

Abstract
The correct splicing of precursor-mRNA depends on the actual splice sites plus exonic and intronic regulatory elements recognized by the splicing machinery. Surprisingly, an increasing number of examples reveal that exonic mutations disrupt the binding of splicing factors to these sequences or generate new splice sites or regulatory elements, causing disease. This contradicts the general assumption that missense mutations disrupt protein function and that synonymous mutations are merely polymorphisms. Autosomal dominant polycystic kidney disease (ADPKD) is a common inherited disorder caused mainly by mutations in the PKD1 gene. Recently, we analyzed a substantial number of PKD1 missense or synonymous mutations to further characterize their consequences on pre-mRNA splicing. Our results showed that one missense and 2 synonymous mutations induce significant defects in pre-mRNA splicing. Thus, it appears that aberrant splicing as a result of exonic mutations is a previously unrecognized cause of ADPKD.
Keywords: ADPKD, disease-causing variant, exonic mutations, missense mutation, mRNA analysis, minigene assay, pre-mRNA splicing, PKD1, synonymous mutation, splice site mutation
Abbreviations
- ADPKD
autosomal dominant polycystic kidney disease
- ESE
exonic splicing enhancer
- ESS
exonic splicing silencer
- ISE
intronic splicing enhancer
- ISS
intronic splicing silencer
- NMD
nonsense-mediated mRNA decay
- RT-PCR
reverse-transcribed polymerase chain reaction
- SR proteins
serine/arginine-rich proteins
- SRE
splicing regulatory element
Introduction
Most human precursor-mRNAs (pre-mRNAs) contain noncoding sequences or introns that must be deleted to generate the correct joining of exons in the mature mRNAs. This highly regulated step in gene expression is known as pre-mRNA splicing. The spliceosome, a large complex comprising 5 types of small nuclear ribonucleoproteins (snRNPs) and many additional proteins, catalyzes the splicing process.1 The high accuracy of this process is accomplished through identification of cis-acting elements within the pre-mRNA and a large number of RNA-RNA, RNA-protein, and protein-protein interactions.2 The spliceosome assembles on intron-exon boundaries and, through U6 snRNA catalyzes 2 consecutive trans-esterification reactions that result in intron removal and joining of exons.3 In addition to canonical splice sites, the pre-mRNA molecules contain, both in the exons and the introns, sequence motifs that regulate constitutive and alternative splicing.
Splice Sites
The most conserved splicing sequences are the donor and acceptor splice sites located at the 5' and 3' of the intron ends, respectively.4 The donor splice site is mainly composed of an intronic GU dinucleotide preceded by an exonic AG dinucleotide, and the acceptor splice site contains the intronic branch point sequence (YNYURAY), a polypyrimidine tract and an AG at the intron boundary. These sequences are detected repeatedly by components of the spliceosome during the processes of intron removal and exon joining. The donor splice site is initially recognized by the U1 snRNP, whereas the branch point sequence and the acceptor splice site are bound by the U2 snRNP and the U2 auxiliary factor (U2AF), respectively (Fig. 1A).2 Successive interactions with other snRNPs result in formation of an active complex that carries out the transesterification reactions leading to mRNA.
Figure 1.
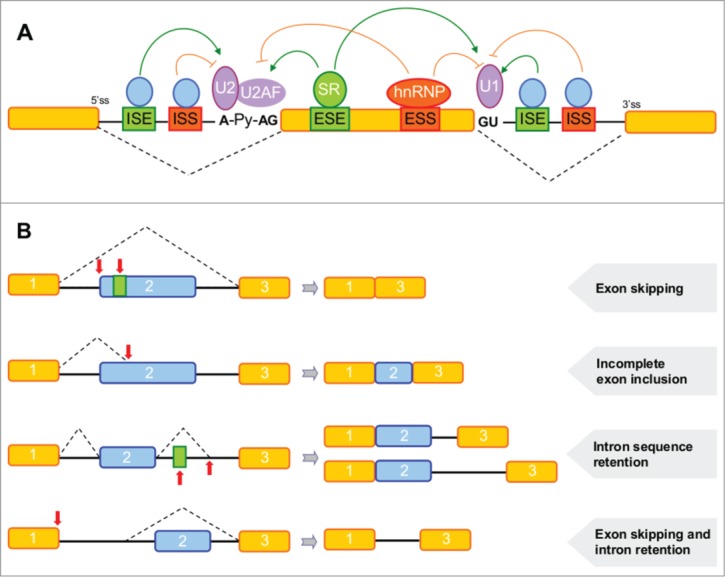
Splice sites, regulatory elements and splicing alterations caused by mutations. (A) Yellow or blue boxes and black lines symbolize exons and introns respectively. The acceptor (AG) and donor (GU) sites, branch site (A), polypyrimidine (Py) tract and splicing regulatory elements (ESE, ESS, ISE, ISS) are indicated. Mutations in any of these sites could disturb pre-mRNA splicing by disruption of binding of the respective splicing factors. An SR protein and a splicing inhibitor (hnRNP) bind to an exonic splicing enhancer (ESE) and an exonic splicing silencer, respectively. The U2AF splicing factor binds to the Py tract, and this promotes binding of U2 snRNP to the branch site. U1 snRNP binds to the donor splice site. Arrows indicate protein-protein interactions between the splicing factors. (B) Examples of splicing alterations caused by mutations (indicated by vertical arrows) that affect an acceptor splice site, ESE or ISE, a mutation that generates a new acceptor site or activates a cryptic one, and a mutation that affects a donor splice site.
