

Abstract
Ribosome biogenesis governs protein synthesis. NIFK is transactivated by c-Myc, the key regulator of ribosome biogenesis. The biological function of human NIFK is not well established, except that it has been shown to interact with Ki67 and NPM1. Here we report that NIFK is required for cell cycle progression and participates in the ribosome biogenesis via its RNA recognition motif (RRM). We show that silencing of NIFK inhibits cell proliferation through a reversible p53-dependent G1 arrest, possibly by induction of the RPL5/RPL11-mediated nucleolar stress. Mechanistically it is the consequence of impaired maturation of 28S and 5.8S rRNA resulting from inefficient cleavage of internal transcribed spacer (ITS) 1, a critical step in the separation of pre-ribosome to small and large subunits. Complementation of NIFK silencing by mutants shows that RNA-binding ability of RRM is essential for the pre-rRNA processing and G1 progression. More specifically, we validate that the RRM of NIFK preferentially binds to the 5′-region of ITS2 rRNA likely in both sequence specific and secondary structure dependent manners. Our results show how NIFK is involved in cell cycle progression through RRM-dependent pre-rRNA maturation, which could enhance our understanding of the function of NIFK in cell proliferation, and potentially also cancer and ribosomopathies.
Keywords: cell cycle, Ki67, ribosome biogenesis, nucleolar stress, RNA recognition motif
Abbreviations
- 5S RNP
5S ribonucleoprotein particle
- CDK1
cyclin dependent kinase 1
- DFC
dense fibrillar component
- ETS/ITS
external/internal transcribed spacers
- GSK3
glycogen synthase kinase 3
- Ki67FHAID
Ki67-FHA interaction domain
- LSU
large subunit
- MDM2
murine double minute 2
- NIFK
Nucleolar protein Interacting with the FHA domain of pKi-67
- Noprecipitation
- NPM1/B23
nucleophosmin
- PAR-CLIP
Photo-Activatable-Ribonucleoside-Enhanced Crosslinking and Immu-pre-rRNAs, rRNA precursors
- REMSA
RNA electrophoresis mobility shift assay
- rNIFK
recombinant NIFK
- RNP1 and 2
ribonucleoprotein motif 1 and 2
- RPL5 and RPL11
large ribosomal protein 5 and 11
- RRM
RNA recognition motif
- snoRNP
small nucleolar ribonucleoprotein
Introduction
Ki67 is a well-known cell proliferation marker that has been correlated with aggressiveness of tumor and considered as a prognostic parameter.1,2 A human nucleolar protein interacting with the forkhead associated (FHA) domain of Ki67, named NIFK, was identified through 2-hybrid screening by using the FHA domain of Ki67 as the bait.3 Human NIFK protein consists of a putative RNA recognition motif (RRM) and a Ki67-FHA interaction domain (Ki67FHAID). Phosphorylation of Thr-234 and Thr-238 of the Ki67 interaction domain of NIFK was shown to be responsible for Ki67 interaction during mitosis.3 We previously showed that the phosphorylation of Thr-238 of NIFK by cyclin dependent kinase 1 (CDK1) primes the phosphorylation of Thr-234 by glycogen synthase kinase 3 (GSK3), and demonstrated the molecular detail of the NIFK-Ki67 interaction.4 In addition, NIFK was reported to be transcriptionally up-regulated by both c-Myc5 and estrogen,6 suggesting a role of NIFK in cell proliferation. In agreement, NIFK was shown to maintain the proliferation and pluripotency of embryonic stem cells by interacting with nucleophosmin 1 (NPM1/B23), a multi-functional protein with endoribonuclease activity.7,8 However, specific functions of NIFK and their molecular mechanisms remain poorly understood.
Ribosome biogenesis is a multifaceted process initiated by the transcription of rRNA precursors (pre-rRNAs), followed by subsequent processing steps to remove external/internal transcribed spacers (ETS/ITS) for the maturations of 18S, 25S/28S and 5.8S rRNAs.9-11 Strictly monitored ribosome biogenesis ensures the concerted coordination for cell growth and proliferation, while its dysregulation may lead to ribosomopathies and cancer.12,13 c-Myc, a proto-oncogene, was shown to enhance cell growth and tumorigenesis14 directly through transactivation of factors that are involved in rRNA synthesis, ribosome biogenesis, and protein translation.15,16 In addition, perturbation of ribosome biogenesis stimulates nucleolar stress that subsequently activates p53-dependent G1 arrest,17-19 and this activation relies on the 5S ribonucleoprotein particle (5S RNP), a trimeric complex composed of large ribosomal proteins 5, 11 (RPL5, RPL11) and 5S rRNA.20,21 Ribosomal 5S RNP is essential for large subunit (LSU) rRNA maturation during cell growth, whereas nonribosomal 5S RNP directly interacts with and suppresses the E3 ubiquitin ligase activity of MDM2 (murine double minute 2, or HDM2 for its human ortholog) upon nucleolar stress,20,21 through which p53 is stabilized.
Recently, systematic screenings have suggested association of NIFK with ribosome biogenesis22 and requirement of NIFK for rRNA processing.23 In agreement, Nop15, the yeast ortholog of NIFK that consists of a RRM but lacks the Ki67-FHA interaction motif, is required for yeast 5.8S and 25S rRNA maturation,24 presumably through remodeling of the ITS2 structure.25,26 These observations collectively point to NIFK as a participant in ribosome biogenesis. However, the underlying molecular mechanisms and the relationship with the role of NIFK in cell growth and proliferation are still unknown. In this work, we tested the possibility that NIFK functions as a key adaptor that bridges cell proliferation, presumably through Ki67FHAID, to cell growth, through the RRM-mediated pre-rRNA processing. By using siRNA silencing and phenotypic rescue, we showed that NIFK functions in cell cycle progression, checkpoint signaling, and ribosome biogenesis. To be more specific, we verified the contribution of RRM in the above processes and pinpointed critical residues that are involved in these functions. In addition, we also mapped the putative pre-rRNA binding region recognized by NIFK. We therefore propose that NIFK is required for LSU rRNA maturation, likely through the RRM-mediated pre-rRNA binding.
Results
Silencing of NIFK inhibited cell proliferation through a reversible p53-dependent G1 arrest
Since Ki67 is a well-known cell proliferation marker,2 we asked whether its interacting protein NIFK is also involved in cell proliferation. We first generated NIFK deficient phenotypes in U2OS cells by siRNA silencing. The most effective siRNA (#1 in Fig. S1A-B) was chosen for the rest of this work unless otherwise specified. As shown in Fig. 1A, cell proliferation was significantly repressed in NIFK-silenced cells, suggesting that NIFK is required during cell proliferation. To characterize whether slower cell proliferation rate is due to apoptosis or arrested cell cycle, we analyzed NIFK-knockdown cells using flow cytometry. No significant change in apoptosis from NIFK-knockdown cells was detected (Fig. S1C), while an increase in G1 phase population resulting from NIFK knockdown was observed in the DNA content histogram (Fig. 1B, traces 1–2). The effect became more pronounced when cells were trapped in the G2/M phase by synchronizing with nocodazole (Fig. 1B, traces 3–4). The results suggest that the slower cell proliferation in NIFK-knockdown cells is more likely caused by perturbation in the G1-S progression of the cell cycle. Similar phenotypes were confirmed in another cell line MCF7 (Fig. S1D-G).
Figure 1.

Silencing of NIFK induces reversible p53 dependent G1 arrest. (A) Cellular proliferation of U2OS cells transfected with siNIFK. (B) Flow cytometry analyses of asynchronous and G2/M synchronous U2OS cells transfected with siNIFK. (C) Same as B, with indicated siRNA alone or in combination. The quantifications of 2 repeats are also shown in the right panel. (D) Western blot analysis of the expressions of NIFK, p53, and p21 in asynchronous U2OS cells transfected with indicated siRNA.
To further understand how NIFK knockdown leads to G1 arrest, we next examined the effect of NIFK knockdown on p53 since p53 has been suggested to choose between cell cycle and apoptosis.27 As shown in Fig. 1D and S1E, the protein levels of both p53 and cyclin-dependent kinase inhibitor p21 are more pronounced upon NIFK down-regulation, suggesting that the p53-p21 axis is responsible for the NIFK-dependent G1 arrest. This observation was further supported by the reversal of the NIFK-dependent G1 arrest upon p53 and p21 co-knockdown with NIFK (Fig. 1C). Interestingly, the elevated protein level of p21 in response to NIFK downregulation was eliminated when p53 was also silenced, but not vice versa (Fig. 1D, lanes 3–5), indicating that the NIFK knockdown-mediated up-regulation of p21 occurs through activation of p53. Such upregulation of p21 likely directs the NIFK-dependent G1 arrest. We therefore conclude that p53 and p21 are the key mediator and the major effector, respectively, that control the G1 arrest in response to NIFK down-regulation.
