

Abstract
Merlin ((Moesin-ezrin-radixin-like protein, also known as schwannomin) is a tumor suppressor protein encoded by the neurofibromatosis type 2 gene NF2. Loss of function mutations or deletions in NF2 cause Neurofibromatosis type 2 (NF2), a multiple tumor forming disease of the nervous system. NF2 is characterized by the development of bilateral vestibular schwannomas. Patients with NF2 can also develop schwannomas on other cranial and peripheral nerves, as well as meningiomas and ependymomas. The only potential treatment is surgery/radiosurgery which often results in loss of function of the involved nerve. There is an urgent need for chemotherapies that slow or eliminate tumors and prevent their formation in NF2 patients. Interestingly NF2 mutations and merlin inactivation also occur in spontaneous schwannomas and meningiomas, as well as other types of cancer including mesothelioma, glioma multiforme, breast, colorectal, skin, clear cell renal cell carcinoma, hepatic and prostate cancer. Except for malignant mesotheliomas, the role of NF2 mutation or inactivation has not received much attention in cancer, and NF2 might be relevant for prognosis and future chemotherapeutic approaches.
This review discusses the influence of merlin loss of function in NF2-related tumors and common human cancers. We additionally discuss the NF2 gene status and merlin signaling pathways affected in the different tumor types and the molecular mechanisms that lead to tumorigenesis, progression and pharmacological resistance.
Keywords: merlin, NF2, schwannoma, tumorigenesis, tumor progression, cancer
INTRODUCTION
Merlin is the protein encoded by the tumor suppressor gene, NF2, located on chromosome 22q12. Deletion or loss-of-function mutation of NF2 causes Neurofibromatosis type 2 (NF2), a nervous system tumor-forming disease (1–3). The incidence of NF2 is 1 in 25,000 and it is dominantly inherited, although in 50% to 60% of cases it is caused by de novo mutations with high frequency of somatic mosaicism (1, 4). NF2 is characterized by formation of bilateral vestibular schwannomas. In addition, patients commonly develop multiple schwannomas on other cranial and peripheral nerves, on spinal nerve roots and as intracutaneous plaques, as well as meningiomas and ependymomas. Schwannomas originate from Schwann cells, while meningiomas originate from cells of the arachnoid layer of the leptomeninges that line the brain and spinal cord (5). NF2 patients may also present ependymomas found mostly in the spinal cord. Intramedullary ependymomas arise from ependymal cells that line the central canal of the spinal cord (6, 7). NF2 patients can eventually develop peripheral neuropathy (1, 4, 8). Electrophysiological studies found that 66% of NF2 patients presented neuropathy, mostly of axonal type (9). In many cases, the neuropathy results from compression produced by the tumor, however, non-tumor-related peripheral neuropathy may be caused by the deleterious effect of merlin loss in neurons (4, 10, 11). Interestingly, schwannomas and meningiomas also develop in non-NF2 patients and commonly exhibit NF2 somatic mutations, epigenetic changes or protein inactivation. Notably, NF2 mutations are present in several malignant cancers including mesotheliomas, breast, prostate, colorectal, hepatic, clear cell renal cell carcinoma, and melanomas.
The first symptoms of NF2 usually appear in the late teens or early twenties and include hearing loss with or without tinnitus and dizziness or imbalance caused by the vestibular schwannomas (mostly unilateral at the beginning) that ultimately lead to deafness (1, 4). Other NF2 symptoms depend on the nerve or structure affected by the tumors. Despite the benign nature of schwannomas, their morbidity is based on their location and size and the only currently available treatment is surgical resection. Surgical outcomes, however, are poor because nerve function cannot always be salvaged and some tumors are inoperable due to location. Stereotactic radiosurgery has been explored, but remains controversial because of the potential for subsequent radiation-induced malignant transformation. Thus, the current standard of care, while the subject of continuing debate, consists of initial monitoring followed by surgical schwannoma resection when rapid tumor growth is identified or becomes life threatening (4). Meningiomas, the second most frequent tumors in NF2 patients, are 45–58% intracranial and 20% intradural extramedullary spinal tumors. Similar to schwannomas, associated symptoms relate to tumor location and include headaches (most common) and seizures. On the optic nerve and lower cranial nerves, very small meningiomas may produce compression symptoms and loss of visual acuity (8). Overall, the presence of intracranial meningiomas is associated with poor prognosis and increases the relative risk of mortality (12). The surgical removal of intracranial and spinal meningiomas can be achieved in most cases depending on the anatomical location (4). In line with schwannomas and meningiomas, the recommended treatment for ependymomas is observation while asymptomatic, and maximum surgical resection if symptoms develop or rapid growth is identified. Only for anaplastic ependymomas, rare in NF2, is postoperative adjuvant radiation considered (7, 13).
Identification of NF2 as a member of the Band 4.1 FERM gene family predicted a mechanism of action for merlin’s tumor suppressor function (2, 3). FERM domain proteins link plasma membrane receptors to the cortical actin cytoskeleton (3). Merlin shares 64% sequence similarity with family members in the conserved FERM (4.1, ezrin, radixin, moesin) domain at the N-terminus. The FERM domain is followed by an α-helical domain and a C-terminal domain (Figure 1a). In contrast to the other ERM proteins, merlin lacks the actin-binding site located in the C-terminal domain, instead it has a unique actin binding motif in the N-terminal domain (14). ERM proteins including merlin form head-to-tail homo- and hetero-dimeric intermolecular, and intramolecular associations. Although merlin’s intramolecular association is more dynamic and weaker compared to other ERMs, it is nonetheless critical for regulating its tumor suppressor activity (15, 16). Merlin can be regarded as a scaffold protein indirectly linking F-actin, transmembrane receptors and intracellular effectors to modulate receptor mediated signaling pathways controlling cell proliferation and survival. These include receptor tyrosine kinase (RTK), cell adhesion, small GTPases, mammalian target of rapamycin (mTOR), PI3K/Akt and hippo pathways (17). However, merlin’s tumor suppressor function is not restricted to regulating events at the plasma membrane. Unraveling the myriad of merlin interactions in relation to tumor suppression has proven to be a daunting task.
Figure 1.
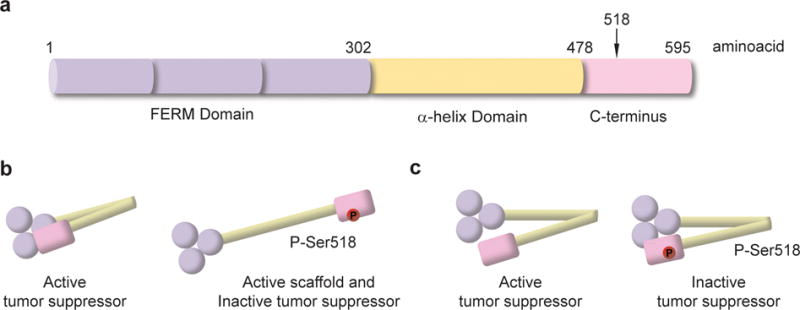
Merlin domain organization and molecular conformation. a) Diagram of merlin structural domains. b) Diagram of merlin molecular conformation based on ERM analogy. c) Diagram of merlin molecular conformation based on experimental data.
Merlin’s conformation and activity is regulated by post-translational modifications. Merlin is phosphorylated at Ser10, Thr230 and Ser315 by Akt (also known as protein kinase B, PKB) and controls merlin’s proteasome-mediated degradation by ubiquitination to prevent its interaction with binding partners (18, 19). Merlin-Ser10 is also a substrate for cyclic AMP-dependent protein kinase, also known as protein kinase A (PKA); Ser10 phosphorylation modulates cell morphology by modifying the actin cytoskeleton (20). The most studied phosphorylation site is Ser518 located in the C-terminal. It is a substrate for both p-21 activated kinase (PAK) and PKA and inactivates merlin tumor suppressor activity (21–24). Conversely, myosin phosphatase MYPT1-PP1δ dephosphorylates Ser518 thereby activating its tumor suppressor function (25). Confusion regarding the relationship between activity and conformation for merlin stems from the initial assumption that merlin’s conformation-activity resembles the ERM proteins. Merlin, similar to ERMs is active as a scaffold protein at the plasma membrane where it is phosphorylated at Ser518 and by this means facilitates or promotes receptor mediated signaling events associated with cell proliferation and survival. Following the ERM model, phosphorylated merlin would adopt an open, active configuration at the plasma membrane. In this way, both merlin and ERMs are active as scaffolds in the open conformation (Figure 1b) (26, 27). Dephosphorylation at S518 and thus transition to a closed conformation inactivates its scaffold function but activates its tumor suppressor activity. The closed, head-to-tail conformation of the active tumor-suppressor was believed to be incapable of interaction with growth factor receptors (28–30). Recent results of fluorescence resonance energy transfer analysis of merlin conformational changes, characterization of a series of merlin variants with different degrees of head-to-tail association, and small-angle neutron scattering studies of full-length merlin, have suggested a new conformational model (31–33). The results indicate that in solution both unphosphorylated merlin and a phospho- mimetic, merlin S518D, adopt closed conformations; and both forms are capable of binding partner proteins. Furthermore, unphosphorylated merlin adopts a more open, but still closed, conformation upon binding phoshatidylinositol 4,5 biphosphate lipid whereas the merlinS518D phosphomimetic remains fully closed (31–33) (Figure 1c). The exact relationship between merlin’s conformation, binding partners and sub-cellular localization critical for tumor suppressor activity has yet to be fully understood.
