

Abstract
RNA family models describe classes of functionally related, non-coding RNAs based on sequence and structure conservation. The most important method for modeling RNA families is the use of covariance models, which are stochastic models that serve in the discovery of yet unknown, homologous RNAs. However, the performance of covariance models in finding remote homologs is poor for RNA families with high sequence conservation, while for families with high structure but low sequence conservation, these models are difficult to built in the first place. A complementary approach to RNA family modeling involves the use of thermodynamic matchers. Thermodynamic matchers are RNA folding programs, based on the established thermodynamic model, but tailored to a specific structural motif. As thermodynamic matchers focus on structure and folding energy, they unfold their potential in discovering homologs, when high structure conservation is paired with low sequence conservation. In contrast to covariance models, construction of thermodynamic matchers does not require an input alignment, but requires human design decisions and experimentation, and hence, model construction is more laborious. Here we report a case study on an RNA family that was constructed by means of thermodynamic matchers. It starts from a set of known but structurally different members of the same RNA family. The consensus secondary structure of this family consists of 2 to 4 adjacent hairpins. Each hairpin loop carries the same motif, CCUCCUCCC, while the stems show high variability in their nucleotide content. The present study describes (1) a novel approach for the integration of the structurally varying family into a single RNA family model by means of the thermodynamic matcher methodology, and (2) provides the results of homology searches that were conducted with this model in a wide spectrum of bacterial species.
Keywords: alphaproteobacteria, cuckoo RNA, family model, homology search, small RNA, structural RNA, thermodynamic matcher
Abbreviations
- sRNA
small non-coding RNA
- RFM
RNA family model
- CM
covariance model
- HMM
hidden Markov model
- MFE
minimum free energy
- TDM
thermodynamic matcher
- dRNA-seq
differential RNA sequencing
- aSD
anti Shine-Dalgarno
- CIN
conserved intergenic neighborhood
- OG
orthologous group of genes
- RBS
ribosome binding site
Introduction
Modeling RNA families
Recent interest in the non-coding part of the genome has created a wealth of new results about the functional repertoire of RNA. RNA-seq experiments based on next-generation sequencing technology have become common practice in discovering novel small non-coding RNAs (sRNAs).1,2 The advantage of homology searches for sRNAs that employ the results of RNA-seq experiments, compared to in-silico de novo sRNA prediction, is that one starts from an experimentally verified transcript, and often, the exact length of the hypothetical RNA gene is known.3 Even when a function of a newly detected transcript is not yet suspected, its conservation in sequence and/or structure in a number of related species can be taken as first evidence that the transcript is a bona-fide RNA gene. As a result of RNA gene hunting efforts, the Rfam database4 has grown from 400 RNA family models (RFMs) in 2004 to 2,208 at the time of this writing.
The Rfam effort has established the creation of covariance models (CMs)5 as the de-facto standard for modeling structural RNA families. A covariance model starts from a sequence alignment that supports a conserved consensus structure, and creates a stochastic model.4,6 It can be used to test individual transcripts for family membership, or to scan complete genomes for RNA gene prediction.
CMs are implemented as stochastic context free grammars, a generalization of profile hidden Markov models (HMMs)7 to incorporate conserved structural information. When sequence conservation is high and structure conservation is weak in a family, CMs behave much like profile HMMs. In contrast, when structure is strongly conserved, but sequence is diverged, CMs generalize better, i.e. they are more successful in finding family members also in remotely related species. However, when creating such a model, we face a chicken-and-egg problem: How do we find a group of sequences in the first place, which are diverged in sequence but not in structure? Even when there are some sequence motifs with high conservation, these may not allow for a BLAST search of high specificity. In this situation, an alternative or complementary route to construct an RFM is desirable. One such strategy is the construction of thermodynamic matchers (TDMs). It is worked out in this study for the “cuckoo” RNA family.
Biology background: the cuckoo family
In recent years, high-throughput sequencing studies of the transcriptome (RNA-seq) in bacteria revealed a multitude of novel sRNAs. In 2009, Berghoff et al.8 used differential RNA sequencing (dRNA-seq) for the identification of sRNAs in the photosynthetic alphaproteobacterium Rhodobacter sphaeroides, which are induced under photooxidative stress. Among other sRNAs, a cluster of 4 paralogous sRNAs, named RSs0680a-d, was discovered and verified by Northern blot experiments. Berghoff et al.8 showed that RSs0680a-d are co-transcribed with an upstream located, so far uncharacterized hypothetical gene, RSP_6037, and expressed under the control of an RpoHI/RpoHII dependent promoter. Besides, a terminator-like hairpin was found downstream of the sRNA cluster. In two complementing studies it could be demonstrated that RSs0680a is induced under heat stress as well, and is directly bound by Hfq.9,10 Despite the characterization efforts, the precise function of the RSs0680 family remains to be elucidated.
Derived from the RS0680a-d sRNAs, the RSs0680 RNA family features a modular secondary structure, which is composed of 2 adjacent stem loops of similar size and a conserved loop motif CCUCCUCCC, resembling an anti Shine-Dalgarno sequence (aSD). Comparative analysis in selected alphaproteobacteria led to the identification of 17 RSs0680a homologs in the orders of the Rhodobacterales and the Rhizobiales.8 For 14 of these sRNAs, a preserved genomic context was observed.