Splicing Regulatory Elements
Splicing regulatory elements (SREs) are binding sites for regulatory factors that modulate splicing efficiency by either increasing or decreasing spliceosomal component recruitment.5-12 These sequences of about 10 nucleotides are referred to as exonic or intronic splicing enhancers (ESE or ISE) or silencers (ESS or ISS). However, depending on their relative location in pre-mRNA, some of these splicing regulators can function as either enhancer or silencers.13,14 Most ESEs are binding sites for members of the serine/arginine-rich (SR) protein family, which are characterized by having one or more RNA binding domains and an arginine/serine-rich domain for protein–protein interactions.15-17 SR proteins promote the initial steps of spliceosome assembly by recruiting U1 snRNP to the donor splice site or by antagonizing the effects of ESSs in the neighborhood.15 In some cases, however, SR proteins can interfere with exon recognition when bound to intronic positions, possibly due to steric hindrance.18
ESSs and ISSs are binding sites for splicing repressors of the heterogenous nuclear ribonucleoprotein (hnRNP) family, a diverse group of proteins containing one or more RNA-binding domains and protein-protein interaction domains.19,20 hnRNPs proteins are mainly implicated in exon skipping and function by different mechanisms.8,11 For instance, hnRNP I can compete with auxiliary factor U2AF for its binding site close to the 3′ splice site, whereas hnRNP A1 binds to ESSs and can prevent binding of SR proteins to their ESEs.6
Genetic Variants that Cause Splicing Defects in Disease
Alterations in pre-mRNA splicing due to gene mutations are the underlying cause of many human diseases.11,4,21,22 For some genes, up to 50% of disease-causing mutations are found to affect splicing.23 These mutations occurring in coding or non-coding regions may disrupt splice sites or SREs (Fig. 1).24 Other exonic or intronic variants are able to generate new splice sites or regulatory sequences that can cause disease. The most easily recognized mechanism of splicing alteration, accounting for 10% of all human disease-causing mutations, involves substitutions in the GU and AG splice site dinucleotides and neighboring bases.23,25 Branch point sequence and polypyrimidine track mutations are rare.26 Over the last 15 y it has been demonstrated that different exonic mutations have an effect on pre-mRNA splicing.10,24,27 In fact, recent studies suggest that approximately 25% of exonic mutations are likely to affect this process.28 The alterations can lead to skipping of exons, inclusion of introns, generation of new splice sites or activation of cryptic splice sites (Fig. 1B).10,29-31 Defective mRNAs produced by these events may include exons or intron sequences with premature stop codons. It is not easy to predict the cytoplasmic fate of these aberrant messages, but some of them are probably degraded by nonsense-mediated decay (NMD).32
Identification of Pre-mRNA Splicing Alterations
Analysis of the mRNA from a patient by RT-PCR and sequencing is probably the most reliable method of determining whether a specific mutation disturbs pre-mRNA splicing. However, in the majority of cases tissue RNA samples are not accessible or unavailable. Even if they are available, the detection of the altered mRNAs might be difficult as they can be degraded rapidly by NMD. An experimental method that is commonly used to examine pre-mRNA splicing defects is the minigene assay.33,34 An exon of interest with part of its intronic flanking regions can be amplified from a patient carrying the mutation and cloned into the minigene vector. Alternatively, specific mutations can be introduced by site-directed mutagenesis of a minigene containing the wild-type exon. The resulting minigenes are then transfected in a suitable cell line. After cell culture, RNA is extracted and analyzed by RT-PCR followed by DNA sequencing. The effectiveness of minigene analyses has been established by different studies, and a high level of consonance between data obtained with these assays and data from patients RNA has been found.35,36
This experimental method can be combined with in silico approaches that predict splicing defects for mutations in splice sites and in splicing regulatory elements. The effects of splice sites mutations on the mature mRNA are relatively easy to deduce. There are several in silico methods that analyze the strength of splice sites and the effect of different mutations on them, including NNSplice (http://www.fruitfly.org/seq_tools/splice.html), Genscan (http://genes.mit.edu/GENSCAN.html), Human Splicing Finder, (http://www.umd.be/HSF/) and MaxEntScan, (http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html).37–40 However, evaluating precisely how disease-causing mutations influence the loss or gain of ESE or ESS motifs and its effects on pre-mRNA splicing is much more difficult. 