Merlin is essential for early embryogenesis as evidenced by the finding that mice embryos carrying homozygous Nf2 mutations failed to initiate gastrulation and generally died at E6.5–E7.0 (34). These embryos lacked extra-embryonic ectoderm and failed to form an identifiable mesodermal layer (34). Other conditional knock-out mouse models partially mimic the multifaceted human disease (35–38). Discrepancies between the models and human disease could be caused by several factors, acting individually or in combination, including differences in biological genome/proteome, physiological properties and lifespans; timing of loss of heterozygosity (LOH) or merlin inactivation; and single cell-type targeting for genetic manipulation when NF2 is active globally (39).
Another area for further investigation that may reveal essential insights to merlin’s mechanism of action is the production of splice variants for merlin. Two major isoforms are produced by alternative splicing of 17 exons of the NF2 gene. Isoform 1 is the predominant and longest, consisting of 595 amino acids and lacking exon 16, while isoform 2 with 590 amino acids contains exon 16, but has a truncated C-terminal domain (40). A recent publication highlights the importance of Schwann cell-axon interactions in preventing schwannoma formation and the contribution of axonal expression of merlin isoform 2 (10, 11, 41).
This review will discuss the contribution of merlin loss of function in NF2-related tumors and common human cancers and the molecular mechanisms that lead to tumorigenesis, tumor progression or pharmacological resistance.
MERLIN IN SCHWANNOMAS, MENINGIOMAS AND EPENDYMOMAS
NF2/merlin status
NF2 mutations cause the development of schwannomas, meningiomas and ependymomas [for an in-depth review on NF2 mutations see ref (1)]. NF2-associated schwannomas exhibit NF2 inactivation and merlin expression is absent (42). Notably, merlin expression assessed by western blot was found to be highly reduced or absent in 11 of 14 sporadic schwannomas, 16 of 19 sporadic meningiomas and 3 of 8 sporadic ependymomas (43). Mutational NF2 inactivation and LOH is the cause of most NF2-associated schwannomas and sporadic schwannomas; however, transcriptional inactivation causes 15.3% of NF2-associated schwannomas and 19.3% of sporadic schwannomas (44). One study reported that methylation of three cis-regulatory elements, CpG sites, in the NF2 promoter elements was found in ~60% of vestibular schwannomas and correlated with decreased promoter activity and mRNA expression (45). By contrast, another study of 35 sporadic vestibular schwannomas reported that in 65% of tumors the NF2 promoter was unmethylated and in the remaining 35% the results were inconclusive (46). At the same time, 50 to 60% of sporadic meningiomas contain NF2 somatic mutations and epigenetic inactivation (47, 48). In another study of 88 sporadic meningiomas, 49% exhibited allelic loss of chromosome 22, 24% had NF2 somatic mutations and 26% had aberrant NF2 promoter methylation, whereas in 17% of the meningiomas, epigenetic NF2 inactivation was the only cause of NF2 deficiency. Similarly, less than 10% of sporadic ependymomas exhibited NF2 promoter methylation (49). In some of the schwannomas and meningiomas without genetic or epigenetic NF2 inactivation, merlin inactivation was caused by μ-calpain constitutive activation that mediated merlin proteolytic cleavage/degradation (50). Finally, a recent meningioma study of 73 patients found a high incidence of TERT promoter activating mutations in meningiomas undergoing malignant transformation in both NF2-related and sporadic meningiomas, whereas no TERT mutations were found in benign tumors. This suggests that TERT independently of NF2 could be used as biomarker of risk of malignant transformation of a meningioma (51).
Because NF2 mutations and inactivation are present in ependymomas and both benign and malignant schwannomas and meningiomas, it is reasonable to speculate that NF2 inactivation could be an early tumorigenic event that disrupts key signaling pathways in a specific molecular context leading to tumor development. Although a complete understanding of the biological functions of merlin is lacking, current knowledge of merlin-related signaling pathways and merlin binding partners shed some light on its role in cellular proliferation, survival and tumor development.
Signaling pathways regulated by merlin
Membrane receptors and RhoGTPase family
Merlin’s tumor suppressor activity was initially related to a role in contact inhibition of cell proliferation. Merlin levels in various cell types were shown to be two- to three-fold higher in high density as opposed to low density cells in cultures. Thus merlin appears necessary for suppression and/or inactivation of receptor-dependent mitogenic signaling pathways. (52, 53). There are several studies supporting this mechanism of action for merlin. First, merlin co-localizes and binds CD44, a recognized cell-cell, cell-substrate adhesion receptor and the major receptor for hyaluronan, an abundant extracellular matrix (ECM) component (54, 55). Binding of merlin unphosphorylated at Ser518 with the cytoplasmic tail of CD44 mediates contact inhibition at high cell density. Consistent with this phenomenon, treatment of sub-confluent cells of various cell types with a soluble glycosaminoglycan hyaluronan rapidly induced merlin dephosphorylation and inhibition of growth and thus activated merlin tumor suppressor activity (52). Furthermore, merlin overexpression in Tr6BC1 mouse schwannoma cells inhibited the binding of fluorescein-labeled hyaluronan to CD44 and inhibited subcutaneous tumor growth in immunocompromised mice, and overexpression of a merlin mutant lacking the CD44 binding domain was unable to inhibit schwannoma growth (56). Collectively, these results indicate that merlin inhibits cell growth by contact inhibition in part by binding CD44 and negatively regulating CD44 function (Figure 2).
Figure 2.
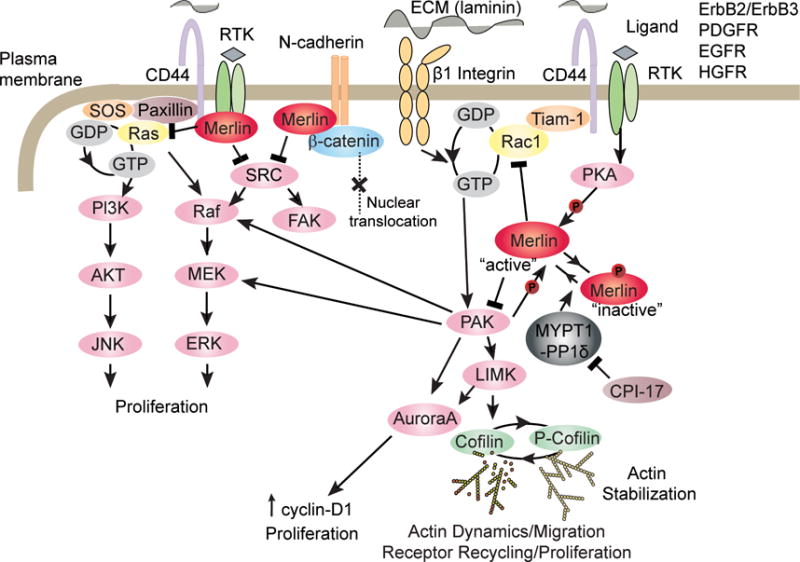
Merlin inhibits membrane receptors and RhoGTPase family signaling cascade.
CD44 signaling through Rac1activation is demonstrated in multiple cell types. Hyaluronan-CD44 interaction in astrocytes and an immortalized mouse mammary epithelial cell line, EpH4, leads to Rac1 signaling activation and actin cytoskeleton rearrangement (57, 58). The activation of Rac1 through CD44 was identified via the interaction of CD44 with Tiam-1, a Rac1 guanine nucleotide exchange factor (GEF) that catalyzes the replacement of the tightly-bound GDP with GTP. In various cell types, the binding of hyaluronan to CD44 stimulates Tiam1-dependent Rac1 signaling and cytoskeleton-mediated tumor cell migration (59, 60). Moreover, expression of constitutively active Rac mutants, as well as activated cell division cycle-42 (Cdc42), induced phosphorylation of merlin at Ser518 and decreased merlin’s association with the actin cytoskeleton. Merlin phosphorylation at Ser518 inactivated its growth inhibitory function (Figure 2) (21). In fibroblasts, merlin overexpression inhibited Rac1-induced signaling whereas a merlin S518D phosphomimetic did not. As expected, increased levels of active Rac1 were found in NF2-deficient Schwann cells and human schwannoma samples compared to controls (61–63).
Independent of merlin-CD44 interactions, merlin has been shown to modulate Rac signaling by specifying its subcellular localization. For example, in confluent human umbilical vein endothelial cells, merlin suppressed recruitment of Rac to the plasma membrane, and its silencing promoted recruitment of Rac1 to sites of extracellular matrix adhesion, and promoted cell growth (64). In addition, merlin has been shown to inhibit Rac1 signaling responsible for suppression of PAK activation by binding through its FERM domain to the PAK-Cdc42/Rac binding domain (65). In turn, Rac/Cdc42-dependent activation of PAK leads to merlin phosphorylation at Ser518, which inhibits merlin translocation to the plasma membrane in a paxillin-dependent manner, mitigating its inhibitory effect on Rac/Cdc42, and hence, its tumor suppressor activity (Figure 2) (22, 66, 67).