Besides, several studies using computational and experimental approaches identified sRNAs in Sinorhizobium meliloti, which represent structural variants of RSs0680. The first structural variants, C14 and A6, were described by del Val et al.11 who predicted a total set of 32 S. meliloti sRNAs by means of eQRNA12 and RNAz.13 C14 was further confirmed by Northern analysis under various conditions except for the stationary growth phase. Different from RSs0680, the secondary structures of C14 and A6 harbor 3 and 4 adjacent stem loops, respectively, each carrying again the sequence motif CCUCCUCCC. Based on sRNAPredictHT, Valverde et al.14 rediscovered C14 and predicted the sm7 sRNA, which is structurally similar to C14. In addition, it could be demonstrated in a microarray experiment that C14 is induced under salt stress. The dRNA-Seq study of Schlüter et al.15 revealed numerous sRNAs in S. meliloti. In addition to the previously known sRNAs, A6 (SmelA099), C14 (SmelC397), and sm7 (SmelC398), they identified 3 new sRNAs, SmelA075, SmelB161, and SmelC025, which consist of 3 CCUCCUCCC-modules. Experimentally determined transcription start sites by 5′-RACE and RNA-seq for the 6 S. meliloti sRNAs are in good agreement, differing by only a few nucleotides.11,15,16 A first attempt to identify homologs of C14, as part of a study that aimed at kingdom-wide predictions and annotations of trans-encoded sRNA genes, yielded 9 predictions in closely related Rhizobiales.17 Recently, 52 trans-encoded sRNAs of Schlüter et al.15 including SmelA075 and SmelA099, served as the basis for the construction of 39 RNA family models.18 While we mainly used CMs for the compilation of RFMs, in case of SmelA075 and SmelA099 we constructed the respective RFMs, RFMSmelA075 and RFMSmelA099, by means of TDMs. Relatives of SmelA075 and SmelA099 are highly abundant and have been found in multiple copies in the Rhizobiaceae, Phyllobacteriaceae and Brucellaceae. As an example, S. meliloti encodes 5 copies of RFMSmelA075, which are located not only on the chromosome (SmelC025, SmelC397, SmelC398) but are also present on the megaplasmids pSymA (SmelA075) and pSymB (SmelB161). Northern hybridization showed that SmelA075, similar to RSs0680a, is induced under heat stress.15 In total, both families comprise 121 sRNAs (RFMSmelA075 83 sRNAs, RFMSmelA099 38 sRNAs). Due to the presumably paralogous copies on different replicons, fragmented microsynteny was observed for both models. Nevertheless, subsets of homologous RNAs showed a preserved genomic context. Additionally, 3 members of RFMSmelA075 and RFMSmelA099 have been confirmed by RNA-seq studies in Rhizobium etli and Agrobacterium tumefaciens.19,20
In a comparative analysis, del Val et al.21 applied CMs to build RFMs for C14, termed ar14, and 5 other S. meliloti RNAs. The ar14 RFM includes 101 sRNAs. ar14 and RFMSmelA075, which describe the same family of 3 hairpin forming sRNAs, are generally in good agreement, aside from single members that exist exclusively in either of the families. However, as we know today, 33 members of the ar14 family exhibit 4 instead of the 3 annotated stem loops, and thus belong to RFMSmelA099. This is not the only example for misclassification, several sequences that were defined as homologs of RSs0680 actually belong to either RFMSmelA075 or RFMSmelA099.8,18
Regarding the RNA secondary structures and the conserved sequence motif within the loop sequences of the 3 RNA families RSs0680, RFMSmelA075, and RFMSmelA099, it is reasonable to conclude that they all represent structural variants of the same family and that hairpins are the building blocks of their modular RNA architecture (Fig. 1).
Figure 1.
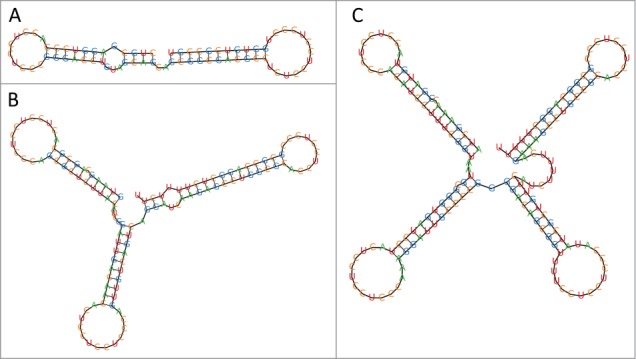
RNA structures of experimentally validated cuckoo RNAs obtained by TDM folding. (A) RSs0680a RNA (position 692386-692458, Rhodobacter sphaeroides 2.4.1), (B) ReC11 RNA (462572-462689, Rhizobium etli CFN 42), (C) L5 RNA (1831446-1831604, Agrobacterium tumefaciens str. C58).
In summary, as of 2013, 145 members belonging to one of the 3 structural variants of this RNA family had been identified by various means, but a general model for the family was lacking (Table S5).
In this study we (1) present a novel approach to integrate the structurally varying RFMs RSs0680, RFMSmelA075, and RFMSmelA099 into a single RFM by means of the thermodynamic matcher methodology, (2) use the novel family model to conduct extensive homology searches and provide an updated list of family members. In the following we refer to the joint family as the cuckoo family. Our name for the family is derived from this sequence motif CCUCCUCCC, which in German is phonetically the same as "Kuckuck" (cuckoo).
Results
Our study has 2 results: One is the considerably extended cuckoo family, and the other one is the TDM developed for its construction and to be used for its maintenance as new bacterial genome sequences become available. The former result is described in this paragraph, the latter in the methods section.
Our new cuckoo RNA family model was used to perform comprehensive screens for cuckoo RNAs on 2,680 prokaryotic genomes downloaded from NCBI's RefSeq database and 9 additional rhodobacterial genomes that were part of the comparative study of Berghoff et al.8 A list of all included bacterial genomes is provided in Table S6. Our approach revealed in total 321 cuckoo RNAs, which were distributed on 156 replicons of 78 alphaproteobacteria (Table 1, Tables S1-S5). 176 of the 321 cuckoo RNAs were previously unknown. Within the alphaproteobacteria, cuckoo RNAs were found in the genomes of 31 Rhodobacteraceae; and in the families of Rhizobiaceae, Brucellaceae, Hyphomicrobiaceae, and Phyllobacteriaceae of the order of the Rhizobiales, cuckoo RNAs occur in 22, 18, 1, and 5 genomes, respectively. Besides, members of the cuckoo family were recovered in the unclassified alphaproteobacterium Polymorphum gilvum.
Table 1.
Distribution of cuckoo RNAs. The table summarizes the number of cuckoo RNAs and their distribution across CINs for each species. The occurrences of cuckoo RNAs divided into structural variants are displayed in columns HP2 to HP4 Columns CIN1–CIN6 show the distribution of cuckoo RNAs within the 6 CINs. Each digit represents a cuckoo RNA within a CIN, while the digit's value reflects the number of modules. Unless denoted by a leading character (c = secondary chromosome, p = plasmid) a CIN and therefore the corresponding cuckoo RNA is located on the primary chromosome of the respective bacterium. The first row, for example, reads in words as follows: “In Brucella abortus A13334, we find 4 cuckoo RNAs, one with 2 hairpins, 2 with 3 hairpins, and one with 4 hairpins. The 4HP cuckoo is found on the main chromosome in neighborhood CIN1; in the same neighborhood, but on the secondary chromosome, we find a 2HP cuckoo. The 3HP cuckoos are found on the main chromosome in neighborhood CIN5, and on the secondary chromosome in neighborhood CIN6.” See Table S1 for complete sequences and detailed results.