NIFK is required for G1 progression by participating in rRNA processing
Having shown that NIFK downregulation triggers p53 activation leading to p21 directed G1/S phase arrest, we next addressed how NIFK is required for cell cycle progression. As a proposed stress sensor, p53 transmits not only genotoxic signals28 but also those of ribosome biogenesis stress.17-21 Since defects in steps of ribosome biogenesis create nucleolar stress that can activate p53 to halt the cell cycle, we hypothesized that NIFK down-regulation activates p53 through 5S RNP-dependent nucleolar stress response. To test this possibility, we examined the effect of 2 stress mediators, RPL5 and RPL11,18,20,21 on the NIFK-mediated stress responses. As shown in Fig. 2A (left panel), the G1 arrest upon NIFK downregulation was relieved in response to co-knockdown of RPL5 or RPL11. Such resumed cell cycle is more likely due to inhibition of stress signals, as the promoted p53 and p21 levels after NIFK knockdown were simultaneously lowered by additional RPL5 or RPL11 co-knockdown (Fig. 2A, right panel; the changes of mRNAs after siRNA transfection are shown in Fig. S2A). This result suggests a possible link between the loss of NIFK to the nucleolar stress that activates p53 for arresting cell cycle. In support of this possibility, we showed that NIFK appears to co-localize with fibrillarin and partially with Ki67 (Fig. 2B), indicating that the subcellular localization of NIFK is in both the dense fibrillar component (DFC) and, to a lesser extent, the outer DFC where Ki67 is located.29,30 In addition, silencing of NIFK does not affect nucleolus organization as indicated by Fig. S2B. Because the majority of cleavage and modification of pre-rRNAs occur in DFC,29 NIFK likely contributes to the fundamental functions of nucleolus through rRNA processing.
Figure 2.
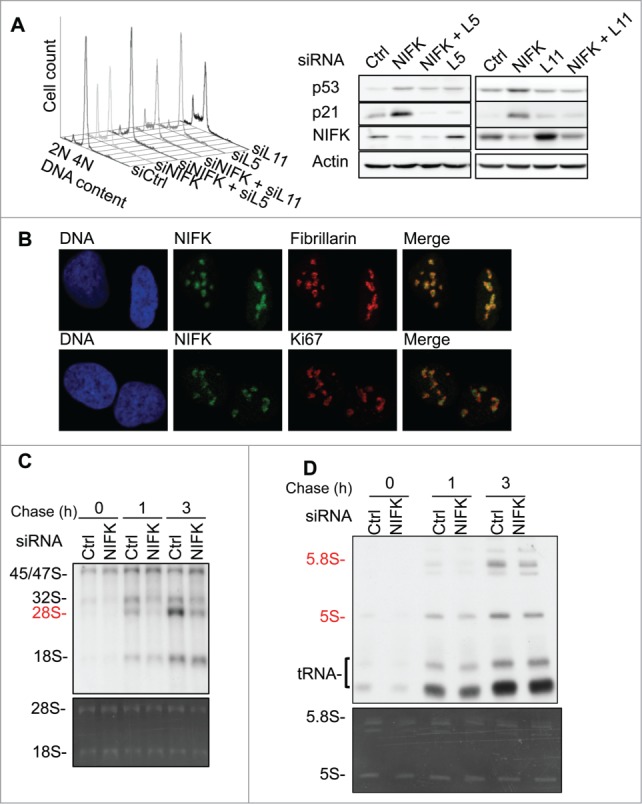
NIFK is required for G1 progression by participating in rRNA processing. (A) Flow cytometry analysis of G2/M synchronous U2OS cells transfected with indicated siRNAs (left panel), and Western blot analysis of indicated proteins of asynchronous cells (right panel). (B) Immunofluorescent staining of U2OS cells showing subnucleolar localization of NIFK (green), Ki67 (red), fibrillarin (red), and nuclei (blue). (C) 32P-orthophosphate based pulse-chase analysis showing the kinetics of nascent rRNA synthesis in U2OS cells transfected with siNIFK. 32P labeled RNAs are separated by 1% agarose-formaldehyde gel and visualized by autoradiography (upper panel). The total RNA is shown by ethidium bromide (EtBr) staining (lower panel). (D) The same as (C) except the RNAs were separated by a 10% polyacrylamide-7 M urea gel. The total RNA is shown by ethidium bromide (EtBr) staining (lower panel and full view in Fig. S2D).
To identify which step of ribosome biogenesis NIFK is involved in, we pulsed the cells with 32P-orthophosphate and chased the nascent rRNA. As shown in Fig. 2C, the 45/47S rRNA precursors representing the phosphate incorporation efficiency were little affected by NIFK-knockdown, implying that NIFK down-regulation may not directly affect rRNA synthesis. On the other hand, the maturations of 28S rRNA (Fig. 2C) and 5.8S rRNAs (Fig. 2D) were markedly delayed in response to NIFK downregulation. The decrease in nascent 28S rRNA synthesis in response to NIFK down-regulation could also result from p53-mediated RNA polymerase I (RNA Pol I) repression, since it has been shown that p53 is able to interfere with the assembly of SL1-UBF-RNA Pol I initiation complex at the rRNA promoter.31 To rule out this possibility, we further showed that p53 knockdown, in addition to NIFK knockdown, had no additional effect on the nascent 28S rRNA synthesis (Fig. S2C). Taken together, our results link the role of NIFK in rRNA processing to its requirement for G1 progression.
It is noteworthy that a decrease in the newly synthesized 5S rRNA was also observed upon silencing of NIFK (Fig. 2D, upper panel), albeit that its steady-state level remains unchanged (Fig. 2D, lower panel). Since 5S rRNA is part of 5S RNP required for the stabilization of p53, the diminished nascent-5S rRNA upon NIFK depletion could contradict to that of promoted p53 level shown in Figures 1D and 2A. However, this inverted correlation between the levels of 5S rRNA and p53 was also observed previously in the depletion of other LSU biogenesis factors,21 suggesting that NIFK may function similarly in supporting LSU biogenesis.21 Furthermore, it was shown that direct depletion of 5S rRNA or inhibition of RNA pol III (TFIIIA) both result in reduced, but not abolished, activation of p53 upon inhibition of RNA pol I (treatment with Actinomycin D),21 suggesting that the remaining amount of 5S rRNA is sufficient to support p53 activation in such condition. In this regard, the inconsistent levels between 5S rRNA and p53 protein upon silencing of NIFK we observed might represent such minor or reduced activation of p53. In agreement, silencing of RPL5 and RPL11 both reverse the activation of p53 caused by silencing of NIFK (Fig. 2A), similar to what was shown previously for the treatment of Actinomycin D.21
NIFK mediated pre-rRNA processing and G1 progression requires RRM but not Ki67FHAID
Given that NIFK consists of a Ki67-FHA interaction domain (Ki67FHAID) in addition to RRM, and that Ki67 is well established to correlate with cell proliferation, it seemed reasonable to expect that the RRM and the Ki67FHAID are responsible for the 2 functions of NIFK, pre-rRNA processing and regulation of G1 progression, respectively. To examine this possibility, the siRNA resistant version of NIFK, RRM deletion (NIFK-dR), Ki67FHAID deletion (NIFK-dK), and alanine substitution of threonine 234 and 238 of NIFK (NIFK-TA) were respectively transduced into U2OS cells (Fig. 3A and S3A left panel) and subjected to siRNA transfection (Fig. S3A right panel) to deplete the endogenous NIFK. Characterization of these cells indicate that NIFK-silenced cells expressing RRM deletion display a dramatic retardation of cell proliferation (Fig. 3B), an irrelievable G1 arrest (Fig. 3C), a significant repression in nascent 28S rRNA synthesis (Fig. 3D), and a marked induction of p53/p21 proteins (Fig. 3E), while NIFK-dK and NIFK-TA expressing cells show only partial delay in proliferation (Fig. 3B). Relevant control experiments are shown in Fig. S3B-D. These observations strongly suggest that RRM is essential for NIFK-mediated rRNA processing and G1 progression, while the Ki67-interacting motif plays only a minor role in these functions.