In primary rat Schwann cells, CD44 was shown to constitutively associate with the heterodimer receptor tyrosine kinase ErbB2/ErbB3 and CD44 enhanced neuregulin-induced ErbB2-activating phosphorylation (68). ErbB2/ErbB3 receptor expression levels and activation were reported to be elevated in human schwannomas and NF2-deficient cell lines (69–71). Accordingly, merlin was shown to reduce the levels of ErbB2/ErbB3 receptor levels at the plasma membrane. Previously, we showed that activation of ErbB2/ErbB3 receptors in primary rat Schwann cells by neuregulin-1 induced merlin phosphorylation at Ser518 via PKA (72). Significantly, ErbB2 activation was also found to be elevated in NF2-ependymomas (73). ErbB2 activation in mouse Nf2-deficient spinal cord neural progenitor cells was shown to be caused by Rac-mediated retention of the receptor at the plasma membrane (73). Merlin re-expression in Nf2−/− Schwann cells similarly reduced the transport of growth factor receptors ErbB2/ErbB3, insulin-like growth factor 1 receptor (IGF1R) and platelet-derived growth factor receptor (PDGFR). Accordingly, the loss of merlin in Nf2−/− peripheral mouse nerves resulted in elevated levels of these growth factor receptors compared to wild-type before the development of tumors, and importantly, similar overexpression was observed in human Schwannomas (Figure 2) (74–77).
In sub-confluent primary Schwann cells, we found that merlin binds to paxillin and mediates merlin localization at the plasma membrane and association with β1-integrin and ErbB2, modifying the organization of the actin cytoskeleton in a cell density-dependent manner (78). We reported that merlin associates with β1-integrin in primary Schwann cells and undifferentiated Schwann cell/neuron co-cultures, and in primary Schwann cell cultures, laminin-1 stimulated integrin signaled though PAK1 and caused merlin Ser518 phosphorylation and inactivation of its tumor suppressor function (72, 79, 80). In sum, multiple lines of evidence have established a feedback regulation loop with merlin being phosphorylated at Ser518 (growth permissive form) via activated Rho small GTPases Rac1/Cdc42 through PAK, and in turn, merlin associating with PAK to inhibit Rac1/Cdc42 signaling (Figure 2). Loss of merlin causes continuous activation of Rac1/Cdc42/PAK signaling, which is associated with cell growth and deregulation of the actin cytoskeleton. Actin cytoskeleton abnormalities have been reported in NF2-deficient human schwannomas and the defects were reversed by reintroduction of merlin (81, 82). Related to that, the PAK substrates, LIM domain kinases 1and 2 (LIMK1/2), were also reported to be elevated in human Schwannomas and mouse Schwann cells in which merlin was inactivated compared to normal controls (83). LIMK1/2 phosphorylates and inactivates cofilin, an F-actin severing and depolymerizing agent. Furthermore, it was shown that overactive PAK/LIMK pathway activity contributed to cell proliferation through cofilin phosphorylation and auroraA activation (83). When merlin was inactivated in immortalized Nf2flox2/flox2 mouse embryonic fibroblasts (MEFs) by Adeno-Cre infection, loss of contact inhibition of growth and increased Rac1 and canonical Wnt signaling were observed compared to controls (84). Pharmacological or genetic inhibition of Rac1 in Nf2−/− MEFs reduced the Wnt signaling activation to basal levels as assessed by reporter assay of transactivation of the nuclear β-catenin-dependent T-cell factor 4 transcription factor (84). These findings were corroborated and further studied in human schwannoma cells, in which over-activation of canonical Wnt signaling with elevated nuclear β-catenin, upregulation of c-myc and cyclin D1 target genes and activation of the Rac1/PAK and PDGFR/Src pathways were observed (Figure 2) (85).
Soon after merlin was cloned, evidence that merlin inhibits another important member of the Rho GTPases family, Ras, was reported in v-Ha-Ras-transformed NIH3T3cells in which merlin overexpression counteracted the oncogenic role of Ras (86). In immortalized nf2−/− rat Schwann cells, reintroduced merlin acted downstream of receptor tyrosine kinase through the growth factor receptor binding protein 2-Son of sevenless (Grb2-SOS) complex (87). SOS is a GEF that activates Ras by catalyzing the nucleotide exchange (88). Merlin action on Ras signaling is indirect by blocking the assembly of ERM proteins, SOS and the Ras complex (87, 89).
Another family of receptor tyrosine kinases activated in schwannomas is the Tyfo3-Axl-Mer (TAM) family. TAM receptors were strongly overexpressed in schwannomas (77). Moreover, increased activation of Axl receptor and Gas6 ligand was also observed in the absence of merlin in primary human schwannoma cells compared to normal Schwann cells, resulting in recruitment and activation of Src and focal adhesion kinase (FAK) signaling (90). Merlin inactivation of Src signaling was also shown in CNS glial cells, where merlin competitively inhibits Src binding to ErbB2 thereby preventing ErbB2-mediated Src phosphorylation and downstream mitogenic signaling. In Nf2−/− glial cells, Src regulated cell growth by sequentially regulating FAK and paxillin activity (91). Primary human schwannoma cells also showed increased cell-matrix adhesion compared to control Schwann cells. Interestingly, it was shown that schwannoma cells release insulin-like growth factor-binding protein 1 which in β1-integrin dependent manner activates Src/FAK signaling (75). In addition, FAK was overexpressed and activated in schwannoma cells where it localized to the nucleus and decreased p53 levels by increasing its export to the cytosol and proteasomal degradation. FAK silencing decreased schwannoma cell proliferation and was associated with increased levels of total and nuclear p53 (75, 92).
PI3K/mTOR/Akt and HDAC signaling
In human schwannomas and meningiomas, loss of merlin leads to activation of the phosphoinositide 3-kinase (PI3K)/Akt pathway and Schwann cell proliferation (93–96). Merlin inhibits PI3K activity by binding phosphatidylinositol 3-kinase enhancer-L (PIKE-L), the GTPase that binds and activates PI3K (94, 97). Merlin association with PIKE-L disrupts PIKE-L binding to and activation of PI3K. Moreover, neuregulin survival signaling through the ErbB2/ErbB3 receptor activates PI3K in rat Schwann cells through the activation of Akt and inhibition of Bad, a pro-apoptotic Blc-2 family protein (98). We recently showed that PI3K inhibition in merlin-deficient mouse Schwann cells selectively decreased their proliferation. A good number of inhibitors showed strong anti-proliferative activity, causing cell death with diverse kinetics via caspase-dependent apoptosis (99). Inhibition of Akt also inhibits schwannoma and meningioma growth (100, 101). Akt phosphorylation is indirectly regulated by histone deacetylases (HDAC) 1 and 6 through their interaction with protein phosphatase 1 (PP1), which functions as an Akt phosphatase (102, 103). HDAC inhibitors disrupt the PP1-HDAC interaction facilitating Akt dephosphorylation and decrease human meningioma and schwannoma cell proliferation and schwannoma growth in an allograft model and meningioma growth in an intracranial xenograft model (102, 104, 105). Lastly, gene-expression profiling of 49 schwannomas and 7 normal vestibular nerves identified overexpression of the PI3K/Akt/mTOR pathway (Figure 3) (106).
Figure 3.
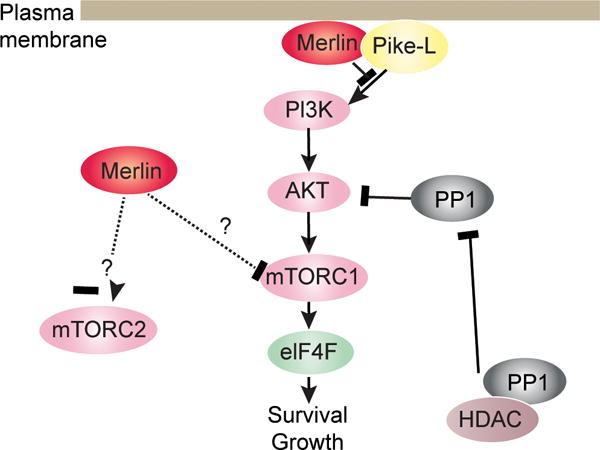
Merlin inhibits PI3K/mTORC1/Akt signaling pathways.
Consistent with this last finding, constitutive activation of the mTORC1 signaling pathway has been reported in NF2-schwannomas and meningiomas. Loss of merlin activated mTORC1 signaling independently of Akt or ERK in these tumor cells; however, the molecular mechanism connecting merlin loss to mTORC1 activation remains to be elucidated (107). In orthotopic sciatic nerve allograft mice, syngeneic subcutaneous grafted mice and transgenic P0-SCH-Δ(39-121)-27 mice, pharmacological inhibition of mTORC1 reduced the growth of NF2-schwannomas (108). Western blot analysis of these tumors indicated a strong reduction of phosphorylation of mTORC1 downstream substrates S6 and 4E-BP1 as well as a small increase of Akt phosphorylation in treated mice, analogous to patients’ response to rapamycin treatment that was characterized by Akt activation resulting from the loss of the p71 S6K1 negative feedback loop (43, 109, 110).. In contrast to mTORC1, in Schwann and arachnoidal cells, merlin activates mTORC2 in response to growth factor stimulation. However, attenuation of mTORC2 signaling was not observed in merlin-deficient schwannomas and meningiomas (Figure 3) (111).
In addition to alterations in kinases and phosphatase activity, we reported that loss of merlin altered the protein acetylation pattern in mouse Schwann cells. Interestingly, merlin loss correlated with increased expression levels of the deacetylase sirtuin 2 (SIRT2). Inhibiting SIRT2 deacetylase activity with small molecules considerably decreased merlin-null Schwann cell viability via a necrotic mechanism while sparing normal Schwann cells (112).