| Species | HP2 | HP3 | HP4 | CIN1 | CIN2 | CIN3 | CIN4 | CIN5 | CIN6 |
|---|---|---|---|---|---|---|---|---|---|
| Polymorphum gilvum SL003B-26A1 | 0 | 1 | 1 | ||||||
| Brucellaceae | |||||||||
| Brucella abortus A13334 | 1 | 2 | 1 | 4;c2 | 3 | c3 | |||
| Brucella abortus S19 | 1 | 2 | 1 | 4;c2 | 3 | c3 | |||
| Brucella abortus bv. 1 str. 9-941 | 1 | 1 | 1 | 4;c2 | 3 | ||||
| Brucella canis ATCC 23365 | 1 | 1 | 1 | 4 | 3 | c2 | |||
| Brucella canis HSK A52141 | 1 | 1 | 1 | 4 | 3 | c2 | |||
| Brucella melitensis ATCC 23457 | 1 | 1 | 1 | 4;c2 | 3 | ||||
| Brucella melitensis M28 | 1 | 1 | 1 | 4;c2 | 3 | ||||
| Brucella melitensis M5-90 | 1 | 1 | 1 | 4;c2 | 3 | ||||
| Brucella melitensis NI | 1 | 1 | 1 | 4;c2 | 3 | ||||
| Brucella melitensis biovar Abortus 2308 | 2 | 1 | 1 | 4;c2 | 3 | c2 | |||
| Brucella melitensis bv. 1 str. 16M | 2 | 1 | 1 | 4 | 3 | c2 | |||
| Brucella microti CCM 4915 | 2 | 1 | 1 | 4;c2 | 3 | c2 | |||
| Brucella ovis ATCC 25840 | 1 | 1 | 1 | 4;c2 | |||||
| Brucella pinnipedialis B2/94 | 1 | 2 | 1 | 4;c2 | 3 | c3 | |||
| Brucella suis 1330 | 1 | 2 | 1 | 4;c2 | 3 | c3 | |||
| Brucella suis ATCC 23445 | 1 | 2 | 1 | 4;c2 | |||||
| Brucella suis VBI22 | 1 | 2 | 1 | 4;c2 | 3 | c3 | |||
| Ochrobactrum anthropi ATCC 49188 | 0 | 4 | 1 | 4;c3 | |||||
| Hyphomicrobiaceae | |||||||||
| Pelagibacterium halotolerans B2 | 0 | 2 | 0 | ||||||
| Phyllobacteriaceae | |||||||||
| Chelativorans sp. BNC1 | 2 | 4 | 0 | 3 | |||||
| Mesorhizobium australicum WSM2073 | 0 | 5 | 0 | 333 | |||||
| Mesorhizobium ciceri biovar biserrulae WSM1271 | 0 | 6 | 0 | 333 | |||||
| Mesorhizobium loti MAFF303099 | 0 | 6 | 0 | ||||||
| Mesorhizobium opportunistum WSM2075 | 0 | 5 | 0 | 333 | |||||
| Rhizobiaceae | |||||||||
| Agrobacterium tumefaciens str. C58 | 0 | 1 | 2 | ||||||
| Agrobacterium radiobacter K84 | 0 | 3 | 3 | 3 | 3 | 3 | |||
| Agrobacterium sp H13-3 | 0 | 2 | 2 | l4 | |||||
| Agrobacterium vitis S4 | 0 | 3 | 3 | c4 | |||||
| Rhizobium etli CFN 42 | 0 | 4 | 2 | p4 | 3 | 3 | |||
| Rhizobium etli CIAT 652 | 1 | 3 | 3 | p4 | 3 | ||||
| Rhizobium etli bv. mimosae str. Mim1 | 0 | 4 | 1 | p4 | 3 | 3 | |||
| Rhizobium leguminosarum bv. trifolii WSM1325 | 0 | 4 | 4 | 3;p4;p4 | 3 | 3 | |||
| Rhizobium leguminosarum bv. trifolii WSM2304 | 1 | 4 | 2 | 3;p4 | 3 | 3 | |||
| Rhizobium leguminosarum bv. viciae 3841 | 0 | 4 | 1 | p4 | 3 | ||||
| Rhizobium tropici CIAT 899 | 0 | 3 | 1 | p4 | 3 | 3 | |||
| Sinorhizobium fredii HH103 | 0 | 6 | 0 | 33 | p3 | ||||
| Sinorhizobium fredii NGR234 | 0 | 4 | 0 | 33 | p3 | ||||
| Sinorhizobium fredii USDA 257 | 0 | 6 | 0 | 33 | 3 | ||||
| Sinorhizobium medicae WSM419 | 1 | 4 | 2 | 33;p4 | p3 | ||||
| Sinorhizobium meliloti 1021 | 0 | 5 | 1 | ||||||
| Sinorhizobium meliloti 2011 | 0 | 5 | 1 | 3 | p3 | ||||
| Sinorhizobium meliloti AK83 | 0 | 5 | 1 | 3 | 3 | c3 | |||
| Sinorhizobium meliloti BL225C | 0 | 4 | 1 | 33 | 3 | p3 | |||
| Sinorhizobium meliloti GR4 | 0 | 4 | 1 | 33 | 3 | p3 | |||
| Sinorhizobium meliloti Rm41 | 0 | 5 | 1 | 3 | p3 | ||||
| Sinorhizobium meliloti SM11 | 0 | 4 | 1 | 33 | 3 | p3 | |||
| Rhodobacteraceae | |||||||||
| Parvibaculum lavamentivorans DS-1 | 1 | 0 | 0 | ||||||
| Dinoroseobacter shibae DFL 12 | 1 | 0 | 0 | ||||||
| Jannaschia sp. CCS1 | 3 | 0 | 0 | 222 | |||||
| Ketogulonicigenium vulgare WSH-001 | 0 | 1 | 0 | ||||||
| Ketogulonicigenium vulgare Y25 | 0 | 1 | 0 | ||||||
| Loktanella vestfoldensis DSM 16212 | 2 | 1 | 0 | ||||||
| Loktanella vestfoldensis SKA53 | 2 | 1 | 0 | 223 | |||||
| Oceanicola batsensis HTCC2597 | 2 | 0 | 1 | 24 | |||||
| Oceanicola granulosus HTCC2516 | 4 | 1 | 0 | 22223 | |||||
| Octadecabacter antarcticus 307 | 2 | 0 | 4 | ||||||
| Octadecabacter arcticus 238 | 3 | 1 | 3 | ||||||
| Paracoccus aminophilus JCM 7686 | 0 | 0 | 2 | 44 | |||||
| Paracoccus denitrificans PD1222 | 4 | 0 | 1 | 22224 | |||||
| Phaeobacter gallaeciensis 2.10 | 2 | 0 | 0 | 22 | |||||
| Phaeobacter gallaeciensis DSM 17395 | 2 | 0 | 0 | 22 | |||||
| Phaeobacter gallaeciensis DSM 26640 | 2 | 0 | 0 | ||||||
| Pseudovibrio sp FO-BEG1 | 0 | 0 | 1 | ||||||
| Rhodobacter capsulatus SB 1003 | 4 | 0 | 0 | 2222 | |||||
| Rhodobacter sphaeroides 2.4.1 | 7 | 0 | 0 | 2222222 | |||||
| Rhodobacter sphaeroides ATCC 17025 | 4 | 0 | 0 | 2222 | |||||
| Rhodobacter sphaeroides ATCC 17029 | 9 | 0 | 0 | 222222222 | |||||
| Rhodobacter sphaeroides KD131 | 7 | 0 | 0 | 2222222 | |||||
| Roseobacter denitrificans OCh 114 | 2 | 0 | 0 | 22 | |||||
| Roseobacter litoralis OCh 149 | 2 | 0 | 0 | 22 | |||||
| Roseovarius nubinhibens ISM | 2 | 0 | 0 | 2 | |||||
| Roseovarius sp. 217 | 2 | 0 | 0 | 22 | |||||
| Ruegeria pomeroyi DSS-3 | 3 | 0 | 0 | 222 | |||||
| Ruegeria sp TM1040 | 2 | 0 | 0 | 22 | |||||
| Sagittula stellata E-37 | 3 | 1 | 0 | 2223 | |||||
| Sulfitobacter sp. EE-36 | 1 | 0 | 1 | 24 | |||||
| Sulfitobacter sp NAS-14.1 | 2 | 0 | 1 | 24 |
Regarding the distribution of the different structures which cuckoo RNAs exhibit, the group of 150 cuckoo members with 3 hairpins (HP3) represents the most abundant structural variant, followed by 105 cuckoo RNAs exhibiting 2 hairpins (HP2) and 66 RNAs that form 4 stem loops (HP4). Despite their high abundance, HP3 cuckoo RNAs are limited mainly to the Rhizobiales and are additionally present as a single copy only in few Rhodobacteraceae. The distribution of HP4 cuckoo RNAs is similar except for the genera of Paracoccus and Octadecabacter, which harbor 2 to 4 RNAs of this structural variant. In contrast, HP2 cuckoo RNAs were identified in all families of the Rhizobiales but are predominant in the Rhodobacteraceae where they are found in clusters with highly variable numbers of up to 9 copies.