2,11,24 ESEs, ESSs, e intronic GGGs can be detected using informatics tools like Human Splicing Finder (http://www.umd.be/HSF/), ESEfinder (http://rulai.cshl.edu/tools/ESE/), Rescue-ESE (http://genes.mit.edu/burgelab/rescue-ese), or ExonScan (http://genes.mit.edu/exonscan/).40–43 These tools still present considerable degrees of inaccuracy in the identification of pre-mRNA splicing defects; therefore, their findings need to be confirmed by using experimental methods.44,45 More recently, 2 novel machine-learning approaches have been developed to predict the impact of genetic variants on pre-mRNA splicing: MutPredSplice (http://mutdb.org/mutpredsplice/submit.htm) and SPANR (http://tools.genes.toronto.edu/).46-47 A combination of various predictive procedures is, up to now, the best chance of identifying potential splicing mutations.45 In order to improve the in silico prediction of ESEs, one can evaluate the strength of the splice sites of the exons. If the donor or acceptor sites were weak, then the likelihood of the prediction being accurate would increase.4
As described below, we contributed to this topic by identifying the first reported PKD1 exonic mutations that induce aberrant mRNAs.
Summary of Our Paper
In a recent study, we have analyzed a significant number of PKD1 exonic mutations, previously designated as missense or synonymous in the literature or databases, for their consequences on pre-mRNA splicing.48 These mutations were selected from the Autosomal Dominant Polycystic Kidney Disease (ADPKD) Mutation Database (http://pkd.mayo.ed). Mutations in PKD1, which encodes polycystin-1, are responsible for the majority and most severe cases with ADPKD, one of the most common monogenic disorders, characterized by progressive cysts development and enlargement leading to end-stage renal disease (ESRD) and a variety of extrarenal manifestations.49 ADPKD shows high phenotypic variability as well as locus and allelic heterogeneity.
Our study included a total 416 PKD1 mutations (258 missense and 158 synonymous) that were initially investigated using several bioinformatics tools (NNSplice, Human Splicing Finder, Rescue-ESE and ESEfinder) in order to predict their potential effect on the existing splice sites and SREs. We found that 28 missense mutations and 4 synonymous mutations had a potential effect on pre-mRNA splicing. These mutations were then investigated in a minigene system, and the results indicated that 3 of them, one missense, p.R3719Q (c.11156G > A) and 2 synonymous, c.327A > T (p.G109G) and c.11257C > A (p.R3753R), induced pre-mRNA splicing defects.
Mutation p.R3719Q, which affects the last nucleotide of exon 38, results in skipping of exon 39 and incorporation of part of intron 38 in the mRNA. The location of such substitution usually has a negative effect on the recognition of the splice site by the cellular machinery.50 Based on our data, we suggested that the donor splice site of intron 38 is disrupted by the mutation and a cryptic site downstream in intron 38 is activated. We concluded that mutation p.R3719Q is pathogenic because it generates an altered mRNA molecule instead of the normal mRNA in the minigene-based assay. Synonymous mutation c.11257C > A, located within exon 39, induces the incorporation of a shortened exon in the mRNA. Our data show that this mutation creates a new donor splice site that is used by the spliceosome. Also, this mutation decreases the score of a putative SRSF6 binding sequence and generates ESS sequences. In contrast to mutation p.R3719Q, with c.11257C>A a certain amount of full-length mRNA is produced by the minigene that could be translated into wild-type polycystin-1. However, previous studies have shown that a decrease in PKD1 expression can generate the disease onset or affect the severity of the phenotype.51,52 Consequently, the reduction of PKD1 expression due to the splicing defect in patients with mutation c.11156G > A could cause the disease. Similarly to c.11257C > A, synonymous mutation c.327A > T, situated within exon 3, results in incorporation of a truncated exon in the mRNA. This mutation generates a new donor splice site that is recognized by the splicing machinery. Additionally, no full-length mRNA was detected in our analysis. Thus, we concluded that c.11257C > A and c.327A > T could be responsible for the disease phenotype by eliminating the expression of polycystin-1. Furthermore, the altered mRNA forms generated by these 3 mutations present premature stop codons and, therefore, they probably undergo degradation by the NMD pathway.32