Figure 3.
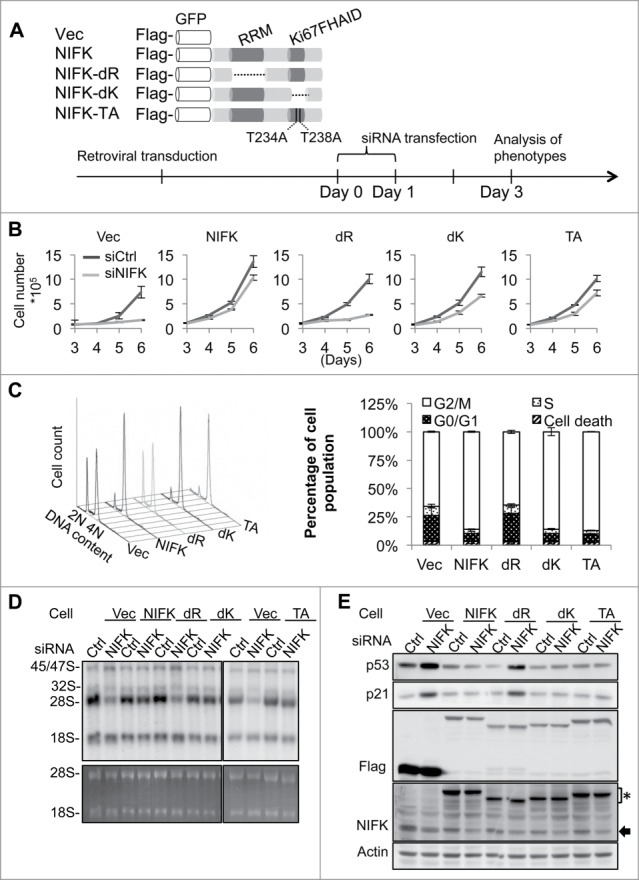
NIFK-mediated pre-rRNA processing and G1 progression require RRM but not Ki67FHAID. (A) Schematic representation of NIFK functional domains and designing of ectopic NIFK expression constructs (upper panel). For immuno and fluorescent detection, Flag-tag epitope and GFP were fused upstream of NIFK cDNA. dR indicates RRM deletion; dK, Ki67FHAID deletion; TA, T234AT238A; Vec, Flag-GFP vector. The procedure and time line for phenotypic rescue experiments are shown in the bottom panel. (B) Cell proliferation assay for cells phenotypically rescued by NIFK wild-type and mutants. (C) Flow cytometry analyses (left) and quantification (right) of rescued cells after G2/M synchronization. (D) 32P-orthophosphate based pulse-chase analysis showing the nascent rRNA synthesis in phenotypically rescued cells. 32P labeld RNAs at 4.5 h chasing time were visualized by autoradiography (upper panel) and EtBr staining (lower panel). (E) Western blot analysis showing p53 and p21 levels in the phenotypically rescued cells described in (D). Anti-NIFK antibody (NIFK) detects both endogenous and ectopically expressed NIFK. The asterisk indicates ectopic NIFK and the arrow, endogenous NIFK.
To identify specific residues responsible for the functions of RRM, we constructed site-specific mutants for further characterization. Based on the structural topology of RRM,32 there are 2 conserved sequences in NIFK-RRM, namely ribonucleoprotein 1 and 2 (RNP1 and 2), which comprises mainly aromatic and positively charged residues. We selected candidate residues out of these 2 RNPs by sequence comparison (Y48 and F93), structural comparison (R75), or both (K86, Y88 and F90)33 (Fig. 4A and S4A). A series of alanine-substituted mutants were generated accordingly, including Y48AR75AY88AF90A (designated as NIFK-4Y), Y48AR75AY88AF90AK86A (NIFK-4YK), Y48AR75AY88AF90AK86AF93A (NIFK-4YKF), and K86AF93A (NIFK-KF), and subjected to retroviral transduction and functional analyses. According to cell cycle analysis, all NIFK-RRM mutants except NIFK-4Y exhibit lesser capacity than that of wild-type NIFK in rescuing G1 arrest upon NIFK silencing (Fig. 4B and control experiments in Fig. S4B-C). 32P-based pulse chase experiments also indicated that the aforementioned NIFK-RRM mutants except NIFK-4Y fail to rescue the deficiency in 28S rRNA maturation upon NIFK silencing (Fig. 4C and S4D). Similar pattern was also observed in the evaluation of nucleolar stress markers (Fig. 4D and S4E). Considering that NIFK-4Y is still proficient while NIFK-4YK is deficient, the residue K86 appears to be critical in NIFK function. Paradoxically, it requires double mutation K86AF93A to fully exert the stress effect as shown in Fig. 4C-D and S4D-E.
Figure 4.
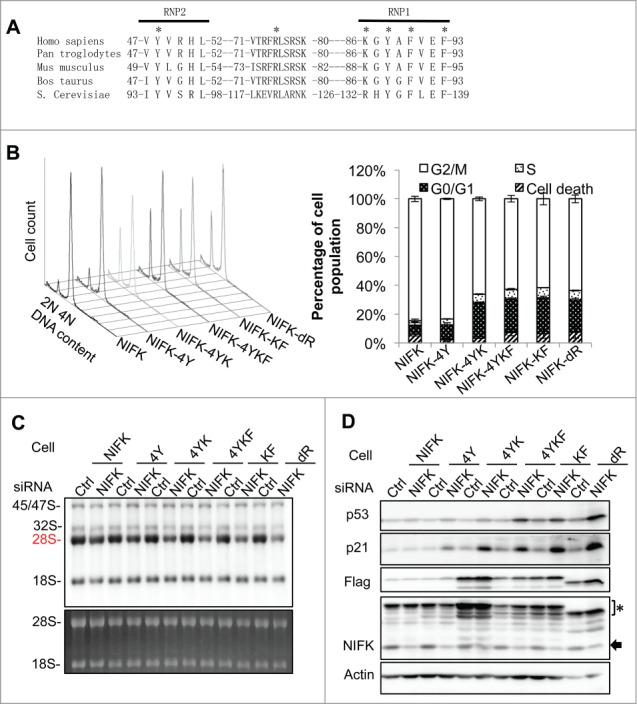
Identification of specific residues responsible for the RRM function in rRNA processing and G1 progression. (A) Sequence alignment of NIFK-RRM orthologues from different species. Asterisks indicate residues selected for alanine substitution. (B) Cell cycle analyses, (C) nascent rRNA synthesis, and (D) Western blot analyses of the phenotypically rescued cells described in Fig. 3, showing defects of NIFK-RRM mutants in rescuing NIFK silencing. The asterisk indicates ectopic NIFK and the arrow, endogenous NIFK.
NIFK regulates 28S rRNA maturation through processing at ITS1 site 2
After initial transcription, maturation of pre-rRNAs relies on the excision of transcribed spacers through a complicated cascade as shown in Fig. 5A and S5.34,35 Any defect in pre-rRNA processing can lead to accumulation of incomplete precursor and intermediates of rRNA. To elucidate why the maturations of 28S and 5.8S rRNA are impaired in the absence of NIFK and, more specifically, how RRM directs the pre-rRNA processing, we analyzed RNA samples derived from NIFK-knockdown cells with Northern blot. As indicated by radiolabeled probe P1 (Fig. 5B), we observed accumulation of early precursor 45S/47S and reduction of 30S/26S pre-rRNAs upon silencing of NIFK. The same trend was also observed using probes P2 and P3 (Fig. 5C-D), suggesting a delayed processing of ITS1 at site 2 upon silencing of NIFK (see Fig. 5A and S5A for steps of pre-rRNA processing, cleavage sites, and intermediate rRNA species). In agreement, probes P2 and P3 also revealed increased 41S and decreased 21S pre-rRNAs upon silencing of NIFK (Fig. 5C-D). In parallel, hybridization with probe P4 (between 5.8S and ITS2 site 4) showed the same increase of 45S/47S/41S/36S rRNA and similar decrease in 32S/12S rRNA (Fig. 5E). The inverse correlation between the levels of 45/47/41/36S and 32/30/21/12S pre-rRNAs suggests that the endonucleolytic cleavage at site 2 was affected. In addition, the inverse relationship between the levels of 36S and 32S pre-rRNA likely points to a NIFK-dependent defect in the conversion of 36S to 32S pre-rRNA, which is presumably through XRN2 exonucleolytic cleavage of ITS1.34,36 Because 32S pre-rRNA is the precursor of 28S and 5.8S rRNA, decrease in 32S pre-rRNA in response to NIFK silencing apparently causes insufficient production of 28S and 5.8S rRNAs (Fig. 5F).