Synteny
The analysis of the genomic context of cuckoo RNA loci indicated several conserved intergenic neighborhoods (CIN). A CIN refers to a conserved genomic segment containing a cuckoo RNA or a cluster of cuckoo RNAs which is furthermore flanked at least on one side by an orthologous group of genes (OG).22 See Methods for a detailed definition of CIN. The most prominent CIN, CIN1, harbors nearly half of all cuckoo RNAs (148) and is present in 63 out of 78 bacteria, representing almost the entire taxonomic range of cuckoo RNAs (Table 1, Fig. 2, Table S2). CIN1 is characterized by its association with an orthologous gene of unknown function (OG1) and is the only CIN that was discovered in the Rhodobacteraceae and the Phyllobacteriaceae. On the other end, CIN1 is flanked either by genes for which no orthology could be determined or by OGs, which are conserved among specific taxa. For example, 27 cuckoo RNAs in the Rhodobacteraceae were identified between a benzoate transporter gene (OG18) and OG1 while 14 cuckoo RNAs associated with OG1 and a leucine tRNA gene were found in members of the Rhizobiaceae. The association of OG1 and a tRNA-methyltransferase (OG7) was observed for 28 cuckoo RNA across the families Brucellaceae, Phyllobacteriaceae, and Rhizobiaceae. All kinds of structural variants of cuckoo RNAs were found in CIN1 and it is the only CIN that harbors clusters of cuckoo RNAs. While the Brucellaceae lack cuckoo RNA clusters, CIN1 in Rhizobiaceae and Phyllobacteriaceae species often exhibit 2 and 3 HP3 cuckoo RNAs, respectively. A different picture is found for CIN1 in the Rhodobacteraceae. Here, the HP2 cuckoo RNA constitutes the most common variant within cuckoo clusters that are located in CIN1. Besides, next to a small number of species that have pure HP4 cuckoo RNA clusters, clusters in CIN1 exist that start with a series of HP2 cuckoo RNAs and end with different structural variants.
Figure 2.

Conserved intergenic neighborhoods of cuckoo RNAs. CINs are drawn as graphs. Nodes and edges depict conserved flanking features of cuckoo RNAs and represent single and combinations of OGs, respectively. Nodes and edges are annotated with the number of involved cuckoo. Each node is a pie chart to display the phylogenetic distribution. The area of the nodes is proportional to the number of flanked cuckoo RNAs. The colors green, yellow, purple and red correspond to the taxa Brucellaceae, Rhizobiaceae, Phyllobacteriaceae, and Rhodobacterales.
Interestingly, no other CIN could be identified among cuckoo RNAs in the Rhodobacteraceae and the Phyllobacteriaceae. However, we discovered 5 CINs within the Rhizobiaceae (CIN2, CIN3, CIN4), the Brucellaceae (CIN6) or within both families (CIN5). CIN2, CIN3, and CIN5 reside only on primary chromosomes while CIN6 is located on the secondary chromosome of the Brucellaceae. CIN4, in contrast, represents a cuckoo RNA locus which is located on the plasmids of Sinorhizobium species with 2 exceptions with CIN4 residing on a chromosome and a secondary chromosome, respectively. Nearly all cuckoo RNAs associated with CIN2 to CIN6 consist of 3 hairpins. An exception is CIN6, which harbors HP3 as well as HP2 cuckoo RNAs.
The analysis of the genomic neighborhoods of cuckoo RNAs shows an occurrence pattern that is biased toward a primary conserved neighborhood (CIN1) accompanied by several smaller CINs, with respect to the number of associated cuckoo RNAs that are limited to smaller taxa within the Rhizobiales. In summary, we developed a unifying model of structurally varying cuckoo RNAs and using our approach, we discovered 176 previously unknown cuckoo RNAs, thereby extending the cuckoo family to 321 members.
Conclusions
In this study we have built an integrative model of the cuckoo RNA family based on TDMs, which is capable to distinguish between different cuckoo RNA structure variants. Applying the cuckoo family model, we have performed systematic homology searches and have identified 321 cuckoo RNAs, 176 of which were previously undescribed. According to our data, cuckoo RNAs are distributed across the Rhodobacteraceae, Phyllobacteriaceae, Brucellaceae, and the Rhizobiaceae. The primary locus of cuckoo RNAs is found in an intergenic region next to a protein homolog of unknown function (OG1) and is present in almost all prokaryotes that harbor cuckoo RNAs.
Homology search for cuckoo RNAs is difficult not only because of the modular nature of the family's secondary structure, which leads to different variants in structure and size, but also of the lack of sequence conservation (beyond the genus level). This is caused by a high number of compensatory mutations, which dominate the stem-loops. The only conserved sequence pattern is the cuckoo loop motif CCUCCUCCC, which in part represents an aSD motif. In such a scenario TDMs prove their strengths, as TDMs can be tailored to multiple competing consensus structures and can emphasize single motifs.
The posttranscriptional regulation mechanism that represses the translation of target mRNAs by occluding the ribosome binding site (RBS) is widespread among sRNA.23 Consistently, C-rich loop sequences are a common feature among sRNAs and it was postulated that these motifs generally are signatures of sRNAs that repress the translation of target mRNAs by pairing with the RBS.24 An example is RNAIII, a sRNA which is predominant in Staphylococcus species. RNAIII harbors a conserved motif, UCCC, in 3 of its hairpin loops. In Staphylococcus aureus these hairpins are suggested to facilitate the initial binding between RNAIII and the RBS of the target mRNAs of several virulence factor mRNAs and the mRNA of the transcriptional regulator rot.25 Moreover, in a comparative study in S. aureus strains, several distinct sRNAs were identified that carry the identical C-rich loop motif.24 The authors hypothesized that these sRNAs are the product of convergent evolution. A similar picture is found in S. meliloti where several representatives of RNA families occur which are either similar to cuckoo HP2 (RFMSmelA003), cuckoo HP3 (RFMSmelB008, RFMSmelC416, RFMSmelC601) or cuckoo HP4 (RFMSmelC023) RNAs.18 Pairwise alignment of these sRNAs in our study (data not shown) with cuckoo members of S. meliloti revealed their sequence dissimilarity and therefore they likely do not share a common history but might act on their target mRNAs in an analogous way. Furthermore, sRNAs with sequence motifs reminding of the cuckoo motif are sX13 RNA and RepG RNA, which are highly conserved in the Xanthomonadaceae family and in Helicobacter pylori strains, respectively.26,27
So far homologs of neither family could be discovered in other species that would establish a phylogenetic connection between the above mentioned RNA families and thus no homology seems to exists. This is in concordance with observations that only a small proportion of RNA families are widely distributed.28
Concerning the aSD pattern of the cuckoo RNAs, the question arises, if cuckoo RNAs act as global translational repressors or if they occlude specific RBS more effectively thus repressing the respective genes. A comparative target analysis conducted to investigate this question could start from our compilation of cuckoo RNAs.