Since we started our study, more mutations have been added to the ADPKD Mutation Database, which contains now 780 missense and 352 synonymous PKD1 mutations. Therefore, we updated the previous report analyzing another 337 missense and 99 synonymous mutations. The same selection criteria and bioinformatics tools were used, including mutations that create, activate or disrupt splice sites. Mutations located in exon 15 were not included in the study since this exon is rather large (3.6 kb) and is well defined. We selected 90 missense and 49 synonymous mutations with potential effects on pre-mRNA splicing. The results are summarized in Tables S1 and S2, and a few illustrations of the results obtained with ESEfinder and Rescue-ESE are shown in Fig. S1 (Supplementary Material). Thirty-two missense and 25 synonymous mutations affected existing splice sites or activated or created new ones, whereas 47 missense and 19 synonymous mutations disturbed potential ESE sequences. For instance, synonymous mutations c.1209C > T and c.7866C > T, located in poorly-defined exons 6 and 21, respectively, reduce the score of the acceptor splice sites in introns 5 and 20 according to NNSplice prediction, and c.1209C > T also disrupted a potential ESE sequence present in exon 6 (Table S2). On the other hand, synonymous mutation c.11007C > T generated a new donor splice site (score: 0.74) in exon 37 that could compete with the functional donor site (score: 0.67), and synonymous mutation c.11313G > A activated a cryptic acceptor splice site in exon 40 (score: 0.44) that could compete with the functional acceptor site (score: 0.34) (Table S2). Two missense mutations, p.D3469N and p.D3469Y, which affect the last nucleotide of exon 33, reduced the score of the functional donor splice site from 0.99 to 0.68 and 0.60, respectively (Table S1). Missense mutations p.P404R, p.R454C, p.R454P, located in poorly defined exon 6, and p.L3610P, and synonymous mutation c.11523C>T affected potential ESE sequences according to ESEfinder analysis (Fig. S1A). Rescue-ESE analysis of c.11523C>T and p.L3610P also revealed disruption of ESE sequences (Fig. S1B). As shown in Figure S1B, Rescue-ESE analysis also predicted disruption of ESE sequences with mutations p.N101D, p.D3649Y and p.D3649N.
Open Questions
In our study, the assessment of the potential effects of PKD1 gene mutations on pre-mRNA splicing was performed by minigene analysis, which is a standard test to confirm splicing mutations. A major issue with the use of minigene constructs is whether they accurately recapitulate the in vivo situation. The minigene we used to study the effect of exons 38 and 39 mutations contained the genomic region between exons 37 and 39 with its intronic flanking regions. It would be important to establish if these mutations have a similar impact in the context of their natural gene using patient-derived RNA or a renal epithelial cell line, and to determine if they have additional effects such as multiple exon skipping.53 It would also be possible to analyze a more complex exon–intron composition, since natural 5′ and 3′ neighbor exons might regulate the splicing reaction. PKD1 exons 35 to 46 are clustered in a 3.8 kb region that would be feasible to clone entirely. Although for diagnostic procedures it is usually sufficient to determine whether there is a splicing abnormality, such a minigene construct could be used to confirm splicing outcomes with mutations located in these exons.
The wild-type minigenes used in our assay also produced alternative mRNAs forms lacking exons 3 or 39, which have not been previoulsly reported. Additional work is needed to determine whether these mRNAs are produced in vivo. The mRNA deletion of the entire exon 3 is in frame and, therefore, it would not activate the NMD pathway. In this case, a polycystin-1 polypeptide lacking amino acids residues 96 to 119, including part of a leucine-rich repeat involved in protein-protein interaction, would be generated. Our finding raises additional questions concerning the potential function of this protein isoform.
The defective mRNA products detected in our study with PKD1 mutations emphasize the need for functional analysis at the mRNA level of hypomorphic mutations in ADPKD, in order to establish an accurate mutation classification for future studies of genotype-phenotype correlation, potential therapeutic targeting, and to uncover other molecular mechanisms of disease development. Functional characterization of more mutations continues to be a great challenge. Further experimental work is needed to determine the potential effect on pre-mRNA splicing of the additional 139 PKD1 mutations selected here with bioinformatics tools, to identify the exact mechanism involved, and to correlate the results with the bioinformatics predictions. Understanding the ways in which mutations can affect pre-mRNA splicing is important if we are to make additional progress in the genetic analysis of ADPKD. We should point out that our work and others in this topic has generated new hypothesis that need to be verified. The real test will be to determine if the defective pre-mRNA splicing induces a phenotype. This is a much more challenging problem that needs real innovative thinking from the field.