Mammalian hippo pathway
Studies in primary mouse Schwann cells and in vitro assays highlighted the role of merlin modulation of the Schwann cell microtubule cytoskeleton (113). Further studies showed that wild-type merlin is transported throughout the cell by microtubule motors and merlin mutants or depletion of the microtubule motor kinesin-1 suppressed merlin transport and was associated with accumulation of yorkie, a Drosophila homolog of the hippo pathway transcriptional co-activator Yes-associated protein (YAP), in the nucleus (114). It is significant to note that phosphorylation of YAP excludes it from the nucleus in part by binding 14-3-3, and, functional inactivation of YAP seems to be cell-type specific (115).
In the canonical hippo pathway, mammalian Ste20-like kinases (Mst1/2; hippo homolog) phosphorylate large tumor suppressor kinases (LATS 1/2), which in turn phosphorylate and inactivate YAP/TAZ, blocking their role as TEAD/MEAD transcription factor co-activators (116–118). Proteomic screening of 68 human schwannomas showed that YAP and YAP transcriptional targets such as Her2, Her3 and PDGFRβ were commonly activated (119). Studies in human meningioma tumors and in paired cell lines—KY21MG1 or MENII-1 meningioma cell lines and AC1 arachnoidal cells—demonstrated that merlin loss was associated with increased YAP expression and nuclear localization. Merlin loss increased arachnoidal cell proliferation, entry into S-phase and cyclinE1 levels, whereas YAP silencing reversed the effect of merlin loss on cell proliferation (120). Merlin negatively regulates the hippo pathway at two cellular locations, the nucleus and cell cortex. Prominent staining of merlin in the nucleus in non-neoplastic human Schwann cells, HEI286, among multiple cell types, was observed using an N-terminal merlin antibody and an optimized immunostaining protocol for detection of nuclear proteins (121). Translocation of merlin to the nucleus allows merlin to bind and inhibit the E3 ubiquitin ligase CRL4DCAF1 (DDB1- and Cul4-Associated Factor 1) (121, 122). CRL4DCAF1 regulates cell proliferation, survival, DNA repair and genomic integrity via ubiquitination of critical regulators. Only unphosphorylated merlin binds DCAF1; transfected merlinS518A bound DCAF1 in HEK293T cells whereas merlinS518D did not bind. Further genetic experiments in HEI286 cells showed that merlin association with DCAF1 suppressed proliferation. In addition, in vitro DCAF1 silencing in mouse Nf2−/− FC-1801 Schwannoma and FH-912 normal control cells caused selective growth inhibition in merlin-null cells compared to control. In support of a proliferative role of DCAF1 in cells with loss of merlin, DCAF1 silencing in FC-1801 cells grown subcutaneously in mouse xenografts decreased tumor growth. Either expression of merlin or DCAF1silencing in FC-1801 schwannoma cells reduced expression of a group of genes regulated by YAP (121). Significantly, silencing of DCAF1 in schwannoma cells isolated from NF2 patients also reduced their proliferation (123). Additional studies showed that CRL4DCAF1 is responsible for LATS1 polyubuquitylation and LATS2 oligoubiquitylation, thereby promoting LATS1proteosomal degradation and inhibition of LATS2 kinase activity, which in turn suppresses YAP phosphorylation. This provides a molecular pathway by which the absence of merlin results in YAP activation (Figure 4) (124).
Figure 4.
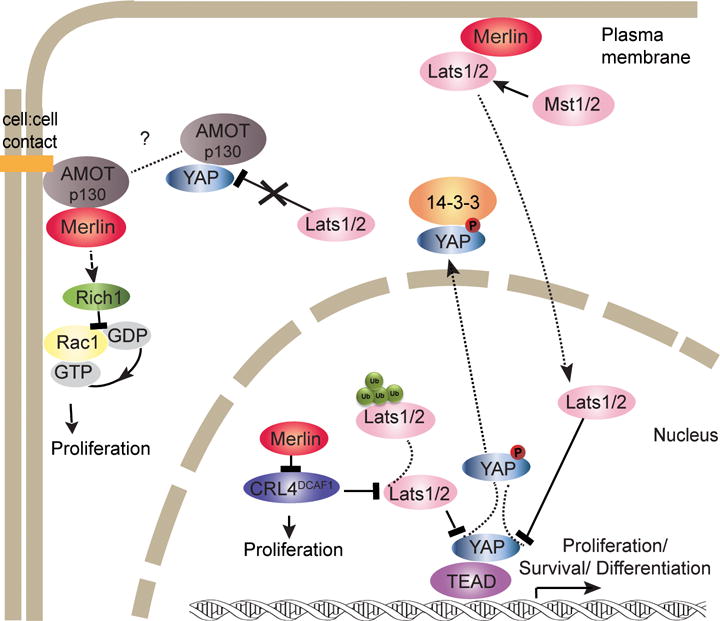
Role of merlin in the activation of the mammalian hippo pathway.
In the mammalian hippo pathway, merlin appears to act upstream and independent of Mst1/2 at the cell cortex. Merlin promoted LATS association to the plasma membrane in mouse Schwann cells. Studies in Nf2−/− mouse Schwann cells (FC-912) revealed low LATS phosphorylation levels and LATS1/2 enrichment in the cytoplasm relative to the membrane fraction compared to control Nf2flox2/flox2 mouse FH-912 Schwann cells. Re-expression of merlin in Nf2−/− FC-912 cells rescued LATS plasma membrane localization (125). Direct binding of wild-type, full-length merlin to the plasma membrane and recruitment of LATS1/2 promoted its phosphorylation by Mst1/2 without regulating intrinsic Mst1/2 enzymatic activity (Figure 4) (125). Deregulation of the actin cytoskeleton resulting from merlin loss also induces cell proliferation signaling through the hippo pathway, and YAP/TAZ have been recently identified as mechanosensors (63, 76, 81–83, 126, 127). Mechanical factors such as stretching, geometry, cell density and rigidity of the substrate influence YAP/TAZ activity (128, 129). Interestingly, F-actin capping and severing proteins including cofilin restrained YAP/TAZ activity in contact inhibited cells that have low mechanical stress via a mechanism that depended on merlin (130). Consistent with this phenomenon, disruption of F-actin stress fibers with LatrunculinA inhibited YAP nuclear localization and activity (131). The relationship between mechanical factor signaling in schwannomas, meningiomas and ependymomas and merlin loss warrants further study.
Additionally, at the cell cortex, components of the cell-cell contact complexes, including angiomotins (Amot), α-catenin and E-cadherin among others, can complex with YAP and TAZ and inhibit their activity by keeping them at the cell junctions and preventing their nuclear localization (132–134). Amot-p130 isoform bound to the WW domains of YAP and blocked LATS1 access to YAP. Interestingly, in 10 human schwannoma samples, Amot staining was abundant by IHC whereas normal human nerve had weak Amot expression (Figure 4) (135).
In peripheral nerves, merlin and Amot colocalize in paranodes and Schimidt-Lantermann incisures. Moreover, in cultured Schwann cells, merlin interaction with Amot was demonstrated by co-immunoprecipitation of the endogenous proteins (136). Merlin-Amot interaction was required for merlin regulation of mitogenic MAPK signaling. Consistently, in RT4-67 cells, loss of merlin or presence of patient-derived merlin mutants that fail to bind Amot resulted in a lack of inhibition of c-Raf and ERK phosphorylation. Silencing of Amot in Nf2−/− Schwann cells (SC4) selectively reduced cell proliferation because it did not change the proliferation rate of SC4 with merlin re-expression. Similarly, Amot knockdown decreased tumor growth of SC4 cells in sciatic nerve orthotopic mice. Furthermore, Amot silencing attenuated Rac1 and Ras/MAPK signaling pathway. In HEK293 transfected cells, merlin competed with Rich1, a small GTPase-activating protein, for Amot binding, releasing Rich1 to inactivate Rac1 (Figure 4) (136).
MERLIN IN MALIGNANT CANCERS
Merlin in mesothelioma
Malignant mesothelioma is an aggressive form of cancer that most commonly develops in the mesothelium of the pleura and peritoneum tissue. It is primarily caused by exposure to asbestos; however, the long latency after exposure suggests that predisposing genetic mutations likely play a role in disease development (137). Interestingly, somatic NF2 mutations are present in a large percentage of malignant mesothelioma patients (138–140). In addition, in human mesothelioma patients without detectable NF2 mutations, merlin was phosphorylated at Ser518 consistent with an inactive tumor suppressor state (141). Furthermore, Nf2 hemizygosity was initially reported to be associated with increased susceptibility to asbestos-induced mesotheliomas in Nf2KO3/+ mice inoculated with crocidolite asbestos fibers. Subsequent DNA analysis of peritoneal mesothelioma cells from these mice showed homozygous Nf2 inactivation (142). Moreover, another Nf2(+/−) mouse line treated with asbestos to induce malignant mesotheliomas phenocopied the human disease (143). The mesothelioma cells from these mice frequently exhibited Nf2 homozygous deletions, supporting a role for LOH in malignant mesothelioma etiology and the possibility that Nf2 heterozygosity increases susceptibility to asbestos-induced mesothelioma. Taken together, such observations support the idea that NF2 mutations or defective merlin tumor suppressor signaling might trigger or contribute to development of mesotheliomas (141).
