caja de Ahorros de Asturias and Consejería de Educación del Principado de Asturias.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website
References
- 1.GLOBOCAN Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2012. 2012 [Google Scholar]
- 2.Colorectal Cancer - Facts and Figures American Cancer Society, 2011–2013:5–9. [Google Scholar]
- 3.Karim-Kos HE, de Vries E, Soerjomataram I, Lemmens V, Siesling S, Coebergh JW. Recent trends of cancer in Europe: a combined approach of incidence, survival and mortality for 17 cancer sites since the 1990s. Eur J Cancer 2008; 44:1345–89; PMID:18280139; http://dx.doi.org/ 10.1016/j.ejca.2007.12.015 [DOI] [PubMed] [Google Scholar]
- 4.Lopez-Abente G, Ardanaz E, Torrella-Ramos A, Mateos A, Delgado-Sanz C, Chirlaque MD, Colorectal Cancer Working G. Changes in colorectal cancer incidence and mortality trends in Spain. Ann Oncol 2010; 21(Suppl 3):iii76–82; PMID:20427364 [DOI] [PubMed] [Google Scholar]
- 5.Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, Gordon JI, Relman DA, Fraser-Liggett CM, Nelson KE. Metagenomic analysis of the human distal gut microbiome. Science 2006; 312:1355–9; PMID:16741115; http://dx.doi.org/ 10.1126/science.1124234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science 2005; 308:1635–8; PMID:15831718; http://dx.doi.org/ 10.1126/science.1110591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto JM, et al.. Enterotypes of the human gut microbiome. Nature 2011; 473:174–80; PMID:21508958; http://dx.doi.org/ 10.1038/nature09944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al.. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010; 464:59–65; PMID:20203603; http://dx.doi.org/ 10.1038/nature08821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maccaferri S, Biagi E, Brigidi P. Metagenomics: key to human gut microbiota. Digest Dis 2011; 29:525–30; PMID:22179207; http://dx.doi.org/ 10.1159/000332966 [DOI] [PubMed] [Google Scholar]
- 10.Zhao L. The gut microbiota and obesity: from correlation to causality. Nat Rev Microbiol 2013; 11:639–47; PMID:23912213; http://dx.doi.org/ 10.1038/nrmicro3089 [DOI] [PubMed] [Google Scholar]
- 11.Shen J, Obin MS, Zhao L. The gut microbiota, obesity and insulin resistance. Mol Aspects Med 2013; 34:39–58; PMID:23159341; http://dx.doi.org/ 10.1016/j.mam.2012.11.001 [DOI] [PubMed] [Google Scholar]
- 12.Vaarala O. Human intestinal microbiota and type 1 diabetes. Curr Diab Rep 2013; 13:601–7; PMID:23934614; http://dx.doi.org/ 10.1007/s11892-013-0409-5 [DOI] [PubMed] [Google Scholar]
- 13.Naseer MI, Bibi F, Alqahtani MH, Chaudhary AG, Azhar EI, Kamal MA, Yasir M. Role of gut microbiota in obesity, type 2 diabetes and Alzheimer's disease. CNS Neurol Disord Drug Targets 2014; 13:305–11; PMID:24059313; http://dx.doi.org/ 10.2174/18715273113126660147 [DOI] [PubMed] [Google Scholar]
- 14.Tilg H, Moschen AR. Microbiota and diabetes: an evolving relationship. Gut 2014; 63(9):1513–21; PMID:24833634; http://dx.doi.org/ 10.1136/gutjnl-2014-306928 [DOI] [PubMed] [Google Scholar]
- 15.Sears CL, Garrett WS. Microbes, microbiota, and colon cancer. Cell Host Microbe 2014; 15:317–28; PMID:24629338; http://dx.doi.org/ 10.1016/j.chom.2014.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rowland IR. The role of the gastrointestinal microbiota in colorectal cancer. Curr Pharm Des 2009; 15:1524–7; PMID:19442169; http://dx.doi.org/ 10.2174/138161209788168191 [DOI] [PubMed] [Google Scholar]