Note added in revision: In the most recent release of Rfam, a RNA family model RF02344 has been added. Its members overlap with the ones of our cuckoo family. As is current practice in Rfam, the family RF02344 is described as a covariance model, which is inherently limited to a fixed consensus structure. It assumes exactly 3 hairpins, and as a consequence, many family members of type HP2 and HP4 are wrongly annotated and some missing. This supports the use of a new, more flexible method (as presented herein) for model construction in the case of significant structural variation.
Methods
Thermodynamic matchers
Let x be an RNA sequence, and F(x) its folding space, i.e., the set of secondary structures it can fold into. The thermodynamic model29 assigns a free energy state E(s) to a secondary structure s. A structure prediction algorithm, such as RNAfold,30 uses this energy model to predict the structure of minimum free energy (MFE),
A thermodynamic matcher31 (see footnote1 ) (TDM) solves the same minimization problem on a specific subset of . Let M be a function that, given sequence x, yields a subset of the folding space, . Our intention is that M somehow captures a class of structures that contains a consensus structure for a family of sequences. Then, a thermodynamic matcher for M computes
This is mathematically strict, with no heuristics involved. The matcher folds the given RNA sequence into the prescribed structure class as good as it – the RNA – can. Should , we find Therefore, it makes sense to compare to . This allows to evaluate whether the matching structure comes close enough to minimal free energy to be plausible from the thermodynamic point of view.
Although notations are quite different, a TDM is similar to a descriptive motif matcher as provided e.g. with RNAMotif.33 However, its implementation is different. It uses dynamic programming during the matching phase, rather than constructing combinatorial matches and evaluating them afterwards. Thus, only the best possible match (if any) for a sequence window is returned. Even with a loosely defined motif, the output is bounded by O(N), where N is the sequence size.
Heretofore, the idea of TDMs has found a variety of applications, but not in RFM construction. A TDM, tuned to a subset of the folding space, is a nontrivial program to construct, similar but with specialized recurrences compared to a standard structure prediction algorithm. In fact, a TDM typically requires more recurrences and dynamic programming tables than a standard RNA folding algorithm. Hand-programming such a matcher, possibly modifying its design several times and debugging the implemented dynamic programming algorithm, would be prohibitive in practice. Fortunately, there is some automated support.
The program RapidShapes34 generates TDMs from abstract shape35 specifications, such as "[[][][]]" for the cloverleaf shape. This allows one to directly compute this shape's contribution to the partition function of x in O(n3), which otherwise requires computing all shapes and has exponential runtime.
The tool Locomotif36 generates TDMs from structure graphics, which a non-programmer can compile from pre-defined building blocks (stems, bulges, multiloops, simple pseudoknots, …). These building blocks can be specialized and decorated with sequence motifs in various ways. But in principle, Locomotif's fixed set of blocks is a restriction of the TDM concept.
The recent Bellman's GAP system37 provides the declarative language GAP-L, which allows to describe TDMs by (tree) grammars and evaluation algebras, and comes with a repository of re-usable components for RNA folding algorithms. In particular, there is an algebra for MFE calculation, which can be used off-the-shelf, and the TDM designer does not have to worry about the intricacies of the energy model. It implements the TDM grammar with the best asymptotic efficiency and a minimal number of dynamic programming tables.38
In this study, we chose to first produce a shape matcher with Locomotif, obtain it's GAP-L source code, and then to further refine the GAP-L program according to our design decisions.
General overview of TDM construction
The TDM construction process, like other descriptor based approaches for modeling RNA families, is neither standardized nor fully automated, but driven by human design decisions. Nevertheless, the steps that are involved in TDM construction follow a general scheme:
parameter derivation from an input RNA or a set of related RNAs,
creation of a graphical description using Locomotif,
compilation of the underlying folding grammar into a TDM,
screening for sequences applying the TDM,
assessment of new candidates.
Steps 1 to 5 are repeated until the final model of the RNA family is obtained. In this study, we started with a structurally varying set of 145 known cuckoo RNAs (Table S5) and derived structure and sequence features of the cuckoo family. We thereby focused on the modular nature of the cuckoo RNAs. See Supplementary Methods for a detailed description of how initial parameters were derived. In order to capture all important features of the cuckoo family we built two complementary TDMs, the skeleton TDM and the cuckoo TDM, which are described in the following sections. The Locomotif editor was used to draw annotated structure graphics and obtain basic grammars for both TDMs. We manually adapted these basic grammars to match variable numbers of consecutive cuckoo modules and introduced restrictive structure constraints and sequence motifs according to the parameters gathered initially. For the screening of genome sequences, we implemented a variant aware search procedure, which integrates the skeleton and the cuckoo TDM and separates structural variants of cuckoo RNAs (see section below). After assessment, in the subsequent modeling process we refined the TDMs by relaxing constraints gradually, by adding new cuckoo motifs, and by the integration of new constraints.
The skeleton TDM
The skeleton TDM aims at evaluating an input RNA sequence to be a potential cuckoo RNA by focusing on two aspects. One is the definition of sequence motifs that are considered instances of the cuckoo motif, the other is the distance between two such motifs. The output is an unfolded RNA sequence, annotated with the genomic coordinates and the predicted cuckoo motifs in upper case letters.
Conceptually, the underlying grammar of the skeleton TDM defines the (valid) sequence of a cuckoo RNA as a series of two or more basic blocks (non-terminal motif), which might be enclosed by leading and/or trailing bases (Fig. 3A). struct, derived from the grammars axiom, is the complete sequence of a cuckoo RNA without a secondary structure. struct is the start point of the grammar and defines alternative productions of leading unpaired bases (sadd) and the first (leftmost) generation of motif, which is concatenated with one or more instances of motif (struct_m) by applying the algebra function cadd. struct_f might add trailing bases before terminating the generation of the cuckoo RNA. The nonterminal motif designates a sequence that is concatenated of an unpaired sequence of length 12, an instance of the cuckoo motif, and an unpaired sequence with variable length of 12 to 34 bases. motif is derived from a set of productions that stem from the set of specified cuckoo motif variants. The sequence motifs are described in IUPAC code.
Figure 3.