Additional evidence supports a link between merlin-dependent regulatory pathways and mesotheliomas. First, protein kinase C-potentiated phosphatase inhibitor (CPI-17), which is frequently overexpressed in mesothelioma tumors, inhibits merlin phosphatase MYPT1-PP1δ, providing one potential pathway by which merlin’s tumor suppressor function might be inactivated through maintenance of phosphorylation at Ser518 (25). Second, similar to NF2 schwannomas, mesothelioma cells with NF2 inactivation, exhibit activated PAK1 and AKT, and re-expression of merlin in merlin-null human mesothelioma cells (Meso-17) decreases PAK1 activity (144, 145). The loss of merlin was linked to increased cell proliferation via a PAK1-induced increase in cyclin-D1, which resulted in activation of Cdk4 and phosphorylation of retinoblastoma protein (141, 144). The observed upregulation of cyclin-D1 in Meso-17 cells was similar to findings in mouse embryonic fibroblasts from Nf2-deficient mice (144, 146). Further supporting the relationship between merlin and cyclin-D1, reintroduction of merlin into Meso-17 cells by NF2-adenovirus infection resulted in reduced cyclin-D1levels. Finally, an independent analysis of eleven malignant mesothelioma cell lines found activation of mTORC1. Loss of merlin results in integrin-mediated activation of mTORC1 through PAK1, which promotes cell cycle progression by inducing translation of cyclin-D1 mRNA and cyclin-D1 expression (80). The mTORC1 inhibitor rapamycin selectively inhibited proliferation of seven merlin-null mesothelioma cell lines, but not merlin-positive cell lines, suggesting a potential pharmacological target for merlin deficient mesotheliomas (80).
An important functional link has been reported in mesothelioma cells between merlin and the hippo pathway (through CRL4-DCAF1). Silencing DCAF1 in Meso-33, merlin-deficient mesothelioma cells reduced their proliferation by arresting the cell cycle in G1 phase. When LATS1/2 was additionally silenced in these cells, cell cycle progression was restored. By contrast, silencing of DCAF1 in normal, Met-5A mesothelial cells resulted only in a moderate reduction of cell proliferation, suggesting that the absence of merlin sensitized cells to DCAF1 inactivation (121). In addition, in a panel of human mesothelioma cell lines, western blot experiments showed that the absence of merlin was accompanied by decreased LATS1 expression and YAP phosphorylation (124). Further confirming merlin’s functional relationship with the hippo pathway in mesothelioma cells, YAP1 expression studies found elevated YAP1 in malignant pleural mesothelioma samples and silencing of YAP1 reduced the growth of NCI-H290 mesothelioma cells carrying NF2 homozygous deletion (147). Notably, NF2 transfection into these cells induced YAP1 phosphorylation at Ser127, YAP1 retention in the cytoplasm and consequent reduction of YAP1 nuclear localization. Moreover, co-immunoprecipitation experiments revealed that merlin interacts with YAP1, although the interaction is not direct (147). Increased proliferation of mesothelioma cells with loss of merlin function was reported to be concurrent with LATS2 inactivation; however, reintroduction of wild-type merlin to Y-Meso-14 cells with homozygous deletion of LATS2 suppressed cell growth. If LATS1 cannot compensate for LATS2 function, the functional link between merlin and the hippo pathway in mesothelioma cells might not be that of a single axis growth suppression pathway (148).
Studies comparing Meso-33 cells lacking merlin and those with reintroduction of merlin also identified merlin as a microtubule stabilizer (149). Merlin interacts with tubulin and acetylated-tubulin and stabilizes the microtubules by attenuating tubulin turnover—lowering the rates of microtubule polymerization and depolymerization. Thus, merlin is a regulator of both the actin and microtubule cytoskeleton, which links merlin localization throughout the cell with its roles in proliferation, morphology, migration, mitosis, and survival.
Loss of merlin in mesotheliomas has been linked not only to increased proliferation, but also increased invasiveness, spreading and migration. Adenoviral transduction of NF2 in Meso-17 and Meso-25 cell lines decreased invasion through Matrigel membranes compared to cells transduced with empty vector. Similarly, re-expression of merlin in these cells decreased the percentage of cells that spread onto fibronectin-coated coverslips and decreased the motility of cells into the scratch area in a classical wound-healing assay (150). Merlin expression in Meso-17 and Meso-25 cells decreased FAK Tyr397 phosphorylation and consequently disrupted FAK-Src and PI3K interaction, providing a mechanism for the observed enhancement of invasion and spreading caused by merlin inactivation(150). Interestingly, research has shown that malignant mesothelioma cells as well as schwannomas and meningiomas with merlin loss have in common deregulation of several pathways (hippo, mTOR, PAK, FAK-Src and PI3K); thus we speculate that treatments for these malignancies might share commonalities.
Merlin in other malignant cancers
Considering that Nf2 heterozygous mice are prone to develop cancer—primarily osteosarcomas, fibrosarcomas and hepatocellular carcinomas with Nf2 LOH that frequently metastasize, it is tempting to assume that NF2 patients commonly develop other malignant cancers (151). However, the transformation of schwannomas into malignant peripheral nerve sheath tumors or development of other common cancers in NF2 patients is very rare. Moreover, the incidence of NF2 mutations in common human cancers is low. Although NF2 mutations have been detected in multiple human tumor types, mutational analysis of 315 human carcinoma samples found a low prevalence of NF2 mutations—e.g., NF2 mutations were found in just 2.2% of hepatocellular, acute myelogenous leukemia and squamous cell lung carcinomas(152, 153). In common cancers, methylation of CpG islands has been reported in promoter regions which are non-methylated in control tissue resulting in transcriptional inactivation of tumor suppressors (154, 155). Comprehensive studies of promoter methylation, methyl-binding proteins, DNA methyltransferases and HDACs have not been addressed in NF2-tumors or common cancers in relation to NF2/merlin.
The role of cancer stem cells has been gaining increased attention and we foresee merlin taking a place in this discussion. The participation of merlin in the mammalian hippo pathway might suggest a role in stem cell maintenance, differentiation or regulation of the stem cell niche (156–160).
Breast and colorectal cancer
A low incidence of NF2 mutations in breast cancer was reported by the Japanese National Cancer Center Research Institute (161). More recently, a systematic mutational analysis of 22 breast and colorectal cancers found NF2 somatic mutations in 4.5% of the samples. Notably, groups of eleven different genes were frequently mutated per tumor demonstrating molecular heterogeneity of cancer (162, 163). Interestingly, PAK1 amplification was identified in ~33% of breast tumor samples, providing a mechanism of MAPK pathway activation and tumorigenesis. PAK1 expression increased merlin Ser518 phosphorylation (inactivation) in a human mammary epithelial cell transformation cell line (HMLE) whereas silencing of NF2 promoted anchorage-independent proliferation (164). The data suggest that despite the low incidence of NF2 somatic mutations in breast cancer, merlin inactivation, together with the inhibitory regulation through PAK1, may contribute to the transformation or maintenance of the tumor state. In the NF2−/− breast cancer MDA-MB-231 cell line, merlin re-expression inhibited YAP/TEAD activity that was eliminated by LATS1/2 silencing. However, LATS1/2 silencing did not rescue YAP/TAZ inhibition by soft extracellular matrix that provides a low mechanical stress, suggesting that cytoskeletal changes caused by merlin loss regulate YAP more powerfully than the core kinase hippo pathway. Overall, YAP signaling is activated in NF2−/− breast cancer cells (130).
After the discovery of the NF2 gene, two independent studies found a low rate of somatic NF2 mutations in colorectal carcinomas—2 of 44 tumors in one study and 2 of 24 in another study exhibited mutations (165, 166). However, a recent study reported that 113 of 185 colorectal carcinoma samples lacked one functional copy of the NF2 gene and there was NF2 LOH in 20% of heterozygous samples, more frequently in larger and less differentiated tumors. Moreover, among all the samples, the level of NF2 mRNA expression measured by RT-PCR was significantly lower in poorly differentiated tumors. Similarly, merlin expression assessed by immunostaining was weaker in less differentiated tumors and the proportional fraction of merlin that was phosphorylated was higher in tumors compared to normal mucous tissue (167). This new data suggests a potential relationship between merlin expression and prognosis.
Hepatobiliar cancer
In hepatobiliary tumor cell lines, FOCUS and HA22TVGH, a study reported two NF2 homozygous deletions, ex1 and ex2-3, respectively (168). Interestingly, in developing and adult mice, Nf2 deletion in the liver significantly expanded liver progenitor cells without affecting differentiated hepatocytes. These liver progenitor cells increased proliferation by increasing EGFR activity independent of the hippo pathway and associates Nf2 absence in liver progenitor cells with tumor development (169).
Deregulation of the hippo pathway occurs in several human carcinomas including liver cancer (170–172). A specific role of merlin in liver was investigated in the Abl-Cre;Nf2flox2/flox2 mice. These mice initially developed bile duct hamartomas and in within a year all developed hepatocellular carcinomas. Their livers showed reduced YAP phosphorylation, increased YAP nuclear localization and reduced LATS membrane association and phosphorylation (125, 173). The data suggest that in hepatocytes merlin is functionally linked to the hippo pathway and acts upstream Mst1/2 by recruiting LATS to the membrane comparable to what has been shown in FH912 Schwann cell line.