- 17.Sofi F, Cesari F, Abbate R, Gensini GF, Casini A. Adherence to Mediterranean diet and health status: meta-analysis. Bmj 2008; 337:a1344; PMID:18786971; http://dx.doi.org/ 10.1136/bmj.a1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Couto E, Boffetta P, Lagiou P, Ferrari P, Buckland G, Overvad K, Dahm CC, Tjonneland A, Olsen A, Clavel-Chapelon F, et al.. Mediterranean dietary pattern and cancer risk in the EPIC cohort. Br J Cancer 2011; 104:1493–9; PMID:21468044; http://dx.doi.org/ 10.1038/bjc.2011.106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marlow G, Ellett S, Ferguson IR, Zhu S, Karunasinghe N, Jesuthasan AC, Han DY, Fraser AG, Ferguson LR. Transcriptomics to study the effect of a Mediterranean-inspired diet on inflammation in Crohn's disease patients. Hum Genomics 2013; 7:24; PMID:24283712; http://dx.doi.org/ 10.1186/1479-7364-7-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, et al.. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014; 505:559–63; PMID:24336217; http://dx.doi.org/ 10.1038/nature12820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahn J, Sinha R, Pei Z, Dominianni C, Wu J, Shi J, Goedert JJ, Hayes RB, Yang L. Human gut microbiome and risk for colorectal cancer. J Natl Cancer Inst 2013; 105:1907–11; PMID:24316595; http://dx.doi.org/ 10.1093/jnci/djt300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu Q, Gao R, Wu W, Qin H. The role of gut microbiota in the pathogenesis of colorectal cancer. Tumour Biol 2013; 34:1285–300; PMID:23397545; http://dx.doi.org/ 10.1007/s13277-013-0684-4 [DOI] [PubMed] [Google Scholar]
- 23.Azcarate-Peril MA, Sikes M, Bruno-Barcena JM. The intestinal microbiota, gastrointestinal environment and colorectal cancer: a putative role for probiotics in prevention of colorectal cancer? Am J Physiol Gastrointest Liver Physiol 2011; 301:G401–24; PMID:21700901; http://dx.doi.org/ 10.1152/ajpgi.00110.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gorbach SL, Thadepalli H. Isolation of Clostridium in human infections: evaluation of 114 cases. J Infect Dis 1975; 131(Suppl):S81–5; PMID:805193; http://dx.doi.org/ 10.1093/infdis/131.Supplement.S81 [DOI] [PubMed] [Google Scholar]
- 25.Klein RS, Recco RA, Catalano MT, Edberg SC, Casey JI, Steigbigel NH. Association of Streptococcus bovis with carcinoma of the colon. N Engl J Med 1977; 297:800–2; PMID:408687; http://dx.doi.org/ 10.1056/NEJM197710132971503 [DOI] [PubMed] [Google Scholar]
- 26.Keusch GT. Opportunistic infections in colon carcinoma. Am J Clin Nutr 1974; 27:1481–5; PMID:4611196 [DOI] [PubMed] [Google Scholar]
- 27.McCoy AN, Araujo-Perez F, Azcarate-Peril A, Yeh JJ, Sandler RS, Keku TO. Fusobacterium is associated with colorectal adenomas. PLoS One 2013; 8:e53653; PMID:23335968; http://dx.doi.org/ 10.1371/journal.pone.0053653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, Ojesina AI, Jung J, Bass AJ, Tabernero J, et al.. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res 2012; 22:292–8; PMID:22009990; http://dx.doi.org/ 10.1101/gr.126573.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marchesi JR, Dutilh BE, Hall N, Peters WH, Roelofs R, Boleij A, Tjalsma H. Towards the human colorectal cancer microbiome. PLoS One 2011; 6:e20447; PMID:21647227; http://dx.doi.org/ 10.1371/journal.pone.0020447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hamer HM, Jonkers D, Venema K, Vanhoutvin S, Troost FJ, Brummer RJ. Review article: the role of butyrate on colonic function. Aliment Pharmacol Ther 2008; 27:104–19; PMID:17973645; http://dx.doi.org/ 10.1111/j.1365-2036.2007.03562.x [DOI] [PubMed] [Google Scholar]
- 31.Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, Clancy TE, Chung DC, Lochhead P, Hold GL, et al.. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013; 14:207–15; PMID:23954159; http://dx.doi.org/ 10.1016/j.chom.2013.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brim H, Yooseph S, Zoetendal EG, Lee E, Torralbo M, Laiyemo AO, Shokrani B, Nelson K, Ashktorab H. Microbiome analysis of stool samples from African Americans with colon polyps. PloS One 2013; 8:e81352; PMID:24376500; http://dx.doi.org/ 10.1371/journal.pone.0081352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weir TL, Manter DK, Sheflin AM, Barnett BA, Heuberger AL, Ryan EP. Stool microbiome and metabolome differences between colorectal cancer patients and healthy adults. PLoS One 2013; 8:e70803; PMID:23940645; http://dx.doi.org/ 10.1371/journal.pone.0070803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sobhani I, Tap J, Roudot-Thoraval F, Roperch JP, Letulle S, Langella P, Corthier G, Tran Van Nhieu J, Furet JP. Microbial dysbiosis in colorectal cancer (CRC) patients. PloS One 2011; 6:e16393; PMID:21297998; http://dx.doi.org/ 10.1371/journal.pone.0016393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, Bewtra M, Knights D, Walters WA, Knight R, et al.. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011; 334:105–8; PMID:21885731; http://dx.doi.org/ 10.1126/science.1208344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ou J, Carbonero F, Zoetendal EG, DeLany JP, Wang M, Newton K, Gaskins HR, O'Keefe SJ. Diet, microbiota, and microbial metabolites in colon cancer risk in rural Africans and African Americans. Am J Clin Nutr 2013; 98:111–20; PMID:23719549; http://dx.doi.org/ 10.3945/ajcn.112.056689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O'Keefe SJD, Chung D, Mahmoud N, Sepulveda AR, Manafe M, Arch J, Adada H, van der Merwe T. Why do African Americans get more colon cancer than native Africans? J Nutr 2007; 137:175S–82S; PMID:17182822 [DOI] [PubMed] [Google Scholar]
- 38.Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, et al.. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 2013; 31:814–21; PMID:23975157; http://dx.doi.org/ 10.1038/nbt.2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al.. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010; 464:59–65; PMID:20203603; http://dx.doi.org/ 10.1038/nature08821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dave M, Higgins PD, Middha S, Rioux KP. The human gut microbiome: current knowledge, challenges, and future directions. Transl Res 2012; 160:246–57; PMID:22683238; http://dx.doi.org/ 10.1016/j.trsl.2012.05.003 [DOI] [PubMed] [Google Scholar]
- 41.Keku TO, Dulal S, Deveaux A, Jovov B, Han X. The gastrointestinal microbiota and colorectal cancer. Am J Physiol Gastrointest Liver Physiol 2014; 308(5):G351-G363; http://dx.doi.org/ 10.1152/ajpgi003602012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen W, Liu F, Ling Z, Tong X, Xiang C. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PloS One 2012; 7:e39743; PMID:22761885; http://dx.doi.org/ 10.1371/journal.pone.0039743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shen XJ, Rawls JF, Randall T, Burcal L, Mpande CN, Jenkins N, Jovov B, Abdo Z, Sandler RS, Keku TO. Molecular characterization of mucosal adherent bacteria and associations with colorectal adenomas. Gut Microbes 2010; 1:138–47; PMID:20740058; http://dx.doi.org/ 10.4161/gmic.1.3.12360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, et al.. Human gut microbiome viewed across age and geography. Nature 2012; 486:222–7; PMID:22699611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, Collini S, Pieraccini G, Lionetti P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A 2010; 107:14691–6; PMID:20679230; http://dx.doi.org/ 10.1073/pnas.1005963107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Turroni F, Duranti S, Bottacini F, Guglielmetti S, Van Sinderen D, Ventura M. Bifidobacterium bifidum as an example of a specialized human gut commensal. Front Microbiol 2014; 5:437; PMID:25191315; http://dx.doi.org/ 10.3389/fmicb.2014.00437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Belenguer A, Duncan SH, Calder AG, Holtrop G, Louis P, Lobley GE, Flint HJ. Two routes of metabolic cross-feeding between Bifidobacterium adolescentis and butyrate-producing anaerobes from the human gut. Appl Envir Microbiol 2006; 72:3593–9; PMID:16672507; http://dx.doi.org/ 10.1128/AEM.72.5.3593-3599.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hill MJ, Drasar BS, Hawksworth G, Aries V, Crowther JS, Williams RE. Bacteria and aetiology of cancer of large bowel. Lancet 1971; 1:95–100; PMID:4099643; http://dx.doi.org/ 10.1016/S0140-6736(71)90837-3 [DOI] [PubMed] [Google Scholar]
- 49.Toprak N, Yagci A, Gulluoglu B, Akin M, Demirkalem P, Celenk T, Soyletir G. A possible role of Bacteroides fragilis enterotoxin in the aetiology of colorectal cancer. Clin Microbiol Infect 2006; 8:782–6; PMID:16842574; http://dx.doi.org/ 10.1111/j.1469-0691.2006.01494.x [DOI] [PubMed] [Google Scholar]