The ability of computer tools to predict point mutations that disrupt real ESEs has yet to be fully assessed. These studies result in a high percentage of false positives as, for example, the presence of an ESE motif in a sequence does not automatically identify that sequence as a splicing enhancer in its natural context. One feasible way of refining the predictive power of these computer tools when applied to a particular exonic sequence is to additionally consider the strength of the splice sites of the exon. A large fraction of the mutations included in our study did not modify the normal splicing pattern, as many of them are actually located in well-defined exons. If either the donor or acceptor site shows to be weak, then the likelihood of the prediction of an ESE being correct would increase. Also, a combination of several predictive algorithms would improve the chances of identifying potential splicing mutations.44,45 Further improvement in these prediction programs is needed so they can be useful in genetic diagnostic laboratories.
Conclusions
Our recent study represents the first experimental and in silico analysis of the effect of reported PKD1 missense and synonymous mutations on splicing of its pre-mRNA. The results show that exonic PKD1 variants causing ADPKD can lead to aberrant splicing, with predicted detrimental effects on polycystin-1 protein production. Based on this, we suggest that pre-mRNA splicing alterations caused by PKD1 exonic mutations are a new mechanism of pathogenesis in ADPKD.
The fact that 2 PKD1 synonymous variants can disrupt normal pre-mRNA splicing emphasizes the importance of considering this type of mutations as mediators of pathogenic effects. Furthermore, our work highlights the need for functional analysis of these types of mutations in ADPKD, be it at the mRNA and/or protein level. Our data also indicates that most PKD1 exonic mutations are pathogenic because of their direct change of the amino acid sequence rather than by altering pre-mRNA splicing. This was not entirely surprising since the majority of the selected mutations are located in exons that may not require enhancers in order to be included in the mRNA. As reported by others, we showed that in silico analyzes, though valuable in giving a preliminary suggestion of the consequence of mutations, presently need to be complemented by experimental analysis. The results of our study have impotant inplications for genotype-phenotype correlation in ADPKD as truncating mutations are significantly more severe than nontruncating mutations. Although these correlations are not entirely understood, a recent ADPKD mutation screening study concludes that PKD1 truncating mutations are associated with much early onset of ESRD than nontruncating mutations.54
Pre-mRNA splicing defects are likely to have an impact on clinical practice as these seem to have a role in practically all known diseases with a genetic origin.23 An understanding of the role of pre-mRNA splicing in disease increases potential chances to explore therapeutic approaches. There are examples of disease-causing exonic mutations that have been studied considerably and have turn out to be key points for novel RNA-based therapeutic strategies.21
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was financed by Fondo de Investigación Sanitaria, Spain (grants PI07/1037 and PI14/00760), and co-financed by the European Regional Development Fund, “A way to build Europe.” FJGP and ERT were supported by FPU fellowship AP2005–2591 (Ministerio de Educación y Ciencia, Spain) and by FIS CA09/00465, respectively.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Wahl MC, Will CL, Lührmann R. The spliceosome: design principles of a dynamic RNP machine. Cell 2009; 136:701–18; PMID:19239890; http://dx.doi.org/ 10.1016/j.cell.2009.02.009 [DOI] [PubMed] [Google Scholar]