Skeleton (A), cuckoo TDM grammar (B), and cuckoo sequence motif constraints (C). In both grammars struct is the axiom. Vertical bars separate alternative productions that start at the same nonterminal. Algebra functions are colored in green and built the tree-like data structure from terminals and nonterminals. In case of the cuckoo TDM grammar, these functions call upon the energy functions of the thermodynamic model to compute free energies for the corresponding substructure. The following terminals (in blue) are used: ε denotes the empty word, b a single base from the RNA alphabet {A,C,G,U}, r a region of unpaired bases, and loc the end-position of a neighbor subword. Numbers depict thresholds for size filters. A single number specifies the maximum size while two numbers determine a size range. dangle applies a base pair filter (in red), requiring at least six base pairs. For each cuckoo motif in C, an alternative production of seqmotif exists, where rcm corresponds to the cuckoo motif. The IPUAC convention is used to express cuckoo motifs.
The cuckoo TDM
The cuckoo TDM provides a single structural model that integrates all variants of cuckoo RNAs and is able to discover also (so far unknown) cuckoo RNAs that consist of more than four hairpins. Given a single RNA sequence, the cuckoo TDM predicts the energetically most stable RNA secondary structure that forms the shape of the cuckoo RNA family structure. Basically, the underlying grammar follows the same rules for the construction of substructures than a standard RNA folding grammar using the thermodynamic model (Fig. 3B). dangle represents closed substructures, which is the term for substructures that start with a base stack. The function drem passes on the energy of the substructure and implements the dangling-end model OverDangle.39 This model handles energies of dangling bases in a simplified form, by accounting energy for dangling bases on both sides of the helix, regardless if a base is available for dangling or not. strong has two production rules that are repeatedly applied to construct a substructure. sr, used in both productions, adds a stacking base pair and either extends a helix (strong) or introduces one of four (allowed) helix interrupting structural motifs (weak) that end with a closing base pair: internal loop (iloop), bulge (leftB, rightB), or hairpin (hairpin). Since cuckoo RNAs naturally do not form multiloops, the production for multiloops is removed from the standard grammar. Hence, dangle defines a single hairpin, which is at the same time the basic structural component of cuckoo RNAs. The axiom struct describes a complete cuckoo RNA folded into the family structure. In combination with struct_m and struct_f, different structural variants are realized, by connecting variable numbers of components by stretches of unpaired sequences.
For tailoring the grammar to the family structure of cuckoo RNAs, we introduced a number of modifications, which involve the introduction of size restrictions, filter, and sequence patterns. The overall size of hairpins is restricted to sizes between 24 and 50 bases, while hairpin loops, represented by motif, are allowed to range between 9 and 27 bases. For each cuckoo motif or set of cuckoo motifs (Fig. 3C), specified in IPUAC code, an alternative production (seqmotif) exists that encloses a cuckoo motif (rcm) with two flanking unpaired regions of bases r. Helix interruptions by bulges and internal loops are limited to six bases. The maximal length of an unpaired sequence connecting two hairpins is 10 bases. Finally, we defined a helix length filter (hlx_len), which defines a minimal number of six base pairs a helix must exhibit.
Variant-aware homology search of cuckoo RNAs
The search for homologs of the cuckoo family in a single genome sequence can be divided into three main stages. In the first stage the skeleton TDM screens the genome in a sliding-window mode for initial candidate homologs that meet the primary sequence constraints. Here, a window of 120 nt size and a step size of 50 nt is used. Overlapping matches, provided that the discovered cuckoo motifs in the overlapping part are at identical genomic positions, are assembled to a single candidate. The assembly step enables the generation of candidates with variable numbers of cuckoo motifs, corresponding to an equal number of stem-loops. The screen in 2,689 bacterial genomes resulted in 42,312 candidates which represented 39,801 HP2, 2,233 HP3, and 278 HP4 cuckoo candidates (Fig. 4).
Figure 4.
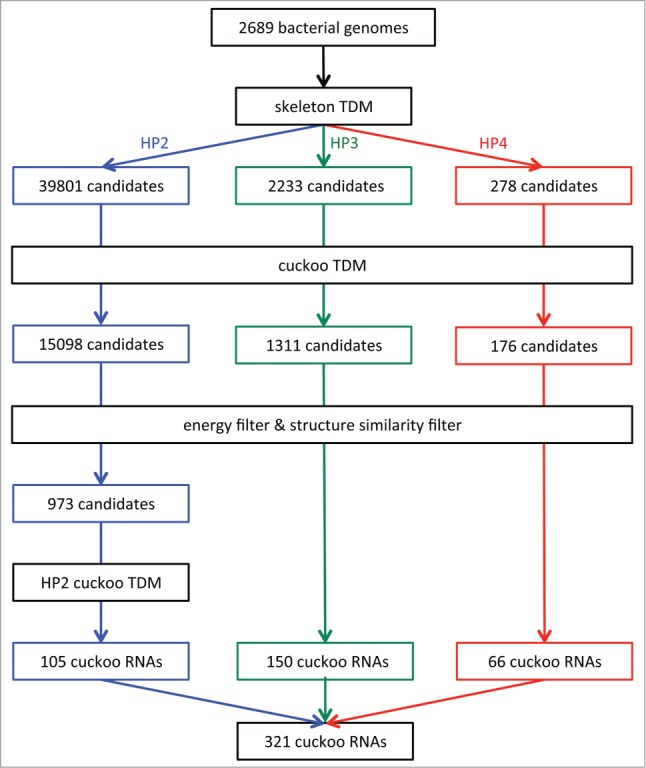
Pipeline for cuckoo RNA discovery based on TDMs. Different colors correspond to structural variants of cuckoo RNAs (HP2 in blue, HP3 in green, HP4 in red). Bacterial genome sequences from the NCBI reference genome database were gathered and consecutively scanned by the skeleton TDM, focusing on primary sequence conservation, and the cuckoo TDM, which incorporates structural constraints. Then the energy filter is applied. HP2 cuckoo candidates that pass the structural filter are processed by the HP2 cuckoo TDM which was adapted to match specifically HP2 cuckoo RNAs.
In the next stage, the cuckoo TDM is used to structurally assess each candidate sequence by attempting to fold it into the cuckoo family structure. Candidates that cannot fold into the cuckoo family structure are discarded. The TDM screen reduced the number of candidate sequences by more than 60% to 16,582, with 15,098 most of them belonging to the HP2 structural variant, followed by 1,311 HP3 and 176 HP4 cuckoo candidates. The purpose of the third stage is to verify that a cuckoo candidate obtained by truly resembles a homolog of the cuckoo family by assessing its structure . For this, the structure must comply to the following two criteria.
The first criterion is the energy filter. It compares to in order to ensures that the candidate folds into the family structure with a free energy similar that of its MFE structure. We apply RNAfold to calculate ; Option –d2 was employed to make sure both programs use the same energy model. Candidate passes the energy filter if the ratio of is equal or greater than 0.85.