Figure 5.

NIFK regulates 28S and 5.8S rRNA maturation through processing of ITS1 site 2. (A) Schematic representation of human rRNA transcripts. Upper, human 47S precursor rRNA showing the transcribed spacers (ETS and ITS), coding sequences, and cleavage sites. Lower left, illustration of 47S pre-rRNA processing pathways. The alternative steps are colored gray. Lower right, overview of pre-rRNA intermediates. Arrowheads indicate the positions of the probes used in Northern blot analysis. (B-F) Northern blot analyses showing the pre-rRNAs derived from siNIFK transfected U2OS cells. The specific rRNA species were detected using probes complementary to the regions downstream of site A0 of 5′ETS (P1, shown in B), between 18S and site 2a (P2, shown in C), between site 2a and 2 of ITS1 (P3, shown in D), between 5.8S and site 4 of ITS2 (P4, shown in E), and 18S, 5.8S, and 28S rRNA (probes 5, 6 and 7, shown in F). (G) Northern blot analysis of the pre-rRNAs derived from phenotypic rescued cells using probe P4. The complementation of NIFK silencing by NIFK and NIFK-KF mutant following that described in Fig. 4. (H) The same as (E), except that the siRPL5 transfected U2OS cells are also compared.
Although the steady-state level of 18S RNA appears to be slightly affected (Fig. 5F, hybridization by probe P5), there was no significant defect in nascent 18S RNA synthesis upon silencing of NIFK (Figs. 2C-D, 3D, and 4C). This might be due to an alternative generation of 18SE (Fig. 5C) through direct cleavage at ITS1 site 2a34. Since Northern blot analysis represents an overview of processed rRNAs while pulse-chase analysis reflects only those de novo rRNAs immediately after processing, one could reason that the difference between steady-state and nascent 18S RNA was due to inefficient production of 30S pre-rRNA upon silencing of NIFK in longer duration. This result collectively suggests that silencing of NIFK creates more significant defects in cleavage at site 2 in contrast to that of site 2a for the maturation of 28S and 5.8S rRNAs.
We also examined whether RRM is required for the rRNA processing at ITS1 site 2. In line with nascent rRNA synthesis and processing results shown above (Fig. 4C), expression of NIFK efficiently rescued the defects in 32S/12S pre-rRNA production upon silencing of NIFK (Fig. 5G, middle panel). As expected, such complementation of NIFK silencing by the NIFK-KF mutant appeared to be relatively inefficient as the levels of 45S/47S/41S/36S increased and those of 32S/12S pre-rRNAs decreased (Fig. 5G, right panel), supporting an indispensable role of RRM in the process. It is noteworthy that, as silencing of RPL5 led to accumulation of 32S/12S rRNA while silencing of NIFK resulted in the reverse (Fig. 5H), the 2 proteins likely function at different stages of ribosome biogenesis. During the preparation of this manuscript, a systematic silencing screening study performed by Tafforeau et al. showed similarly that silencing of NIFK triggers the increase of 41S/18SE rRNA and decrease of 30S/21S/12S rRNA.23 This independently supports the role of NIFK we propose here, though the scopes of the 2 studies are different.
The RRM of NIFK binds to the 5′-end of ITS2 rRNA
Because NIFK lacks enzymatic function, its functional role in pre-rRNA processing should be attributable to the rRNA binding by RRM. To further characterize the binding property of RRM, we mapped the RRM binding region of rRNA. Since accumulated intermediate pre-rRNAs are likely due to inefficient spacer excisions during rRNA maturation in the absence of NIFK (Fig. 5), the spacers are most likely where NIFK-RRM binds. Although its yeast ortholog Nop15 has been shown to preferentially bind with 5′ end of ITS2 rRNA,26 the potential rRNA binding region of NIFK in human pre-rRNA remains unknown. We initially tried to identify the NIFK-bound rRNA sequence using 6-thioguanosine (6SG) based PAR-CLIP (Photoactivatable-Ribonucleoside-Enhanced Crosslinking and Immunoprecipitation) followed by Illumina Solexa sequencing.37 However, the result was inconclusive possibly due to the extremely high GC content in some human pre-rRNA spacers (ca. 80%, compared to ca. 40% in S. cerevisiae) that hampers the library preparation and cluster generation during the sequencing steps. Alternatively, we addressed this issue by preparing a series of biotinylated RNA corresponding to candidate spacers for streptavidin immobilization, including 5′ETS (1–1952, containing site A0), ITS1 (1-509 and 510-1095), ITS2 (1-1155, 1-409, and 880-1155), together with an irrelevant control sequence. These RNA immobilized beads should be able to retrieve endogenous NIFK from cell extracts thus allowing for western blot analysis, if the binding exists. As expected, NIFK did show preference to the spacers, more specifically to the 1st-409th nt region of ITS2 rRNA (Fig. 6A, left panel). We then further truncated ITS2 5′-rRNA and found that NIFK prefers to bind the 50th-150th nt of the ITS2 rRNA (Fig. 6A, right panel and Fig. S6A). Interestingly, NIFK shows preference to the 50th–150th nt but not the 1st–100th nt nor the 100th–200th nt (Fig. 6A, right panel). This observation suggests that the rRNA binding by NIFK should be either sequence specific (to the region covering the 100th nt) or secondary-structure dependent (formed by the 50th–150th nt of ITS2 rRNA).
Figure 6 (See previous page).

The RRM of NIFK binds to the 5′-end of ITS2 rRNA. (A) Western blot analysis of NIFK associated with indicated RNAs. Biotinylated or non-biotinylated RNAs were immobilized on streptavidin beads. RNA coupling efficiency was shown by SYBR green staining (lower panel). The proteins brought down by RNA were eluted, and analyzed by Western blot using NIFK antibody (upper panel). (B) Northern blot of RNase footprinting of ITS2 1-200 RNA protected by rNIFK. Lane 1, RNase T1 digested RNAs. Several G positions are indicated. Lane 2, RNA ladders generated by alkaline hydrolysis. Lanes 3, 0.016 U/μL RNase I digested RNA. Lanes 4-5, RNase I digested RNA in the absence (R) or presence (R+P) of rNIFK. Lanes 6-7, longer exposure of autoradiography shown in lanes 4-5. The bracket marks the position protected by rNIFK. (C) ITS2 50-150 RNA secondary structure detection. Left panel, autoradiography of 5′-end labeled RNA. Lane 1, RNA ladders generated by alkaline hydrolysis. Lanes 2-3, RNase T1 digested RNA with (+) or without (−) urea. Several G positions are indicated. Lanes 4-6, RNAs digested with increasing amount of RNase A. Right panel, predicted secondary RNA structure. The numbers indicate the positions with respective to ITS2 1-200. (D) REMSA analyses of RNAs bound by rNIFK and rNIFK-4YK in vitro. 32P internal-labeled ITS2 1-200 RNA was incubated with increasing concentration of rNIFK (left panel) or rNIFK-4YK (right panel), separated, and detected by autoradiography. The retarded motility corresponding to RNA-protein complex is indicated. (E) Northern blot analysis of NIFK associated rRNA species. Ectopically expressed proteins were immunoprecipitated by Flag-antibody. 10% of immunoprecipitated proteins were analyzed by Western blot (lower panel), and the rest were subjected to RNA extraction followed by Northern blot analysis using probe P4 (upper panel). The asterisk indicates the antibody heavy chain. A non-specific band appears at 28S.
To address the RNA-binding specificity of NIFK and also further narrow down the NIFK binding site in the ITS2, we performed ribonuclease footprinting assay with bacterial expressed recombinant NIFK proteins (rNIFKs) (Fig. S6B). Due to the extreme GC content in ITS2 rRNA, the conventional 5′ end labeling for ITS2 1-200 nt rRNA is proven difficult. To overcome the technical difficulties and obtain the information on the site where rNIFK binds, we performed the RNase footprinting assay with un-labeled ITS2 1-200 nt rRNA with rNIFK followed by Northern blot probing the 3′ terminus of ITS2 1-200 nt rRNA (Fig. 6B). Our result shows the region spanning the 125th–137th nt of ITS2 rRNA is protected from RNase I digestion by rNIFK, suggesting that NIFK likely has sequence specificity to the region. This result is also consistent with the RNA pull down assay that the ITS2 50th–150th nt region is sufficient to bind to NIFK.