- 2.Wang Z, Burge CB. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA 2008; 14:802-13; PMID:18369186; http://dx.doi.org/ 10.1261/rna.876308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fica SM, Tuttle N, Novak T, Li NS, Lu J, Koodathingal P, Dai Q, Staley JP, Piccirilli JA. RNA catalyses nuclear pre-mRNA splicing. Nature 2013; 503:229-34; PMID:24196718; http://dx.doi.org/ 10.1038/nature12734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baralle D, Baralle M. Splicing in action: assessing disease causing sequence changes. J Med Genet 2005; 42:737-48; PMID:16199547; http://dx.doi.org/ 10.1136/jmg.2004.029538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu HX, Zhang M, Krainer AR. Identification of functional exonic splicing enhancer motifs recognized by individual SR proteins. Genes Dev 1998; 12:1998-2012; PMID:9649504; http://dx.doi.org/ 10.1101/gad.12.13.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Z, Rolish ME, Yeo G, Tung V, Mawson M, Burge CB. Systematic identification and analysis of exonic splicing silencers. Cell 2004; 119:831-45; PMID:15607979; http://dx.doi.org/ 10.1016/j.cell.2004.11.010 [DOI] [PubMed] [Google Scholar]
- 7.Wang Y, Xiao X, Zhang J, Choudhury R, Robertson A, Li K, Ma M, Burge CB, Wang Z. A complex network of factors with overlapping affinities represses splicing through intronic elements. Nat Struct Mol Biol 2013; 20:36-45; PMID:23241926; http://dx.doi.org/ 10.1038/nsmb.2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu Y, Maroney PA, Denker JA, Zhang XH, Dybkov O, Lührmann R, Jankowsky E, Chasin LA, Nilsen TW. Dynamic regulation of alternative splicing by silencers that modulate 5' splice site competition. Cell 2008; 135: 1224-36; PMID:19109894; http://dx.doi.org/ 10.1016/j.cell.2008.10.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Culler SJ, Hoff KG, Voelker RB, Berglund JA, Smolke CD. Functional selection and systematic analysis of intronic splicing elements identifies active sequence motifs and associated splicing factors. Nucleic Acids Res 2010; 38: 5152-65; PMID:20385591; http://dx.doi.org/ 10.1093/nar/gkq248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cartegni L, Krainer AR. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet 2002; 30: 377-84; PMID:11925564; http://dx.doi.org/ 10.1038/ng854 [DOI] [PubMed] [Google Scholar]
- 11.Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet 2002; 3:285-98; PMID:11967553; http://dx.doi.org/ 10.1038/nrg775 [DOI] [PubMed] [Google Scholar]
- 12.Goren A, Ram O, Amit M, Keren H, Lev-Maor G, Vig I, Pupko T, Ast G. Comparative analysis identifies exonic splicing regulatory sequences–The complex definition of enhancers and silencers. Mol Cell 2006; 22: 769-81; PMID:16793546; http://dx.doi.org/ 10.1016/j.molcel.2006.05.008 [DOI] [PubMed] [Google Scholar]
- 13.Pandit S, Zhou Y, Shiue L, Coutinho-Mansfield G, Li H, Qiu J, Huang J, Yeo GW, Ares M Jr, Fu XD. Genome-wide analysis reveals SR protein cooperation and competition in regulated splicing. Mol Cell 2013; 50: 223-35; PMID:23562324; http://dx.doi.org/ 10.1016/j.molcel.2013.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang E, Mueller WF, Hertel KJ, Cambi F. G Run-mediated recognition of proteolipid protein and DM20 5' splice sites by U1 small nuclear RNA is regulated by context and proximity to the splice site. J Biol Chem 2011; 286:4059-71; PMID:21127064; http://dx.doi.org/ 10.1074/jbc.M110.199927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graveley BR. Sorting out the complexity of SR protein functions. RNA 2000; 6:1197-211; PMID:10999598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Long JC, Caceres JF. The SR protein family of splicing factors: master regulators of gene expression. Biochem J 2009; 417:15-27; PMID:19061484; http://dx.doi.org/ 10.1042/BJ20081501 [DOI] [PubMed] [Google Scholar]
- 17.Shepard PJ, Hertel KJ. The SR protein family. Genome Biol 2009; 10:242; PMID:19857271; http://dx.doi.org/ 10.1186/gb-2009-10-10-242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ibrahim EC, Schaal TD, Hertel KJ, Reed R, Maniatis T. Serine/arginine-rich protein-dependent suppression of exon skipping by exonic splicing enhancers. Proc Natl Acad Sci U S A. 2005; 102:5002-7; PMID:15753297; http://dx.doi.org/ 10.1073/pnas.0500543102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wagner EJ, Garcia-Blanco MA. Polypyrimidine tract binding protein antagonizes exon definition. Mol Cell Biol 2001; 21:3281-88; PMID:11313454; http://dx.doi.org/ 10.1128/MCB.21.10.3281-3288.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pozzoli U, Sironi M. Silencers regulate both constitutive and alternative splicing events in mammals. Cell Mol Life Sci 2005; 62:1579-604; PMID:15905961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cooper TA, Wan L, Dreyfuss G. RNA and disease. Cell 2009; 136:777-93; PMID:19239895; http://dx.doi.org/ 10.1016/j.cell.2009.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sterne-Weiler T, Sanford JR. Exon identity crisis: disease-causing mutations that disrupt the splicing code. Genome Biol 2014; 15:201; PMID:24456648; http://dx.doi.org/ 10.1186/gb4150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang GS, Cooper TA. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet 2007; 8:749-61; PMID:17726481; http://dx.doi.org/ 10.1038/nrg2164 [DOI] [PubMed] [Google Scholar]