- 50.Sears CL. Enterotoxigenic Bacteroides fragilis: a rogue among symbiotes. Clin Microbiol Rev 2009; 2:349–69; PMID:19366918; http://dx.doi.org/ 10.1128/CMR.00053-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reynolds TY, Rockwell S, Glazer PM. Genetic instability induced by the tumor microenvironment. Cancer Res 1996; 56:5754–7; PMID:8971187 [PubMed] [Google Scholar]
- 52.Hirayama A, Kami K, Sugimoto M, Sugawara M, Toki N, Onozuka H, Kinoshita T, Saito N, Ochiai A, Tomita M, et al.. Quantitative metabolome profiling of colon and stomach cancer microenvironment by capillary electrophoresis time-of-flight mass spectrometry. Cancer Res 2009; 69:4918–25; PMID:19458066; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-4806 [DOI] [PubMed] [Google Scholar]
- 53.Decker MD. Eikenella corrodens. Infect Control 1986; 7:36–41; PMID:3512467 [DOI] [PubMed] [Google Scholar]
- 54.Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner AC, Yu WH, Lakshmanan A, Wade WG. The human oral microbiome. J Bacteriol 2010; 192:5002–17; PMID:20656903; http://dx.doi.org/ 10.1128/JB.00542-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fricke WF, Maddox C, Song Y, Bromberg JS. Human microbiota characterization in the course of renal transplantation. Am J Transplant 2014; 14:416–27; PMID:24373208; http://dx.doi.org/ 10.1111/ajt.12588 [DOI] [PubMed] [Google Scholar]
- 56.Lee SH, Fang YC, Luo JP, Kuo HI, Chen HC. Inflammatory pseudotumour associated with chronic persistent Eikenella corrodens infection: a case report and brief review. J Clin Pathol 2003; 56:868–70; PMID:14600136; http://dx.doi.org/ 10.1136/jcp.56.11.868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yetimoglu C, Rafeiner P, Engel D, Fournier JY. Spinal infections due to Eikenella corrodens: case report and literature review. Neurochirurgie 2014; 60:197–200; PMID:24874721; http://dx.doi.org/ 10.1016/j.neuchi.2014.03.002 [DOI] [PubMed] [Google Scholar]
- 58.Rivas Castillo FJ, Gomez Martinez JR, Lopez Alvarez F, Garcia Velasco F. Eikenella corrodens: a rare cause of deep neck infection. Acta Otorrinolaringol Esp 2014; pii: S0001-6519(14)00125-3; PMID:25308794; http://dx.doi.org/10.1016 [DOI] [PubMed] [Google Scholar]
- 59.Gowda AL, Mease SJ, Dhar Y. Eikenella corrodens septic hip arthritis in a healthy adult treated with arthroscopic irrigation and debridement. Am J Orthop (Belle Mead NJ) 2014; 43:419–21; PMID:25251528 [PubMed] [Google Scholar]
- 60.Garnier F, Masson G, Bedu A, Masson P, Decroisette E, Guigonis V, Fermeaux V, De Barbentane MC, Denis F, Ploy MC. Maternofetal infections due to Eikenella corrodens. J Med Microbiol 2009; 58:273–5; PMID:19141750; http://dx.doi.org/ 10.1099/jmm.0.002568-0 [DOI] [PubMed] [Google Scholar]
- 61.Ohkusa T, Okayasu I, Ogihara T, Morita K, Ogawa M, Sato N. Induction of experimental ulcerative colitis by Fusobacterium varium isolated from colonic mucosa of patients with ulcerative colitis. Gut 2003; 52:79–83; PMID:12477765; http://dx.doi.org/ 10.1136/gut.52.1.79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Warren RL, Freeman DJ, Pleasance S, Watson P, Moore RA, Cochrane K, Allen-Vercoe E, Holt RA. Co-occurrence of anaerobic bacteria in colorectal carcinomas. Microbiome 2013; 1:16; PMID:24450771; http://dx.doi.org/ 10.1186/2049-2618-1-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, Barnes R, Watson P, Allen-Vercoe E, Moore RA, et al.. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res 2012; 22:299–306; PMID:22009989; http://dx.doi.org/ 10.1101/gr.126516.