Prostate cancer
In prostate cancer, the second leading cause of cancer death in males, inactivation of NF2 reportedly contributes to the progression of the cancer toward a highly invasive and chemoresistant state (174, 175). Studies of merlin expression and merlin Ser518 phosphorylation in several prostate cancer cell lines, including LNCaP, PC3, 22RV1 and LAPC-4, have indicated an overall decrease in merlin expression. In the DUI145 cell line, which exhibits merlin expression, phosphorylated merlin-Ser518 (inactive as tumor suppressor) was abundant and the high levels were sustained at high cell density concurrent with PAK1 activation. In sum, absence or inactivation of merlin in prostate cell lines appears to contribute to tumor development and progression (176).
Glioblastomas
In a very aggressive brain tumor, glioblastoma multiforme (GBM), merlin expression was reported to be reduced in 61% (n=23) of tumors. Merlin protein and mRNA levels were decreased by over 50% in GBM compared to normal human astrocytes and brain tissue (177). Another study reported that merlin was absent in 32% (n=53) of tumors and its expression was reduced in the rest, and in one-third of 7 GBM cell lines merlin was missing and re-expression suppressed proliferation (178). In addition, microarray expression analysis of GBM cells revealed that the absence of merlin induced changes in gene expression, decreasing transcripts that activate LATS2 signaling and decreasing expression of transcripts that inhibit the canonical Wnt and RhoA signaling pathway (177). These results were confirmed by RT-PCR, luciferase-reporter assays and western blot, with merlin re-expression resulting in activation of the hippo pathway and inhibition of the canonical (β-catenin) and non-canonical Wnt pathways in GBM cells. Importantly, merlin re-expression decreased GBM cell proliferation in vitro and in subcutaneous and intracranial xenograft mouse models (177). In GBM cell lines that expressed merlin, an alternative mechanism of merlin inactivation was revealed that operated through ezrin interaction. Ezrin was found to be expressed at higher levels in GBM cells and associated with merlin, removing merlin from its cortical localization, prohibiting its inhibitory action on Rac1, and thereby enhancing cell growth (178). Interestingly, a study of U251 glioblastoma cells identified different cell subpopulations, one with a high proliferation rate, spindle morphology, and EGFR and NOTCH1 expression (U251-S), and another with a lower proliferation rate, rhomboid morphology, no NOTCH1 expression and low EGFR expression (U251-R). Notably, both subpopulations exhibited similar merlin expression levels as measured by western blot; however, merlin-Ser518 phosphorylation was higher in U251-S cells regardless of cell density. The disrupted cell-contact inhibition signaling and merlin phosphorylation correlated with increased expression of NOTCH1 and its downstream target gene, HES1, which represses the transcription factor E2F in cell-contact growth arrest. Also increased was the EGFR effector CCND1, the Cdk1 gene, which is essential in the G1-S phase transition, providing an alternative mechanism for glioblastoma cell proliferation regulated by merlin phosphorylation when NF2 is not mutated or epigenetically inactivated (179).
Medullary thyroid carcinoma
NF2 allelic loss has also been reported to be of potential value in determining the risk of recurrence of rare metastatic medullary thyroid carcinomas. Among a tumor suppressor gene panel, NF2 was found to be one of the most frequent allelic losses (75%) in 11 medullary thyroid carcinomas. When considered in association with age and disease stage, NF2 allelic loss helped define high- and low-risk recurrence groups with excellent prognostic certainty (180).
Melanoma
After an initial report of NF2 somatic mutations in 6 of 20 human melanoma samples, a larger study reported only a 5% incidence of NF2 somatic mutation in melanoma (152, 181). Merlin inactivation in WM1552C and MeWo human melanoma cell lines in vitro and in vivo enhanced migration and invasion in addition to cell proliferation. Of interest, transduction of NF2 wild-type in MeWo cells activated Mst1/2 kinases (hippo pathway) as evidenced by increased levels of active phosphorylated Mst1/2 (181). An additional molecular mechanism that contributes to increased melanoma cell motility observed with merlin loss was identified in merlin-deficient MV3 human melanoma cells. Despite decreased Na+/H+ exchanger 1 (NHE1) expression, NH4Cl pre-pulse experiments revealed increased NHE1 activity and over-expression of NHE1 further increased migration, suggesting a functional link between merlin and NHE1 activity (182). Importantly, treatment of the human melanoma cell line A375 (this cell line carries the BRAF gain-of-function V600E mutation) with the RAF inhibitor vemurafenib allowed screening for genes involved in development of drug resistance in surviving cells by negative selection with the new CRISPR-Cas9 technology. NF2 was found, among 18,080 genes screened in the genome-scale CRISPR-Cas9 knockout (GeCKO) library, to have a role in vemurafenib resistance. Experiments comparing resistance between NF2 knockout and knockdown in vemurafenib-treated cells demonstrated that merlin absence was necessary for resistance development and the presence of merlin even at very low levels was sufficient to maintain sensitivity (183). In agreement, a whole exome sequencing and pharmacological response analysis of sixty cell lines of the US National Cancer Institute (NCI-60) showed that cancer cell lines that harbor NF2 mutations were resistant to vemurafenib treatment (184). Taken together, these data implicate NF2 mutations in the development of drug resistance in melanoma rather than oncogenesis and indicate that mutations are typically not of the non-functional passenger alteration variety. This makes treatment of melanoma tumors with loss of merlin function particularly challenging.
Interestingly, the Genomics of Drug Sensitivity in Cancer project (a collaboration between the Cancer Genome Project at the Wellcome Trust Sanger Institute - U.K. and the Center for Molecular Therapeutics, Massachusetts General Hospital Cancer Center -U.S.A.) has compiled information about NF2 mutations and drug sensitivity in a large panel of cancer cell lines (185, 186). Supporting the results from the GeCKO library and the NCI-60 screens with vemurafenib, this project found that all cancer cell lines carrying NF2 mutations (22 of 641 lines tested) were less sensitive to SB590885, a B-RAF inhibitor, irrespective of the tissue of origin. In a similar fashion, NF2 mutations increased the resistance to dihydrofolate reductase inhibitors methotrexalate and pyremethamine as well as the JNK inhibitor JNK-9L. However, this collaborative project also found instances in which NF2 mutations increased sensitivity to drugs such as dasatinib (ABL, SRC, KIT and PDGFR inhibitor), WH-4-023 (SRC family and ABL inhibitor), PHA-665752 (Hepatocyte Growth Factor Receptor MET inhibitor), 6881640 (WEE1 and CHK1 inhibitor) and FH535 (unknown target) (185). The information can be accessed on their website: http://www.cancerrxgene.org/translation/Gene/1268. Therapeutic resistance is a complex problem. Drug resistance may be related to several factors, i.e., the genomic tumor landscape, development of secondary mutations in the drug target, changes in the tumor microenvironment and activation of bypass or feedback signaling loops. In vemurafenib-resistant melanoma tumors and tumor-matched short-term cultures from clinical trial patients, resistance to vemurafenib was found to develop due to up-regulation of PDGFRβ or an NRas mutation (187). We speculate that NF2 mutations in B-Raf mutated melanomas may help confer resistance to vemurafenib because the absence of merlin per se activates the PDGFR signaling assisting in establishment of a compensatory route. In contrast, NF2 mutations may confer sensitivity to Src, KIT and PDGFR inhibitors because cells with only NF2 inactivation when treated with these inhibitors decreased proliferation and increased apoptosis (76, 91, 188–190). Thus targeting pathways known to be activated by loss of merlin function in combination with drugs targeting the primary gene mutation in cancer cells may decrease development of drug resistance.
CONCLUSION AND PERSPECTIVES
Merlin’s role as a scaffolding protein can explain how it regulates multiple signaling pathways and integrates extracellular signals to modulate morphology, motility, proliferation and survival. Here we summarized the role of NF2/merlin in NF2-related tumors and common human cancers. Although NF2 mutations are not frequent in common human malignant cancers, when present can affect disease progression, therapeutic approach or prognosis. Moving forward, larger studies evaluating transcriptional NF2 inactivation in common human cancers might require reassessment of its participation. In addition, the multiplicity of signaling pathways affected by merlin loss/inactivation in tumor cells and the differential influence of the microenvironment might guide selection of the most appropriate molecular target/s for therapy. Consequently, combinatorial molecular therapy might represent the most successful pharmacological approach; however, further research and a better understanding of molecular cross-signaling are still required to identify the best therapeutic strategies.
A better understanding of the role of merlin loss/inactivation in tumorigenesis and tumor progression is still required. The fact that loss of merlin in humans mostly leads to tumor formation from Schwann, meningeal and ependymal cells, whereas other cell types do not transform despite ubiquitous merlin expression in normal tissues, suggests tissue specific molecular context and microenvironment dependence that requires further elucidation. In addition, the significance of NF2/merlin inactivation in common human cancers deserves more investigation for its potential contribution to progression, prognosis and therapeutics.