- 24.Pagani F, Baralle FE. Genomic variants in exons and introns: identifying the splicing spoilers. Nat Rev Genet 2004; 5:389-96; PMID:15168696; http://dx.doi.org/ 10.1038/nrg1327 [DOI] [PubMed] [Google Scholar]
- 25.Krawczak M, Thomas NS, Hundrieser B, Mort M, Wittig M, Hampe J, Cooper DN. Single base-pair substitutions in exon-intron junctions of human genes: nature, distribution, and consequences for mRNA splicing. Hum Mutat 2007; 28:150-58; PMID:17001642; http://dx.doi.org/ 10.1002/humu.20400 [DOI] [PubMed] [Google Scholar]
- 26.Lewandowska MA. The missing puzzle piece: splicing mutations. Int J Clin Exp Pathol 2013; 6:2675-82; PMID:24294354 [PMC free article] [PubMed] [Google Scholar]
- 27.Pagani F, Raponi M, Baralle FE. Synonymous mutations in CFTR exon 12 affect splicing and are not neutral in evolution. Proc Natl Acad Sci U S A 2005; 102:6368-72; PMID:15840711; http://dx.doi.org/ 10.1073/pnas.0502288102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sterne-Weiler T, Howard J, Mort M, Cooper DN, Sanford JR. Loss of exon identity is a common mechanism of human inherited disease. Genome Res 2011; 21:1563-71; PMID:21750108; http://dx.doi.org/ 10.1101/gr.118638.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kashima T, Manley JL. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat Genet 2003; 34:460-63; PMID:12833158; http://dx.doi.org/ 10.1038/ng1207 [DOI] [PubMed] [Google Scholar]
- 30.Baralle M, Baralle D, De Conti L, Mattocks C, Whittaker J, Knezevich A, Ffrench-Constant C, Baralle FE. Identification of a mutation that perturbs NF1, a gene splicing using genomic DNA samples and a minigene assay. J Med Genet 2003; 40:220-22; PMID:12624144; http://dx.doi.org/ 10.1136/jmg.40.3.220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Claverie-Martín F, Flores C, Antón-Gamero M, González-Acosta H, García-Nieto V. The Alu insertion in the CLCN5 gene of a patient with Dent's disease leads to exon 11 skipping. J Hum Genet. 2005; 50:370-4; PMID:16041495; http://dx.doi.org/ 10.1007/s10038-005-0265-5 [DOI] [PubMed] [Google Scholar]
- 32.Nicholson P, Mühlemann O. Cutting the nonsense: the degradation of PTC-containing mRNAs. Biochem Soc Trans 2010; 38:1615-20; PMID:21118136; http://dx.doi.org/ 10.1042/BST0381615 [DOI] [PubMed] [Google Scholar]
- 33.Gaildrat P, Killian A, Martins A, Tournier I, Frébourg T, Tosi M. Use of splicing reporter minigene assay to evaluate the effect on splicing of unclassified genetic variants. Methods Mol Biol 2010; 653:249-57; PMID:20721748; http://dx.doi.org/ 10.1007/978-1-60761-759-4_15 [DOI] [PubMed] [Google Scholar]
- 34.Cooper TA. Use of minigene systems to dissect alternative splicing elements. Methods 2005; 37:331-40; PMID:16314262; http://dx.doi.org/ 10.1016/j.ymeth.2005.07.015 [DOI] [PubMed] [Google Scholar]
- 35.Bonnet C, Krieger S, Vezain M, Rousselin A, Tournier I, Martins A, Berthet P, Chevrier A, Dugast C, Layet V, et al.. Screening BRCA1 and BRCA2 unclassified variants for splicing mutations using reverse transcription PCR on patient RNA and an ex vivo assay based on a splicing reporter minigene. J Med Genet 2008; 45:438-46; PMID:18424508; http://dx.doi.org/ 10.1136/jmg.2007.056895 [DOI] [PubMed] [Google Scholar]
- 36.Tournier I, Vezain M, Martins A, Charbonnier F, Baert-Desurmont S, Olschwang S, Wang Q, Buisine MP, Soret J, Tazi J, et al.. A large fraction of unclassified variants of the mismatch repair genes MLH1 and MSH2 is associated with splicing defects. Hum Mutat 2008; 29:1412-24; PMID:18561205; http://dx.doi.org/ 10.1002/humu.20796 [DOI] [PubMed] [Google Scholar]
- 37.Reese MG, Eeckman FH, Kulp D, Haussler D. Improved Splice Site Detection in Genie. J Comput Biol 1997; 4:311-23; PMID:9278062 [DOI] [PubMed] [Google Scholar]