We observed that, once the TDM had indicated the correct sequence boundaries, the MFE structure often resembles the cuckoo family structure. This led to the implementation of a second filter, which tests if the candidate folds also without structural constraints into the cuckoo RNAs characteristic structure(s). Structural similarity is assessed by using pointed shapes. A pointed shape refers to the abstract shape representation of an RNA structure, which is annotated by hairpin centers, e.g., „[35][66][82][110]."40 A hairpin center depicts the central position of a hairpin loop and is calculated as , where and are the positions of the hairpin closing base pair. Abstract shape analysis is performed by applying RNAshapes.35 Two pointed shapes and are regarded similar if they share the same shape and if hairpin centers at the same relative order positions do not differ by more than 2.5 nt. Only candidates with similar shapes pass the filter. In some cases, the MFE structure of an cuckoo RNA exhibits a small extra stem-loop between 2 cuckoo modules, which is ignored by the structure filter as this stem-loop typically consists of not more than 3 base pairs. The filtering process returned 150 HP3 and 66 HP4 cuckoo RNAs. Table S3 lists the structure properties of the final cuckoo RNAs.
The HP2 motif is statistically the least significant, and the inspection of the remaining 973 HP2 sequences revealed a high number of false positives, in the form of outliers or repeats. Therefore, we built a new TDM, , that is specific to HP2 cuckoo RNAs, only. The parameters used for adapting were derived from already known HP2 cuckoo RNAs and manually selected candidates that showed a high plausibility based on synteny, distribution pattern, and structural properties. See Figure S2 for details on the grammar of . The additional application of on the 973 HP2 candidates narrowed down the number of cuckoo RNAs to 105.
Analysis of preserved genomic context of cuckoo RNAs
We analyzed the genomic context of cuckoo RNAs for conservation. For this purpose we searched for orthologous genes that flank cuckoo RNA loci using Proteinortho.41 Orthologous groups of genes that had more than 5 members were retained. We defined a conserved intergenic neighborhood as the locus of a cuckoo RNA or a cluster of cuckoo RNAs, which is flanked at least on one side by an orthologous group of genes.22
Footnotes
The name “thermodynamic matcher” goes back to Reeder et al.,32 but no formal definition was given there.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1. Vogel J, Bartels V, Tang TH, Churakov G, Slagter-Jäger JG, Hüttenhofer A, Wagner EGH. RNomics in Escherichia coli detects new sRNA species and indicates parallel transcriptional output in bacteria. Nucleic Acids Res 2003; 31:6435-43; PMID:14602901; http://dx.doi.org/ 10.1093/nar/gkg867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Willkomm DK, Minnerup J, Hüttenhofer A, Hartmann RK. Experimental RNomics in Aquifex aeolicus: identification of small non-coding RNAs and the putative 6S RNA homolog. Nucleic Acids Res 2005; 33:1949-60; PMID:15814812; http://dx.doi.org/ 10.1093/nar/gki334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 2009; 10:57-63; PMID:19015660; http://dx.doi.org/ 10.1038/nrg2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Burge SW, Daub J, Eberhardt R, Tate J, Barquist L, Nawrocki EP, Eddy SR, Gardner PP, Bateman A. Rfam 11.0: 10 years of RNA families. Nucleic Acids Res 2013; 41:D226-32; PMID:23125362; http://dx.doi.org/ 10.1093/nar/gks1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Eddy SR, Durbin R. RNA sequence analysis using covariance models. Nucleic Acids Res 1994; 22:2079-88; PMID:8029015; http://dx.doi.org/ 10.1093/nar/22.11.2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nawrocki EP, Eddy SR. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013; 29:2933-5; PMID:24008419; http://dx.doi.org/ 10.1093/bioinformatics/btt509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Durbin R, Eddy SR, Krogh A, Mitchison G. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. Cambridge, UK: Cambridge University Press; 1998. [Google Scholar]
- 8. Berghoff BA, Glaeser J, Sharma CM, Vogel J, Klug G. Photooxidative stress-induced and abundant small RNAs in Rhodobacter sphaeroides. Mol Microbiol 2009; 74:1497-512; PMID:19906181; http://dx.doi.org/ 10.1111/j.1365-2958.2009.06949.x [DOI] [PubMed] [Google Scholar]
- 9. Nuss AM, Glaeser J, Berghoff BA, Klug G. Overlapping alternative sigma factor regulons in the response to singlet oxygen in Rhodobacter sphaeroides. J Bacteriol 2010; 192:2613-23; PMID:20304993; http://dx.doi.org/ 10.1128/JB.01605-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Berghoff BA, Glaeser J, Sharma CM, Zobawa M, Lottspeich F, Vogel J, Klug G. Contribution of Hfq to photooxidative stress resistance and global regulation in Rhodobacter sphaeroides. Mol Microbiol 2011; 80:1479-95; PMID:21535243; http://dx.doi.org/ 10.1111/j.1365-2958.2011.07658.x [DOI] [PubMed] [Google Scholar]
- 11. del Val C, Rivas E, Torres-Quesada O, Toro N, Jiménez-Zurdo JI. Identification of differentially expressed small non-coding RNAs in the legume endosymbiont Sinorhizobium meliloti by comparative genomics. Mol Microbiol 2007; 66:1080-91; PMID:17971083; http://dx.doi.org/ 10.1111/j.1365-2958.2007.05978.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rivas E. Evolutionary models for insertions and deletions in a probabilistic modeling framework. BMC Bioinformatics 2005; 6:63; PMID:15780137; http://dx.doi.org/ 10.1186/1471-2105-6-63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Washietl S, Hofacker IL, Stadler PF. Fast and reliable prediction of noncoding RNAs. Proc Natl Acad Sci USA 2005; 102:2454-9; PMID:15665081; http://dx.doi.org/ 10.1073/pnas.0409169102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Valverde C, Livny J, Schlüter J-P, Reinkensmeier J, Becker A, Parisi G. Prediction of Sinorhizobium meliloti sRNA genes and experimental detection in strain 2011. BMC Genomics 2008; 9:416; PMID:18793445; http://dx.doi.org/ 10.1186/1471-2164-9-416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schlüter J-P, Reinkensmeier J, Daschkey S, Evguenieva-Hackenberg E, Janssen S, Jänicke S, Becker JD, Giegerich R, Becker A. A genome-wide survey of sRNAs in the symbiotic nitrogen-fixing alpha-proteobacterium Sinorhizobium meliloti. BMC Genomics 2010; 11:245; PMID:20398411; http://dx.doi.org/ 10.1186/1471-2164-11-245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schlüter J-P, Reinkensmeier J, Barnett MJ, Lang C, Krol E, Giegerich R, Long SR, Becker A. Global mapping of transcription start sites and promoter motifs in the symbiotic alpha-proteobacterium Sinorhizobium meliloti 1021. BMC Genomics 2013; 14:156; PMID:23497287; http://dx.doi.org/ 10.1186/1471-2164-14-156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Livny J, Teonadi