Acknowledgments
This work was supported by the National Institute of Health 5R01DC10189 and 5R01NS062825 grants. We thank A.B. Knott for editorial assistance and Dr. C. Vivacharawongse for reviewing the manuscript and helpful discussions.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
References
- 1.Evans DG. Neurofibromatosis type 2 (NF2): a clinical and molecular review. Orphanet journal of rare diseases. 2009;4:16. doi: 10.1186/1750-1172-4-16. Epub 2009/06/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rouleau GA, Merel P, Lutchman M, Sanson M, Zucman J, Marineau C, et al. Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2. Nature. 1993;363(6429):515–21. doi: 10.1038/363515a0. Epub 1993/06/10. [DOI] [PubMed] [Google Scholar]
- 3.Trofatter JA, MacCollin MM, Rutter JL, Murrell JR, Duyao MP, Parry DM, et al. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell. 1993;72(5):791–800. doi: 10.1016/0092-8674(93)90406-g. Epub 1993/03/12. [DOI] [PubMed] [Google Scholar]
- 4.Asthagiri AR, Parry DM, Butman JA, Kim HJ, Tsilou ET, Zhuang Z, et al. Neurofibromatosis type 2. Lancet. 2009;373(9679):1974–86. doi: 10.1016/S0140-6736(09)60259-2. Epub 2009/05/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wiemels J, Wrensch M, Claus EB. Epidemiology and etiology of meningioma. Journal of neuro-oncology. 2010;99(3):307–14. doi: 10.1007/s11060-010-0386-3. Epub 2010/09/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bydon M, Mathios D, Aguayo-Alvarez JJ, Ho C, Gokaslan ZL, Bydon A. Multiple primary intramedullary ependymomas: a case report and review of the literature. The spine journal : official journal of the North American Spine Society. 2013;13(10):1379–86. doi: 10.1016/j.spinee.2013.06.037. Epub 2013/08/31. [DOI] [PubMed] [Google Scholar]
- 7.Plotkin SR, O’Donnell CC, Curry WT, Bove CM, MacCollin M, Nunes FP. Spinal ependymomas in neurofibromatosis Type 2: a retrospective analysis of 55 patients. Journal of neurosurgery Spine. 2011;14(4):543–7. doi: 10.3171/2010.11.SPINE10350. Epub 2011/02/08. [DOI] [PubMed] [Google Scholar]
- 8.Lloyd SK, Evans DG. Neurofibromatosis type 2 (NF2): diagnosis and management. Handbook of clinical neurology. 2013;115:957–67. doi: 10.1016/B978-0-444-52902-2.00054-0. Epub 2013/08/13. [DOI] [PubMed] [Google Scholar]
- 9.Sperfeld AD, Hein C, Schroder JM, Ludolph AC, Hanemann CO. Occurrence and characterization of peripheral nerve involvement in neurofibromatosis type 2. Brain : a journal of neurology. 2002;125(Pt 5):996–1004. doi: 10.1093/brain/awf115. Epub 2002/04/19. [DOI] [PubMed] [Google Scholar]
- 10.Schulz A, Baader SL, Niwa-Kawakita M, Jung MJ, Bauer R, Garcia C, et al. Merlin isoform 2 in neurofibromatosis type 2-associated polyneuropathy. Nature neuroscience. 2013;16(4):426–33. doi: 10.1038/nn.3348. Epub 2013/03/05. [DOI] [PubMed] [Google Scholar]
- 11.Schulz A, Zoch A, Morrison H. A neuronal function of the tumor suppressor protein merlin. Acta neuropathologica communications. 2014;2(1):82. doi: 10.1186/s40478-014-0082-1. Epub 2014/07/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aboukais R, Zairi F, Baroncini M, Bonne NX, Schapira S, Vincent C, et al. Intracranial meningiomas and neurofibromatosis type 2. Acta neurochirurgica. 2013;155(6):997–1001. doi: 10.1007/s00701-013-1692-2. discussion Epub 2013/04/06. [DOI] [PubMed] [Google Scholar]
- 13.Gilbert MR, Ruda R, Soffietti R. Ependymomas in adults. Current neurology and neuroscience reports. 2010;10(3):240–7. doi: 10.1007/s11910-010-0109-3. Epub 2010/04/29. [DOI] [PubMed] [Google Scholar]
- 14.Xu HM, Gutmann DH. Merlin differentially associates with the microtubule and actin cytoskeleton. Journal of neuroscience research. 1998;51(3):403–15. doi: 10.1002/(SICI)1097-4547(19980201)51:3<403::AID-JNR13>3.0.CO;2-7. Epub 1998/03/05. [DOI] [PubMed] [Google Scholar]
- 15.Gronholm M, Sainio M, Zhao F, Heiska L, Vaheri A, Carpen O. Homotypic and heterotypic interaction of the neurofibromatosis 2 tumor suppressor protein merlin and the ERM protein ezrin. Journal of cell science. 1999;112(Pt 6):895–904. doi: 10.1242/jcs.112.6.895. Epub 1999/02/26. [DOI] [PubMed] [Google Scholar]
- 16.Nguyen R, Reczek D, Bretscher A. Hierarchy of merlin and ezrin N- and C-terminal domain interactions in homo- and heterotypic associations and their relationship to binding of scaffolding proteins EBP50 and E3KARP. The Journal of biological chemistry. 2001;276(10):7621–9. doi: 10.1074/jbc.M006708200. Epub 2000/12/07. [DOI] [PubMed] [Google Scholar]
- 17.Stamenkovic I, Yu Q. Merlin, a “magic” linker between extracellular cues and intracellular signaling pathways that regulate cell motility, proliferation, and survival. Current protein & peptide science. 2010;11(6):471–84. doi: 10.2174/138920310791824011. Epub 2010/05/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang X, Jang SW, Wang X, Liu Z, Bahr SM, Sun SY, et al. Akt phosphorylation regulates the tumour-suppressor merlin through ubiquitination and degradation. Nature cell biology. 2007;9(10):1199–207. doi: 10.1038/ncb1641. Epub 2007/09/25. [DOI] [PubMed] [Google Scholar]
- 19.Laulajainen M, Muranen T, Nyman TA, Carpen O, Gronholm M. Multistep phosphorylation by oncogenic kinases enhances the degradation of the NF2 tumor suppressor merlin. Neoplasia. 2011;13(7):643–52. doi: 10.1593/neo.11356. Epub 2011/07/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laulajainen M, Muranen T, Carpen O, Gronholm M. Protein kinase A-mediated phosphorylation of the NF2 tumor suppressor protein merlin at serine 10 affects the actin cytoskeleton. Oncogene. 2008;27(23):3233–43. doi: 10.1038/sj.onc.1210988. Epub 2007/12/12. [DOI] [PubMed] [Google Scholar]
- 21.Shaw RJ, Paez JG, Curto M, Yaktine A, Pruitt WM, Saotome I, et al. The Nf2 tumor suppressor, merlin, functions in Rac-dependent signaling. Developmental cell. 2001;1(1):63–72. doi: 10.1016/s1534-5807(01)00009-0. Epub 2001/11/13. [DOI] [PubMed] [Google Scholar]
- 22.Xiao GH, Beeser A, Chernoff J, Testa JR. p21-activated kinase links Rac/Cdc42 signaling to merlin. The Journal of biological chemistry. 2002;277(2):883–6. doi: 10.1074/jbc.C100553200. Epub 2001/11/24. [DOI] [PubMed] [Google Scholar]
- 23.Surace EI, Haipek CA, Gutmann DH. Effect of merlin phosphorylation on neurofibromatosis 2 (NF2) gene function. Oncogene. 2004;23(2):580–7. doi: 10.1038/sj.onc.1207142. Epub 2004/01/16. [DOI] [PubMed] [Google Scholar]
- 24.Alfthan K, Heiska L, Gronholm M, Renkema GH, Carpen O. Cyclic AMP-dependent protein kinase phosphorylates merlin at serine 518 independently of p21-activated kinase and promotes merlin-ezrin heterodimerization. The Journal of biological chemistry. 2004;279(18):18559–66. doi: 10.1074/jbc.M313916200. Epub 2004/02/26. [DOI] [PubMed] [Google Scholar]
- 25.Jin H, Sperka T, Herrlich P, Morrison H. Tumorigenic transformation by CPI-17 through inhibition of a merlin phosphatase. Nature. 2006;442(7102):576–9. doi: 10.1038/nature04856. Epub 2006/08/04. [DOI] [PubMed] [Google Scholar]
- 26.Pearson MA, Reczek D, Bretscher A, Karplus PA. Structure of the ERM protein moesin reveals the FERM domain fold masked by an extended actin binding tail domain. Cell. 2000;101(3):259–70. doi: 10.1016/s0092-8674(00)80836-3. Epub 2000/06/10. [DOI] [PubMed] [Google Scholar]
- 27.Scoles DR. The merlin interacting proteins reveal multiple targets for NF2 therapy. Biochimica et biophysica acta. 2008;1785(1):32–54. doi: 10.1016/j.bbcan.2007.10.001. Epub 2007/11/06. [DOI] [PubMed] [Google Scholar]
- 28.Curto M, Cole BK, Lallemand D, Liu CH, McClatchey AI. Contact-dependent inhibition of EGFR signaling by Nf2/Merlin. The Journal of cell biology. 2007;177(5):893–903. doi: 10.1083/jcb.200703010. Epub 2007/06/06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.James MF, Beauchamp RL, Manchanda N, Kazlauskas A, Ramesh V. A NHERF binding site links the betaPDGFR to the cytoskeleton and regulates cell spreading and migration. Journal of cell science. 2004;117(Pt 14):2951–61. doi: 10.1242/jcs.01156. Epub 2004/05/27. [DOI] [PubMed] [Google Scholar]