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tahara T, Yamamoto E, Suzuki H, Maruyama R, Chung W, Garriga J, Jelinek J, Yamano HO, Sugai T, An B, et al.. Fusobacterium in colonic flora and molecular features of colorectal carcinoma. Cancer Res 2014; 74:1311–8; PMID:24385213; http://dx.doi.org/ 10.1158/0008-5472.CAN-13-1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Geng J, Fan H, Tang X, Zhai H, Zhang Z. Diversified pattern of the human colorectal cancer microbiome. Gut Pathog 2013; 5:2; PMID:23497613; http://dx.doi.org/ 10.1186/1757-4749-5-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dejea CM, Wick EC, Hechenbleikner EM, White JR, Mark Welch JL, Rossetti BJ, Peterson SN, Snesrud EC, Borisy GG, Lazarev M, et al.. Microbiota organization is a distinct feature of proximal colorectal cancers. Proc Natl Acad Sci U S A 2014; 111:18321–6; PMID:25489084; http://dx.doi.org/ 10.1073/pnas.1406199111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, Ojesina AI, Jung J, Bass AJ, Tabernero J, et al.. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res 2011; 146:1489–99; PMID:22009990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen HM, Yu YN, Wang JL, Lin YW, Kong X, Yang CQ, Yang L, Liu ZJ, Yuan YZ, Liu F, et al.. Decreased dietary fiber intake and structural alteration of gut microbiota in patients with advanced colorectal adenoma. Am J Clin Nutr 2013; 97:1044–52; PMID:23553152; http://dx.doi.org/ 10.3945/ajcn.112.046607 [DOI] [PubMed] [Google Scholar]
- 69.Zackular JP, Rogers MA, Ruffin MTt, Schloss PD. The human gut microbiome as a screening tool for colorectal cancer. Cancer Prev Res (Phila) 2014; 7:1112–21; PMID:25104642; http://dx.doi.org/ 10.1158/1940-6207.CAPR-14-0129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Burns MB, Lynch J, Starr TK, Knights D, Bleckham R. Virulence genes are a signature of the microbiome in the colorectal tumor microenvironment. 2014; http://dx.doi.org/ 10.1101/009431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dutilh BE, Backus L, van Hijum SA, Tjalsma H. Screening metatranscriptomes for toxin genes as functional drivers of human colorectal cancer. Best Pract Res Clin Gastroenterol 2013; 27:85–99; PMID:23768555; http://dx.doi.org/ 10.1016/j.bpg.2013.03.008 [DOI] [PubMed] [Google Scholar]
- 72.Devine AA, Gonzalez A, Speck KE, Knight R, Helmrath M, Lund PK, Azcarate-Peril MA. Impact of ileocecal resection and concomitant antibiotics on the microbiome of the murine jejunum and colon. PloS One 2013; 8:e73140; PMID:24015295; http://dx.doi.org/ 10.1371/journal.pone.0073140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Edwards U, Rogall T, Blocker H, Emde M, Bottger EC. Isolation and direct complete nucleotide determination of entire genes. Characterization of a gene coding for 16S ribosomal RNA. Nucleic Acids Res 1989; 17:7843–53; PMID:2798131; http://dx.doi.org/ 10.1093/nar/17.19.7843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fierer N, Hamady M, Lauber CL, Knight R. The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proc Natl Acad Sci U S A 2008; 105:17994–9; PMID:19004758; http://dx.doi.org/ 10.1073/pnas.0807920105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, et al.. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 2010; 7:335–6; PMID:20383131; http://dx.doi.org/ 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Reeder J, Knight R. Rapidly denoising pyrosequencing amplicon reads by exploiting rank-abundance distributions. Nat Methods 2010; 7:668–9; PMID:20805793; http://dx.doi.org/ 10.1038/nmeth0910-668b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010; 26:2460–1; PMID:20709691; http://dx.doi.org/ 10.1093/bioinformatics/btq461 [DOI] [PubMed] [Google Scholar]
- 78.Price MN, Dehal PS, Arkin AP. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One 2010; 5:e9490; PMID:20224823; http://dx.doi.org/ 10.1371/journal.pone.0009490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lozupone C, Hamady M, Knight R. UniFrac–an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics 2006; 7:371; PMID:16893466; http://dx.doi.org/ 10.1186/1471-2105-7-371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Abubucker S, Segata N, Goll J, Schubert AM, Izard J, Cantarel BL, Rodriguez-Mueller B, Zucker J, Thiagarajan M, Henrissat B, et al.. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput Biol 2012; 8:e1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.