To examine whether structural elements in ITS2 RNA also contribute to rNIFK binding, we adopted ribonuclease mediated mapping assay in combination with the RNA secondary structure prediction tool38 to identify the potential RNA structure of the 50th–150th nt of ITS2 rRNA used in the RNA pull down assay. We used an increasing amount of RNase A to cleave the single stranded region of the 5′-end labeled ITS2 50-150 rRNA (Fig. 6C, left panel) and found that the stem-like secondary structure does exist covering the 125th–137th nt regions of ITS2 rRNA (Fig. 6C, right panel). It is noteworthy that, the sequence identity between the 125th–137th nt, GCCGCCGCGC, could also be found in ITS1 at 38th–48th nt region but likely shows low affinity to rNIFK (Fig. 6A), suggesting that the sequence identity is not the major requisite for NIFK binding. These observations collectively suggest the rRNA binding by NIFK cannot be non-specific but depends on both the sequence identity and secondary structure.
To validate whether the function of NIFK in vivo is correlated with the RNA binding ability, the REMSA (RNA Electrophoresis Mobility Shift Assay) were performed with 32P internal labeled ITS2 1-200 RNA with either rNIFK or rNIFK-4YK, a RRM mutant with impaired rRNA processing ability in vivo (Fig. 4C and S4D) and is obtainable in a soluble form in vitro (Fig. S6B). The result indicates that rNIFK showed 2 sequential protein binding events to the ITS2 1-200 RNA with 50% of the first rNIFK-RNA complex formed at 3 nM of rNIFK (Fig. 6D, left panel). On the other hand, the second protein-RNA complex of rNIFK-4YK was not observed and the 50% protein-RNA complex formation only occurs when 10 nM rNIFK-4YK is used (Fig. 6D, right panel, and S6C). In addition, the in vivo 32S/12S pre-rRNAs brought down by NIFK immuno-precipitants were significantly attenuated when RRM is mutated (Fig. 6E, pull down efficiency of proteins in the lower panel and pre-rRNAs in the upper panel). The in vivo evidence appears to correlate with the phenotypic complementary efficiency shown earlier (Figs. 3 and 4). This consistency therefore aligns the NIFK in vivo functions closely with the RNA binding ability of RRM.
Discussion
We previously demonstrated that the high-affinity binding between FHA domain of human Ki67 and a fragment of NIFK requires sequential phosphorylation by CDK1 and GSK3.4 The present study pursues potential roles of NIFK in cell proliferation through facilitating pre-rRNA processing during G1 progression. Between the 2 functional domains of NIFK, we demonstrate the essentiality of RRM in G1 progression, a requisite for successful cell proliferation, by participating in ITS1 site 2 processing to support LSU biogenesis. In agreement, we show that NIFK preferentially binds to the 5' end of ITS2 rRNA, likely in both sequence and secondary structure dependent manner. In contrast, the Ki67 interacting motif appears to be nonessential for this pre-rRNA processing.
Emerging evidences have raised the discrepancy of ribosome biogenesis steps between human and yeast.23,34,35,39,40 It was suggested that yeast ITS1 sites A2 and A3 resemble human ITS1 site 2a and 2, respectively,34,36,40 and that endonucleolytic cleavage at yeast ITS1 site A2 splits the pre-ribosome to SSU (small subunit) and LSU, in analogy to that of human ITS1 site 2.34 Our results suggest that NIFK is one of the LSU biogenesis factors required for such cleavage, similar to what was reported for Bop1.34,41 On the other hand, Nop15 appears to be dispensable for SSU and LSU separation but essential for downstream Rat1 and Rrp17 directed exonucleolytic processing of 27SA3 on the 5′-end of 5.8S rRNA,24-26 apparently distinct from its human ortholog NIFK. Collectively, both NIFK and Nop15 are conserved in terms of LSU biogenesis, but the steps involved are different.
In yeast, A3-cluster protein complex, formed by non-enzymatic ribonucleoproteins, is responsible for ITS1 site A3 processing by recruiting exonuclease Rat1 and Rrp17.25,26 Among A3 cluster, Nop15 was shown to maintain the flexibility of ITS2 by binding with ITS2 5'-end to prevent the premature structural transition from open-ring to thermo-stable hairpin. This rRNA structural flexibility potentially controls the timing of pairing between 5.8S and 25S allowing subsequent ITS2 cleavage.26 To further dissect into the detailed RNA-protein interaction underlying rRNA biogenesis described above, the systematic sequencing of protein-bound small RNA ligands shall be performed. A state-of-the-art experiment called CLIP has recently been applied to comprehensively evaluate the rRNA binding sites of small nucleolar ribonucleoprotein (snoRNP).42 Presumably due to the greater sequence complexity of human pre-RNA than that of yeast,35 such structural remodeling of pre-ribosome and the systematic mapping of corresponding pre-rRNA binding sites have not been elucidated in human previously. Our study provides implications that the aforementioned exo-nuclease recruitment and structural reorganization models in yeast are relevant in human, as suggested by the following results. First, both NIFK and Nop15 are essential for ITS1 processing (Fig. 5) and NIFK exhibits similar preference to 5′-end of ITS2 (Fig. 6A) as that of Nop15.26 Next, the mild accumulation of 36S pre-rRNA in the absence of NIFK (Fig. 5D and E) likely suggests an involvement of NIFK in XRN2-directed exonucleolytic cleavage of 36S to 32S pre-rRNA,36 a similar event to Rat1 and Rrp17-mediated processing of 27SA3 to 27SBs in yeast.25,26 Finally, since the core folding architecture of ITS2 is conserved in eukaryotes,43,44 the 3 dimensional structural remodelling suggested in yeast26 is likely to be relevant in human and requires the NIFK-associated complex. In conclusion, our results support the ITS1 processing model that the efficient processing of ITS1 is decided by an accurate ITS2 remodelling.26
p53 is activated in response to nucleolar stress, and this activation is likely to be the molecular basis underlying the coordination of cell growth and division through faithful ribosome biogenesis.17-21,45 In agreement, we showed that mutations on the RRM domain of NIFK lead to not only rRNA processing defect but also the activation of p53 signaling as well as G1 arrest. This observation evidently links NIFK deficiency with nucleolar stress that in turn activates p53 (Figs. 1D and 2A). More specifically, as the silencing of p21 readily resumes the G1 arrest caused by NIFK deficiency (Fig. 1C-D), p21 is more likely to be the predominant effector downstream of p53-mediated stress response that is responsible for regulation of cell cycle under suboptimal growth condition.46,47 Paradoxically, although disruption of nucleolus often correlates with p53 stability in response to stresses,28 we were not able to observe collapsed nucleolar structure upon such stress (Fig. S2B). This atypical nucleolar stress is similar to what was observed in the absence of small ribosomal protein 6 (S6).48 As the disintegrated nucleolus usually results from inhibition of rRNA synthesis or early rRNA processing,49 we propose that NIFK likely participates in the later rather than early stage of rRNA maturation.
In this study, we observed no functional defect in 28S rRNA maturation or G1 progression from NIFK lacking the Ki67 interacting domain, but we did observe a delayed cell proliferation (Fig. 3B). This minor effect on proliferation may suggest an alternative regulation mediated by the interaction between NIFK and Ki67. Because yeast does not have an ortholog for Ki67 and the C-terminus Ki67FHAID is absent from Nop15,3 one may consider such alternative regulation evolutionarily reinforced. In line with our observation, a most recent study of Ki67 showed that the nuclear localization of GFP-NIFK in interphase remains the same no matter Ki67 is silenced or not, but the localization is greatly altered in early mitosis and metaphase in the absence of Ki67.50 We additionally propose that NIFK is required for G1 progression through the RRM-mediated ribosome biogenesis and mitotically interacts with Ki67 to facilitate M to G1 transition through regulation of post-mitotic nucleolar reassembly.50,51
The complexity of human ribosome biogenesis has been addressed by systematic identification of trans-acting factors and comprehensive analysis of rRNA processing pathways, which broadened our current knowledge on ribosomopathies and cancer.23,34 Our in-depth analysis may provide the molecular basis of these processes and enlighten their disease relevance. In this regard, we have identified that NIFK also correlates with poor survival and severe metastasis in lung cancer presumably due to its indispensable role in ribosome biogenesis during cell proliferation (in submission). It is worth mentioning that, although the in vivo expression level of NIFK varies in diseases,52 no single nucleotide polymorphism (SNP) has been identified in the RRM domain of NIFK based on our search for RRM SNP on NCBI. Taken together with the essential function in ribogenesis presented in this study, the RRM domain of NIFK could be a potential therapeutic target against ribosomopathies and cancer.