H, Livny M, Waldor MK. High-throughput, kingdom-wide prediction and annotation of bacterial non-coding RNAs. PLoS ONE 2008; 3:e3197; PMID:18787707; http://dx.doi.org/ 10.1371/journal.pone.0003197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Reinkensmeier J, Schlüter J-P, Giegerich R, Becker A. Conservation and occurrence of trans-encoded sRNAs in the Rhizobiales. Genes [Internet] 2011; 2:925-56. Available from: http://www.mdpi.com/2073-4425/2/4/925; PMID:24710299; http://dx.doi.org/ 10.3390/genes2040925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vercruysse M, Fauvart M, Cloots L, Engelen K, Thijs IM, Marchal K, Michiels J. Genome-wide detection of predicted non-coding RNAs in Rhizobium etli expressed during free-living and host-associated growth using a high-resolution tiling array. BMC Genomics 2010; 11:53; PMID:20089193; http://dx.doi.org/ 10.1186/1471-2164-11-53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wilms I, Overlöper A, Nowrousian M, Sharma CM, Narberhaus F. Deep sequencing uncovers numerous small RNAs on all four replicons of the plant pathogen Agrobacterium tumefaciens. RNA Biol 2012; 9:446-57; PMID:22336765; http://dx.doi.org/ 10.4161/rna.17212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. del Val C, Romero-Zaliz R, Torres-Quesada O, Peregrina A, Toro N, Jiménez-Zurdo JI. A survey of sRNA families in α-proteobacteria. RNA Biol 2012; 9:119-29; PMID:22418845; http://dx.doi.org/ 10.4161/rna.18643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kuzniar A, van Ham RCHJ, Pongor S, Leunissen JAM. The quest for orthologs: finding the corresponding gene across genomes. Trends Genet 2008; 24:539-51; PMID:18819722; http://dx.doi.org/ 10.1016/j.tig.2008.08.009 [DOI] [PubMed] [Google Scholar]
- 23. Storz G, Vogel J, Wassarman KM. Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell 2011; 43:880-91; PMID:21925377; http://dx.doi.org/ 10.1016/j.molcel.2011.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Geissmann T, Chevalier C, Cros M-J, Boisset S, Fechter P, Noirot C, Schrenzel J, François P, Vandenesch F, Gaspin C, et al. A search for small noncoding RNAs in Staphylococcus aureus reveals a conserved sequence motif for regulation. Nucleic Acids Res 2009; 37:7239-57; PMID:19786493; http://dx.doi.org/ 10.1093/nar/gkp668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boisset S, Geissmann T, Huntzinger E, Fechter P, Bendridi N, Possedko M, Chevalier C, Helfer AC, Benito Y, Jacquier A, et al. Staphylococcus aureus RNAIII coordinately represses the synthesis of virulence factors and the transcription regulator Rot by an antisense mechanism. Genes Dev 2007; 21:1353-66; PMID:17545468; http://dx.doi.org/ 10.1101/gad.423507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schmidtke C, Abendroth U, Brock J, Serrania J, Becker A, Bonas U. Small RNA sX13: a multifaceted regulator of virulence in the plant pathogen Xanthomonas. PLoS Pathog 2013; 9:e1003626; PMID:24068933; http://dx.doi.org/ 10.1371/journal.ppat.1003626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pernitzsch SR, Tirier SM, Beier D, Sharma CM. A variable homopolymeric G-repeat defines small RNA-mediated posttranscriptional regulation of a chemotaxis receptor in Helicobacter pylori. Proc Natl Acad Sci USA 2014; 111:E501-10; PMID:24474799; http://dx.doi.org/ 10.1073/pnas.1315152111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hoeppner MP, Gardner PP, Poole AM. Comparative analysis of RNA families reveals distinct repertoires for each domain of life. PLoS Comput Biol 2012; 8:e1002752; PMID:23133357; http://dx.doi.org/ 10.1371/journal.pcbi.1002752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mathews DH, Sabina J, Zuker M, Turner DH. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J Mol Biol 1999; 288:911-40; PMID:10329189; http://dx.doi.org/ 10.1006/jmbi.1999.2700 [DOI] [PubMed] [Google Scholar]
- 30. Lorenz R, Bernhart SHF, Höner zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL. ViennaRNA Package 2.0. Algorithms Mol Biol 2011; 6:26; PMID:22115189; http://dx.doi.org/ 10.1186/1748-7188-6-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Höchsmann T, Höchsmann M, Giegerich R. Thermodynamic matchers: strengthening the significance of RNA folding energies. Comput Syst Bioinformatics Conf 2006; 111-21; PMID:17369630; http://dx.doi.org/ 10.1142/9781860947575_0018 [DOI] [PubMed] [Google Scholar]
- 32. Reeder J, Giegerich R. Design, implementation and evaluation of a practical pseudoknot folding algorithm based on thermodynamics. BMC Bioinformatics 2004; 5:104; PMID:15294028; http://dx.doi.org/ 10.1186/1471-2105-5-104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Macke TJ, Ecker DJ, Gutell RR, Gautheret D, Case DA, Sampath R. RNAMotif, an RNA secondary structure definition and search algorithm. Nucleic Acids Res 2001; 29:4724-35; PMID:11713323; http://dx.doi.org/ 10.1093/nar/29.22.4724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Janssen S, Giegerich R. Faster computation of exact RNA shape probabilities. Bioinformatics 2010; 26:632-9; PMID:20080511; http://dx.doi.org/ 10.1093/bioinformatics/btq014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Giegerich R, Voss B, Rehmsmeier M. Abstract shapes of RNA. Nucleic Acids Res 2004; 32:4843-51; PMID:15371549; http://dx.doi.org/ 10.1093/nar/gkh779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Reeder J, Reeder J, Giegerich R. Locomotif: from graphical motif description to RNA motif search. Bioinformatics 2007; 23:i392-400; PMID:17646322; http://dx.doi.org/ 10.1093/bioinformatics/btm179 [DOI] [PubMed] [Google Scholar]
- 37. Sauthoff G, Möhl M, Janssen S, Giegerich R. Bellman's GAP-a language and compiler for dynamic programming in sequence analysis. Bioinformatics 2013; 29:551-60; PMID:23355290; http://dx.doi.org/ 10.1093/bioinformatics/btt022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sauthoff G, Giegerich R. Yield grammar analysis and product optimization in a domain-specific language for dynamic programming. Sci Comput Program 2014; 87:2-22; http://dx.doi.org/ 10.1016/j.scico.2013.09.011 [DOI] [Google Scholar]
- 39. Janssen S, Schudoma C, Steger G, Giegerich R. Lost in folding space? Comparing four variants of the thermodynamic model for RNA secondary structure prediction. BMC Bioinformatics 2011; 12:429; PMID:22051375; http://dx.doi.org/ 10.1186/1471-2105-12-429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Huang J, Backofen R, Voss B. Abstract folding space analysis based on helices. RNA 2012; 18:2135-47; PMID:23104999; http://dx.doi.org/ 10.1261/rna.033548.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lechner M, Findeiss S, Steiner L, Marz M, Stadler PF, Prohaska SJ. Proteinortho: detection of (co-)orthologs in large-scale analysis. BMC Bioinformatics 2011; 12:124; PMID:21526987; http://dx.doi.org/ 10.1186/1471-2105-12-124 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.