- 38.Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J Mol Biol 1997; 268:78-94; PMID:9149143; http://dx.doi.org/ 10.1006/jmbi.1997.0951 [DOI] [PubMed] [Google Scholar]
- 39.Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 2004; 11:377-94; PMID:15285897 [DOI] [PubMed] [Google Scholar]
- 40.Desmet FO, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, Béroud C. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 2009; 37:e67; PMID:19339519; http://dx.doi.org/ 10.1093/nar/gkp215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res 2003; 31:3568-71; PMID:12824367; http://dx.doi.org/ 10.1093/nar/gkg616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fairbrother WG, Yeo GW, Yeh R, Goldstein P, Mawson M, Sharp PA, Burge CB. RESCUE-ESE identifies candidate exonic splicing enhancers in vertebrate exons. Nucleic Acids Res 2004; 32:W187-90 PMID:15215377; http://dx.doi.org/ 10.1093/nar/gkh393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fairbrother WG, Yeh RF, Sharp PA, Burge CB. Predictive identification of exonic splicing enhancers in human genes. Science 2002; 297: 1007-13; PMID:12114529; http://dx.doi.org/ 10.1126/science.1073774 [DOI] [PubMed] [Google Scholar]
- 44.Hartmann L, Theiss S, Niederacher D, Schaal H. Diagnostics of pathogenic splicing mutations: does bioinformatics cover all bases? Front Biosci 2008;13:3252-72; PMID:18508431; http://dx.doi.org/ 10.2741/2924 [DOI] [PubMed] [Google Scholar]
- 45.Houdayer C, Dehainault C, Mattler C, Michaux D, Caux-Moncoutier V, Pagès-Berhouet S, d'Enghien CD, Laugé A, Castera L, Gauthier-Villars M, et al.. Evaluation of in silico splice tools for decision-making in molecular diagnosis. Hum Mutat 2008; 29:975-82; PMID:18449911; http://dx.doi.org/ 10.1002/humu.20765 [DOI] [PubMed] [Google Scholar]
- 46.Mort M, Sterne-Weiler T, Li B, Ball EV, Cooper DN, Radivojac P, Sanford JR, Mooney SD. MutPred Splice: machine learning-based prediction of exonic variants that disrupt splicing. Genome Biol 2014; 15(1):R19; PMID:24451234; http://dx.doi.org/ 10.1186/gb-2014-15-1-r19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, Yuen RK, Hua Y, Gueroussov S, Najafabadi HS, Hughes TR, et al.. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 2015; 347(6218):1254806; PMID:25525159; http://dx.doi.org/ 10.1126/science.1254806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gonzalez-Paredes FJ, Ramos-Trujillo E, Claverie-Martin F. Defective pre-mRNA splicing in PKD1 due to presumed missense and synonymous mutations causing autosomal dominant polycystic disease. Gene 2014; 546:243-9; PMID:24907393; http://dx.doi.org/ 10.1016/j.gene.2014.06.004 [DOI] [PubMed] [Google Scholar]
- 49.Harris PC, Torres VE. Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J Clin Invest 2014; 124:2315-24; PMID:24892705; http://dx.doi.org/ 10.1172/JCI72272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Buratti E, Baralle M, Baralle FE. Defective splicing, disease and therapy: searching for master checkpoints in exon definition. Nucleic Acids Res 2006; 34:3494-510; PMID:16855287; http://dx.doi.org/ 10.1093/nar/gkl498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lantinga-van Leeuwen IS, Dauwerse JG, Baelde HJ, Leonhard WN, van de Wal A, Ward CJ, Verbeek S, Deruiter MC, Breuning MH, de Heer E, et al.. Lowering of PKD1 expression is sufficient to cause polycystic kidney disease. Hum Mol Genet 2004; 13:3069-77; PMID:15496422; http://dx.doi.org/ 10.1093/hmg/ddh336 [DOI] [PubMed] [Google Scholar]
- 52.Hopp K, Ward CJ, Hommerding CJ, Nasr SH, Tuan HF, Gainullin VG, Rossetti S, Torres VE, Harris PC. Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J Clin Invest 2012; 122:4257-73; PMID:23064367; http://dx.doi.org/ 10.1172/JCI64313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baralle M, Skoko N, Knezevich A, De Conti L, Motti D, Bhuvanagiri M, Baralle D, Buratti E, Baralle FE. NF1 mRNA biogenesis: effect of the genomic milieu in splicing regulation of the NF1 exon 37 region. FEBS Lett 2006; 580:4449-56; PMID:16870183; http://dx.doi.org/ 10.1016/j.febslet.2006.07.018 [DOI] [PubMed] [Google Scholar]
- 54.Cornec-Le Gall E, Audrézet MP, Chen JM, Hourmant M, Morin MP, Perrichot R, Charasse C, Whebe B, Renaudineau E, Jousset P, et al.. Type of PKD1 mutation influences renal outcome in ADPKD. J Am Soc Nephrol 2013; 24:1006-13; PMID:23431072; http://dx.doi.org/ 10.1681/ASN.2012070650 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.