Materials and Methods
Cell culture, stable lines preparation, and siRNA transfection
Detailed descriptions for maintenance, retroviral transduction, and siRNA transfection of cells are provided in the Supplementary Materials. After siRNA transfection, real time-PCR was performed accordingly to determine the remnant endogenous mRNAs (Fig. S1A and S2A).
Cell proliferation, cell cycle analysis, antibodies, Western blot analysis, immunoprecipitation experiments, and Immunofluorescent staining
Please see the Supplementary Materials for detailed information.
Total RNA extraction and quantitative PCR (RT-qPCR)
When indicated, protein bound RNAs or total RNAs were isolated using Trizol® reagent following manufacturer's protocols (Invitrogen) prior to analysis. When needed, gene specific mRNA were quantitatively measured through LightCycler® 480 real-time PCR system (Roche) after first-strand cDNA synthesis. Detailed procedures are described in the Supplementary Materials.
Phenotypic rescue of NIFK silencing
Phenotypic rescue experiments were performed by silencing of endogenous NIFK in NIFK stably expressing cells as described previously53 with some modifications. NIFK-siRNA #1 was selected in this study for the best knockdown efficiency (Fig. S1A-B) and the most effective rescued phenotype in cell proliferation (Fig. S3B). The design of siRNA #1 resistant NIFK-cDNA was depicted and the expression profile was validated accordingly (Fig. S3A). When indicated, phenotypic rescues of NIFK mutants were performed as described above and these mutants showed indistinct difference in proliferation, cell cycle progression (Fig. S3C and S4C), protein expression (Fig. 3E, 4D and S4E), and localization (Fig. S3D and S4B) upon transfection of control siRNA, justifying unbiased phenotypes.
RNA metabolic labeling and Northern blot analysis
The nascent rRNA was analyzed as previously described with minor modification.54 Briefly, siRNA-transfected cells were pre-cultured with phosphate-free Dulbecco's Modified Eagle Medium (Gibco) in the presence of 10% dialyzed fetal bovine serum (Gibco) for 1 h, then pulsed with 20 μCi/mL 32P-orthophosphate (Perkin Elmer) in the same phosphate-free medium. After 1 h, cells were incubated in complete DMEM and harvested at the indicated time. Total RNAs were extracted as described earlier and separated by 1% agarose-formaldehyde gel electrophoresis. The gel was dried and subjected to autoradiography for visualization of 32P-labeled RNA. When indicated, the steady-state rRNA profiles were analyzed by Northern blot as previously described with minor modification.55 Briefly, total RNA from siRNA-transfected cells were extracted and separated as earlier described. Separated RNA species in gel were treated sequentially with 0.05 M NaOH/1.5 M NaCl, and 1.5 M NaCl/0.5 M Tris.Cl (pH 7.4) before transferring to nylon membrane (Millipore) using a vacuum blotting apparatus. After UV cross-linking, the membrane was then pre-hybridized with hybridization buffer (Ambion) at 42°C for 1 h and then probed with 32P-5′ end labeled DNA oligonucleotides for 16 h. After several washes with 1-fold SSC buffer in the presence of 0.5% SDS, the membrane was subjected to autoradiography. The 5′ end labeled DNA probes was generated by T4 polynucleotide kinase (T4PNK, Thermo Scientific) in the presence of the [γ-32P]ATP. Please see Supplementary Materials for sequences of the probes used in this study.
RNA transcription
cDNAs complementary to human rRNA transcribed spacers 5′ETS (1-1952), ITS1 (1-1095), and ITS2 (1-1155) were synthesized based on the GenBank sequence U13369.135 and cloned into pJET1.2 (Thermo Scientific) in the same orientation of T7 promoter according to manufacturer's information. When indicated, in vitro transcribed rRNAs were prepared with TranscriptAid T7 High Yield Transcription Kit according to manufacturer's protocol (Thermo Scientific) using either Xba1-linearized pJET1.2 vectors or T7 promoter containing PCR products as templates (see Supplementary Materials for specific sequences of PCR primers used in the study), and PCR was performed as previously described.56 When indicated, UTP-biotinylated RNAs were in vitro transcribed in the same protocol except in the presence of biotinylated-UTP (Ambion). All Transcribed RNAs were treated with DNase I before extraction by TRIzol® to remove free nucleotides.
RNA pull-down assay
To immobilize UTP-biotinylated RNA, 20 μL magnetic streptavidin beads were pre-blocked by binding buffer [5 mM Tris-Cl pH 7.5, 0.5 mM EDTA, 1 M NaCl, and 20 μg/ml yeast tRNA (Ambion)] supplemented with 5% BSA and then incubated with 2 μg in vitro transcribed UTP-biotinylated RNA in the same binding buffer at room temperature for 1 h. When indicated, 40% of bead-bound immobilized RNA were cooked at 90°C for 2 min in the presence of 1 mM EDTA and 95% formamide and analyzed by 1% agarose gel electrophoresis to show the coupling efficiencies. To examine RNA-bound proteins, cell extracts were prepared with the same protocol as Western blot except the DEPC-water was present in NP-40 lysis buffer and pre-cleared by magnetic streptavidin beads (Invitrogen) at 4°C for 1 h before pull-down. Pre-cleared cell extracts were incubated with bead-bound immobilized RNAs in DEPC-NP-40 buffer supplemented with 1 mM DTT, 0.2 U/μL RNaseOUT, and 20 μg/mL yeast tRNA for 16 h at 4°C. After washing with NP-40 buffer, RNA-bound proteins were eluted by 2.5x Laemmli protein sample buffer and subjected to Western blot analysis.
RNA electrophoresis mobility shift assay (REMSA)
To prepare RNA probes for REMSA, 32P internal labeled RNAs were in vitro transcribed as previously described except in the presence of [α-32P]CTP (50 μCi). RNA Transcripts were separated by 6% polyacrylamide native gel and subjected to autoradiography in order to locate the complete RNA products for extraction. Gels containing desired RNAs were sliced and incubated with elution buffer (10 mM Tris, pH 7.5, 300 mM NaCl, and 1 mM EDTA) overnight at room temperature, and RNAs were isolated through isopropanol precipitation. The REMSA was conducted by incubation of purified 32P internal labeled RNAs with recombinant proteins in NP-40 buffer at room temperature for 20 min. Protein-RNA samples were mixed with glycerol before separation by 6% polyacrylamide native gel supplemented with 45 mM Tris, pH 7.4, and 45 mM boric acid at 4°C. The gels were dried and subjected to autoradiography. For detailed procedures of recombinant protein expression and purification, please see the Supplementary Materials.
RNA footprinting and secondary structure detection
In vitro transcribed ITS2 1-200 RNA (40 nM) was incubated with rNIFK (120 nM) and digested with 0.008 U/μL RNase I (Invitrogen) in 250 μL (final volume) of the NP-40 buffer supplemented with 0.08 U/μL RNase inhibitor (Invitrogen) at 30°C for 15 min. When indicated, RNA without rNIFK was incubated with 0.016 U/μL RNase I. RNase I digested RNAs were extracted by TRIzol®, separated by 8% polyacrylamide-7 M urea gels, and analyzed by Northern blot using probe complementary to ITS2 172-200. To indicate RNA size, RNA ladders were prepared by partially digesting RNA with alkaline hydrolysis or RNase T1 (Invitrogen) following RNA Structure-Function Protocols from Life Technologies. For ITS2 50-150 secondary structure detection, in vitro transcribed RNA was dephosphorylated by FastAP (Thermo Scientific), and 5′ end-labeled with [γ-32P]ATP by T4PNK. The labeled RNAs were suspended in NP40 buffer and treated with 6.25, 0.625, and 0.0625 μg/μL of RNase A for 5 min on ice. The T1 ladder was prepared by partially digesting RNA with RNase T1 in NP40 buffer with or without 7 M urea. The reactions were quenched and the RNAs were extracted using TRIzol® and separated by 12% polyacrylamide-7M urea gels.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgment
We thank John Y.-J. Shyy (UCSD, USA) and Mau-Sun Chang, Ching-Jin Chang, Ruey-Hwa Chen, and Jui-Hung Weng (IBC, Academia Sinica) for useful discussion. We also thank Frederick Y. Luh for preparation of recombinant proteins and Haiyan Song for initial construction of NIFK cDNA. Finally we thank Chin-Chun Hung for technical support (IBC Imaging and Cell Biology Core Facility).
Funding
This work was supported by funding from Academia Sinica [Thematic Project AS-101-TP-B02 and AS Investigator Award] and from National Health Research Institutes [NHRI-EX103-10002NI].
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Gerdes J, Lemke H, Baisch H, Wacker HH, Schwab U, Stein H. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol 1984; 133:1710–5; PMID:6206131 [PubMed] [Google Scholar]
- 2.Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cellular Physiol 2000; 182:311–22; PMID:10653597; http://dx.doi.org/ 10.1002/(SICI)1097-4652(200003)182:3%3c311::AID-JCP1%3e3.0.CO;2-9 [DOI] [PubMed] [Google Scholar]
- 3.Takagi M, Sueishi M, Saiwaki T, Kametaka A, Yoneda Y. A novel nucleolar protein, NIFK, interacts with the forkhead associated domain of Ki-67 antigen in mitosis. J Biol Chem 2001; 276:25386–91; PMID:11342549; http://dx.doi.org/ 10.1074/jbc.M102227200 [DOI] [PubMed] [Google Scholar]
- 4.Byeon IJ, Li H, Song H, Gronenborn AM, Tsai MD. Sequential phosphorylation and multisite interactions characterize specific target recognition by the FHA domain of Ki67. Nat Struct Mol Biol 2005; 12:987–93; PMID:16244663; http://dx.doi.org/ 10.1038/nsmb1008 [DOI] [PubMed] [Google Scholar]
- 5.Schlosser I, Holzel M, Murnseer M, Burtscher H, Weidle UH, Eick D. A role for c-Myc in the regulation of ribosomal RNA processing. Nucleic Acids Res 2003; 31:6148–56; PMID:14576301; http://dx.doi.org/ 10.1093/nar/gkg794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Musgrove EA, Sergio CM, Loi S, Inman CK, Anderson LR, Alles MC, Pinese M, Caldon CE, Schutte J, Gardiner-Garden M, et al.. Identification of functional networks of estrogen- and c-Myc-responsive genes and their relationship to response to tamoxifen therapy in breast cancer. PloS One 2008; 3:e2987; PMID:18714337; http://dx.doi.org/ 10.1371/journal.pone.0002987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abujarour R, Efe J, Ding S. Genome-wide gain-of-function screen identifies novel regulators of pluripotency. Stem Cells 2010; 28:1487–97; PMID:20629179; http://dx.doi.org/ 10.1002/stem.472 [DOI] [PubMed] [Google Scholar]
- 8.Savkur RS, Olson MO. Preferential cleavage in pre-ribosomal RNA byprotein B23 endoribonuclease. Nucleic Acids Res 1998; 26:4508–15; PMID:9742256; http://dx.doi.org/ 10.1093/nar/26.19.4508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petfalski E, Dandekar T, Henry Y, Tollervey D. Processing of the precursors to small nucleolar RNAs and rRNAs requires common components. Mol Cell Biol 1998; 18:1181–9; PMID:9488433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hadjiolova KV, Nicoloso M, Mazan S, Hadjiolov AA, Bachellerie JP. Alternative pre-rRNA processing pathways in human cells and their alteration by cycloheximide inhibition of protein synthesis. Eur J Biochem 1993; 212:211–5; PMID:8444156; http://dx.doi.org/ 10.1111/j.1432-1033.1993.tb17652.x [DOI] [PubMed] [Google Scholar]
- 11.Castle CD, Cassimere EK, Lee J, Denicourt C. Las1L is a nucleolar protein required for cell proliferation and ribosome biogenesis. Mol Cell Biol 2010; 30:4404–14; PMID:20647540; http://dx.doi.org/ 10.1128/MCB.00358-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Montanaro L, Trere D, Derenzini M. Nucleolus, ribosomes, and cancer. Am J Pathol 2008; 173:301–10; PMID:18583314; http://dx.doi.org/ 10.2353/ajpath.2008.070752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fumagalli S, Thomas G. The role of p53 in ribosomopathies. Semin Hematol 2011; 48:97–105; PMID:21435506; http://dx.doi.org/ 10.1053/j.seminhematol.2011.02.004 [DOI] [PubMed] [Google Scholar]
- 14.Barna M, Pusic A, Zollo O, Costa M, Kondrashov N, Rego E, Rao PH, Ruggero D. Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature 2008; 456:971–5; PMID:19011615; http://dx.doi.org/ 10.1038/nature07449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Riggelen J, Yetil A, Felsher DW. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat Rev Cancer 2010; 10:301–9; PMID:20332779; http://dx.doi.org/ 10.1038/nrc2819 [DOI] [PubMed] [Google Scholar]
- 16.Ruggero D, Pandolfi PP. Does the ribosome translate cancer? Nat Rev Cancer 2003; 3:179–92; PMID:12612653; http://dx.doi.org/ 10.1038/nrc1015 [DOI] [PubMed] [Google Scholar]
- 17.Golomb L, Volarevic S, Oren M. p53 and ribosome biogenesis stress: The essentials. FEBS letters 2014; 588:2571–9; PMID:24747423; http://dx.doi.org/ 10.1016/j.febslet.2014.04.014 [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y, Lu H. Signaling to p53: ribosomal proteins find their way. Cancer Cell 2009; 16:369–77; PMID: 19878869; http://dx.doi.org/ 10.1016/j.ccr.2009.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pestov DG, Strezoska Z, Lau LF. Evidence of p53-dependent cross-talk between ribosome biogenesis and the cell cycle: effects of nucleolar protein Bop1 on G(1)/S transition. Molecular and cellular biology 2001; 21:4246–55; PMID:11390653; http://dx.doi.org/ 10.1128/MCB.21.13.4246-4255.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Donati G, Peddigari S, Mercer CA, Thomas G. 5S ribosomal RNA is an essential component of a nascent ribosomal precursor complex that regulates the Hdm2-p53 checkpoint. Cell Rep 2013; 4:87–98; PMID:23831031; http://dx.doi.org/ 10.1016/j.celrep.2013.05.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sloan KE, Bohnsack MT, Watkins NJ. The 5S RNP couples p53 homeostasis to ribosome biogenesis and nucleolar stress. Cell reports 2013; 5:237–47; PMID:24120868; http://dx.doi.org/ 10.1016/j.celrep.2013.08.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Havugimana PC, Hart GT, Nepusz T, Yang H, Turinsky AL, Li Z, Wang PI, Boutz DR, Fong V, Phanse S, et al.. A census of human soluble protein complexes. Cell 2012; 150:1068–81; PMID:22939629; http://dx.doi.org/ 10.1016/j.cell.2012.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tafforeau L, Zorbas C, Langhendries JL, Mullineux ST, Stamatopoulou V, Mullier R, Wacheul L, Lafontaine DL. The complexity of human ribosome biogenesis revealed by systematic nucleolar screening of Pre-rRNA processing factors. Molecular cell 2013; 51:539–51; PMID:23973377; http://dx.doi.org/ 10.1016/j.molcel.2013.08.011 [DOI] [PubMed] [Google Scholar]
- 24.Oeffinger M, Tollervey D. Yeast Nop15p is an RNA-binding protein required for pre-rRNA processing and cytokinesis. EMBO J 2003; 22:6573–83; PMID:14657029; http://dx.doi.org/ 10.1093/emboj/cdg616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sahasranaman A, Dembowski J, Strahler J, Andrews P, Maddock J, Woolford JL Jr. Assembly of Saccharomyces cerevisiae 60S ribosomal subunits: role of factors required for 27S pre-rRNA processing. EMBO J 2011; 30:4020–32; PMID:21926967; http://dx.doi.org/ 10.1038/emboj.2011.338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Granneman S, Petfalski E, Tollervey D. A cluster of ribosome synthesis factors regulate pre-rRNA folding and 5.8S rRNA maturation by the Rat1 exonuclease. EMBO J 2011; 30:4006–19; PMID:21811236; http://dx.doi.org/ 10.1038/emboj.2011.256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brooks CL, Gu W. New insights into p53 activation. Cell Res 2010; 20:614–21; PMID:20404858; http://dx.doi.org/ 10.1038/cr.2010.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rubbi CP, Milner J. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J 2003; 22:6068–77; PMID:14609953; http://dx.doi.org/ 10.1093/emboj/cdg579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boisvert FM, van Koningsbruggen S, Navascues J, Lamond AI. The multifunctional nucleolus. Nat Rev Mol Cell Biol 2007; 8:574–85; PMID:17519961; http://dx.doi.org/ 10.1038/nrm2184 [DOI] [PubMed] [Google Scholar]
- 30.Kill IR. Localisation of the Ki-67 antigen within the nucleolus. Evidence for a fibrillarin-deficient region of the dense fibrillar component. J Cell Sci 1996; 109 (Pt 6):1253–63; PMID:8799815 [DOI] [PubMed] [Google Scholar]
- 31.Zhai W, Comai L. Repression of RNA polymerase I transcription by the tumor suppressor p53. Mol Cell Biol 2000; 20:5930–8; PMID:10913176; http://dx.doi.org/ 10.1128/MCB.20.16.5930-5938.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maris C, Dominguez C, Allain FH. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J 2005; 272:2118–31; PMID:15853797; http://dx.doi.org/ 10.1111/j.1742-4658.2005.04653.x [DOI] [PubMed] [Google Scholar]
- 33.Wang X, Tanaka Hall TM. Structural basis for recognition of AU-rich element RNA by the HuD protein. Nat Struct Biol 2001; 8:141–5; PMID:11175903; http://dx.doi.org/ 10.1038/84131 [DOI] [PubMed] [Google Scholar]
- 34.Sloan KE, Mattijssen S, Lebaron S, Tollervey D, Pruijn GJ, Watkins NJ. Both endonucleolytic and exonucleolytic cleavage mediate ITS1 removal during human ribosomal RNA processing. J Cell Biol 2013; 200:577–88; PMID:; http://dx.doi.org/22342225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mullineux ST, Lafontaine DL. Mapping the cleavage sites on mammalian pre-rRNAs: where do we stand? Biochimie 2012; 94:1521–32; PMID:22342225; http://dx.doi.org/ 10.1016/j.biochi.2012.02.001 [DOI] [PubMed] [Google Scholar]
- 36.Wang M, Pestov DG. 5'-end surveillance by Xrn2 acts as a shared mechanism for mammalian pre-rRNA maturation and decay. Nucleic Acids Res 2011; 39:1811–22; PMID:21036871; http://dx.doi.org/ 10.1093/nar/gkq1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr., Jungkamp AC, Munschauer M, et al.. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 2010; 141:129–41; PMID:20371350; http://dx.doi.org/ 10.1016/j.cell.2010.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bellaousov S, Reuter JS, Seetin MG, Mathews DH. RNAstructure: Web servers for RNA secondary structure prediction and analysis. Nucleic Acids Res 2013; 41:W471–4; PMID:23620284; http://dx.doi.org/ 10.1093/nar/gkt290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Henras AK, Soudet J, Gerus M, Lebaron S, Caizergues-Ferrer M, Mougin A, Henry Y. The post-transcriptional steps of eukaryotic ribosome biogenesis. Cell Mol Life Sci 2008; 65:2334–59; PMID:18408888; http://dx.doi.org/ 10.1007/s00018-008-8027-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carron C, O'Donohue MF, Choesmel V, Faubladier M, Gleizes PE. Analysis of two human pre-ribosomal factors, bystin and hTsr1, highlights differences in evolution of ribosome biogenesis between yeast and mammals. Nucleic Acids Res 2011; 39:280–91; PMID:20805244; http://dx.doi.org/ 10.1093/nar/gkq734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Strezoska Z, Pestov DG, Lau LF. Bop1 is a mouse WD40 repeat nucleolar protein involved in 28S and 5. 8S RRNA processing and 60S ribosome biogenesis. Mol Cell Biology 2000; 20:5516–28; PMID:10891491; http://dx.doi.org/ 10.1128/MCB.20.15.5516-5528.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Granneman S, Kudla G, Petfalski E, Tollervey D. Identification of protein binding sites on U3 snoRNA and pre-rRNA by UV cross-linking and high-throughput analysis of cDNAs. Proc Natl Acad Sci U S A 2009; 106:9613–8; PMID:19482942; http://dx.doi.org/ 10.1073/pnas.0901997106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Joseph N, Krauskopf E, Vera MI, Michot B. Ribosomal internal transcribed spacer 2 (ITS2) exhibits a common core of secondary structure in vertebrates and yeast. Nucleic Acids Res 1999; 27:4533–40; PMID:10556307; http://dx.doi.org/ 10.1093/nar/27.23.4533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Michot B, Joseph N, Mazan S, Bachellerie JP. Evolutionarily conserved structural features in the ITS2 of mammalian pre-rRNAs and potential interactions with the snoRNA U8 detected by comparative analysis of new mouse sequences. Nucleic Acids Res 1999; 27:2271–82; PMID:10325414; http://dx.doi.org/ 10.1093/nar/27.11.2271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Golomb L, Volarevic S, Oren M. p53 and ribosome biogenesis stress: The essentials. FEBS Lett 2014; 588:2571–9; PMID:24747423; http://dx.doi.org/ 10.1016/j.febslet.2014.04.014 [DOI] [PubMed] [Google Scholar]
- 46.Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 1995; 82:675–84; PMID:7664346; http://dx.doi.org/ 10.1016/0092-8674(95)90039-X [DOI] [PubMed] [Google Scholar]
- 47.Waldman T, Kinzler KW, Vogelstein B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res 1995; 55:5187–90; PMID:7585571 [PubMed] [Google Scholar]
- 48.Fumagalli S, Di Cara A, Neb-Gulati A, Natt F, Schwemberger S, Hall J, Babcock GF, Bernardi R, Pandolfi PP, Thomas G. Absence of nucleolar disruption after impairment of 40S ribosome biogenesis reveals an rpL11-translation-dependent mechanism of p53 induction. Nat Cell Biol 2009; 11:501–8; PMID:19287375; http://dx.doi.org/ 10.1038/ncb1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burger K, Muhl B, Harasim T, Rohrmoser M, Malamoussi A, Orban M, Kellner M, Gruber-Eber A, Kremmer E, Holzel M, et al.. Chemotherapeutic drugs inhibit ribosome biogenesis at various levels. J Biol Chem 2010; 285:12416–25; PMID:20159984; http://dx.doi.org/ 10.1074/jbc.M109.074211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Booth DG, Takagi M, Sanchez-Pulido L, Petfalski E, Vargiu G, Samejima K, Imamoto N, Ponting CP, Tollervey D, Earnshaw WC, et al.. Ki-67 is a PP1-interacting protein that organises the mitotic chromosome periphery. Elife 2014; 3:e01641; PMID:24867636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takagi M, Nishiyama Y, Taguchi A, Imamoto N. Ki67 antigen contributes to the timely accumulation of protein phosphatase 1gamma on anaphase chromosomes. J Biol Chem 2014; 289:22877–87; PMID:25012651; http://dx.doi.org/ 10.1074/jbc.M114.556647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rujkijyanont P, Adams SL, Beyene J, Dror Y. Bone marrow cells from patients with Shwachman-Diamond syndrome abnormally express genes involved in ribosome biogenesis and RNA processing. Br J Haematol 2009; 145:806–15; PMID:19438500; http://dx.doi.org/ 10.1111/j.1365-2141.2009.07692.x [DOI] [PubMed] [Google Scholar]
- 53.Grimm T, Holzel M, Rohrmoser M, Harasim T, Malamoussi A, Gruber-Eber A, Kremmer E, Eick D. Dominant-negative Pes1 mutants inhibit ribosomal RNA processing and cell proliferation via incorporation into the PeBoW-complex. Nucleic Acids Res 2006; 34:3030–43; PMID:16738141; http://dx.doi.org/ 10.1093/nar/gkl378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pestov DG, Lapik YR, Lau LF. Assays for ribosomal RNA processing and ribosome assembly. Curr Protoc Cell Biol 2008; 39:22.11.1-22.11.16; PMID:18551418; http://dx.doi.org/ 10.1002/0471143030.cb2211s39 [DOI] [PubMed] [Google Scholar]
- 55.Brown T, Mackey K, Du T. Analysis of RNA by northern and slot blot hybridization. Curr Protoc Mol Biol 2004; 67:4.9.1-4.9.19; PMID:18265351; http://dx.doi.org/ 10.1002/0471142727.mb0409s67 [DOI] [PubMed] [Google Scholar]
- 56.Frey UH, Bachmann HS, Peters J, Siffert W. PCR-amplification of GC-rich regions: 'slowdown PCR'. Nat Protoc 2008; 3:1312–7; PMID:18714299; http://dx.doi.org/ 10.1038/nprot.2008.112 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.