- 30.Yogesha SD, Sharff AJ, Giovannini M, Bricogne G, Izard T. Unfurling of the band 4.1, ezrin, radixin, moesin (FERM) domain of the merlin tumor suppressor. Protein science : a publication of the Protein Society. 2011;20(12):2113–20. doi: 10.1002/pro.751. Epub 2011/10/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hennigan RF, Foster LA, Chaiken MF, Mani T, Gomes MM, Herr AB, et al. Fluorescence resonance energy transfer analysis of merlin conformational changes. Molecular and cellular biology. 2010;30(1):54–67. doi: 10.1128/MCB.00248-09. Epub 2009/11/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sher I, Hanemann CO, Karplus PA, Bretscher A. The tumor suppressor merlin controls growth in its open state, and phosphorylation converts it to a less-active more-closed state. Developmental cell. 2012;22(4):703–5. doi: 10.1016/j.devcel.2012.03.008. Epub 2012/04/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ali Khajeh J, Ju JH, Atchiba M, Allaire M, Stanley C, Heller WT, et al. Molecular conformation of the full-length tumor suppressor NF2/Merlin–a small-angle neutron scattering study. Journal of molecular biology. 2014;426(15):2755–68. doi: 10.1016/j.jmb.2014.05.011. Epub 2014/06/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McClatchey AI, Saotome I, Ramesh V, Gusella JF, Jacks T. The Nf2 tumor suppressor gene product is essential for extraembryonic development immediately prior to gastrulation. Genes & development. 1997;11(10):1253–65. doi: 10.1101/gad.11.10.1253. Epub 1997/05/15. [DOI] [PubMed] [Google Scholar]
- 35.Giovannini M, Robanus-Maandag E, van der Valk M, Niwa-Kawakita M, Abramowski V, Goutebroze L, et al. Conditional biallelic Nf2 mutation in the mouse promotes manifestations of human neurofibromatosis type 2. Genes & development. 2000;14(13):1617–30. Epub 2000/07/11. [PMC free article] [PubMed] [Google Scholar]
- 36.Kalamarides M, Niwa-Kawakita M, Leblois H, Abramowski V, Perricaudet M, Janin A, et al. Nf2 gene inactivation in arachnoidal cells is rate-limiting for meningioma development in the mouse. Genes & development. 2002;16(9):1060–5. doi: 10.1101/gad.226302. Epub 2002/05/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gehlhausen JR, Park SJ, Hickox AE, Shew M, Staser K, Rhodes SD, et al. A murine model of neurofibromatosis type 2 that accurately phenocopies human schwannoma formation. Human molecular genetics. 2014 doi: 10.1093/hmg/ddu414. Epub 2014/08/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kalamarides M, Peyre M, Giovannini M. Meningioma mouse models. Journal of neuro-oncology. 2010;99(3):325–31. doi: 10.1007/s11060-010-0331-5. Epub 2010/08/25. [DOI] [PubMed] [Google Scholar]
- 39.Rangarajan A, Weinberg RA. Opinion: Comparative biology of mouse versus human cells: modelling human cancer in mice. Nature reviews Cancer. 2003;3(12):952–9. doi: 10.1038/nrc1235. Epub 2004/01/23. [DOI] [PubMed] [Google Scholar]
- 40.Gusella JF, Ramesh V, MacCollin M, Jacoby LB. Merlin: the neurofibromatosis 2 tumor suppressor. Biochimica et biophysica acta. 1999;1423(2):M29–36. doi: 10.1016/s0304-419x(99)00005-0. Epub 1999/04/24. [DOI] [PubMed] [Google Scholar]
- 41.Schulz A, Kyselyova A, Baader SL, Jung MJ, Zoch A, Mautner VF, et al. Neuronal merlin influences ERBB2 receptor expression on Schwann cells through neuregulin 1 type III signalling. Brain : a journal of neurology. 2014;137(Pt 2):420–32. doi: 10.1093/brain/awt327. Epub 2013/12/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sainz J, Huynh DP, Figueroa K, Ragge NK, Baser ME, Pulst SM. Mutations of the neurofibromatosis type 2 gene and lack of the gene product in vestibular schwannomas. Human molecular genetics. 1994;3(6):885–91. doi: 10.1093/hmg/3.6.885. Epub 1994/06/01. [DOI] [PubMed] [Google Scholar]
- 43.Gutmann DH, Giordano MJ, Fishback AS, Guha A. Loss of merlin expression in sporadic meningiomas, ependymomas and schwannomas. Neurology. 1997;49(1):267–70. doi: 10.1212/wnl.49.1.267. Epub 1997/07/01. [DOI] [PubMed] [Google Scholar]
- 44.Gonzalez-Gomez P, Bello MJ, Alonso ME, Lomas J, Arjona D, Campos JM, et al. CpG island methylation in sporadic and neurofibromatis type 2-associated schwannomas. Clinical cancer research : an official journal of the American Association for Cancer Research. 2003;9(15):5601–6. Epub 2003/12/05. [PubMed] [Google Scholar]
- 45.Kino T, Takeshima H, Nakao M, Nishi T, Yamamoto K, Kimura T, et al. Identification of the cis-acting region in the NF2 gene promoter as a potential target for mutation and methylation-dependent silencing in schwannoma. Genes to cells : devoted to molecular & cellular mechanisms. 2001;6(5):441–54. doi: 10.1046/j.1365-2443.2001.00432.x. Epub 2001/05/31. [DOI] [PubMed] [Google Scholar]
- 46.Koutsimpelas D, Ruerup G, Mann WJ, Brieger J. Lack of neurofibromatosis type 2 gene promoter methylation in sporadic vestibular schwannomas. ORL; journal for oto-rhino-laryngology and its related specialties. 2012;74(1):33–7. doi: 10.1159/000334968. Epub 2012/01/18. [DOI] [PubMed] [Google Scholar]
- 47.Lomas J, Bello MJ, Arjona D, Alonso ME, Martinez-Glez V, Lopez-Marin I, et al. Genetic and epigenetic alteration of the NF2 gene in sporadic meningiomas. Genes, chromosomes & cancer. 2005;42(3):314–9. doi: 10.1002/gcc.20141. Epub 2004/12/21. [DOI] [PubMed] [Google Scholar]
- 48.Riemenschneider MJ, Perry A, Reifenberger G. Histological classification and molecular genetics of meningiomas. The Lancet Neurology. 2006;5(12):1045–54. doi: 10.1016/S1474-4422(06)70625-1. Epub 2006/11/18. [DOI] [PubMed] [Google Scholar]
- 49.Alonso ME, Bello MJ, Gonzalez-Gomez P, Arjona D, de Campos JM, Gutierrez M, et al. Aberrant CpG island methylation of multiple genes in ependymal tumors. Journal of neuro-oncology. 2004;67(1–2):159–65. doi: 10.1023/b:neon.0000021862.41799.f7. Epub 2004/04/10. [DOI] [PubMed] [Google Scholar]
- 50.Kimura Y, Koga H, Araki N, Mugita N, Fujita N, Takeshima H, et al. The involvement of calpain-dependent proteolysis of the tumor suppressor NF2 (merlin) in schwannomas and meningiomas. Nature medicine. 1998;4(8):915–22. doi: 10.1038/nm0898-915. Epub 1998/08/13. [DOI] [PubMed] [Google Scholar]
- 51.Goutagny S, Nault JC, Mallet M, Henin D, Rossi JZ, Kalamarides M. High incidence of activating TERT promoter mutations in meningiomas undergoing malignant progression. Brain pathology. 2014;24(2):184–9. doi: 10.1111/bpa.12110. Epub 2013/11/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morrison H, Sherman LS, Legg J, Banine F, Isacke C, Haipek CA, et al. The NF2 tumor suppressor gene product, merlin, mediates contact inhibition of growth through interactions with CD44. Genes & development. 2001;15(8):968–80. doi: 10.1101/gad.189601. Epub 2001/04/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shaw RJ, McClatchey AI, Jacks T. Regulation of the neurofibromatosis type 2 tumor suppressor protein, merlin, by adhesion and growth arrest stimuli. The Journal of biological chemistry. 1998;273(13):7757–64. doi: 10.1074/jbc.273.13.7757. Epub 1998/04/29. [DOI] [PubMed] [Google Scholar]
- 54.Sainio M, Zhao F, Heiska L, Turunen O, den Bakker M, Zwarthoff E, et al. Neurofibromatosis 2 tumor suppressor protein colocalizes with ezrin and CD44 and associates with actin-containing cytoskeleton. Journal of cell science. 1997;110(Pt 18):2249–60. doi: 10.1242/jcs.110.18.2249. Epub 1997/10/24. [DOI] [PubMed] [Google Scholar]
- 55.Herrlich P, Morrison H, Sleeman J, Orian-Rousseau V, Konig H, Weg-Remers S, et al. CD44 acts both as a growth- and invasiveness-promoting molecule and as a tumor-suppressing cofactor. Annals of the New York Academy of Sciences. 2000;910:106–18. doi: 10.1111/j.1749-6632.2000.tb06704.x. discussion 18–20. Epub 2000/07/27. [DOI] [PubMed] [Google Scholar]
- 56.Bai Y, Liu YJ, Wang H, Xu Y, Stamenkovic I, Yu Q. Inhibition of the hyaluronan-CD44 interaction by merlin contributes to the tumor-suppressor activity of merlin. Oncogene. 2007;26(6):836–50. doi: 10.1038/sj.onc.1209849. Epub 2006/09/06. [DOI] [PubMed] [Google Scholar]
- 57.Bourguignon LY, Gilad E, Peyrollier K, Brightman A, Swanson RA. Hyaluronan-CD44 interaction stimulates Rac1 signaling and PKN gamma kinase activation leading to cytoskeleton function and cell migration in astrocytes. Journal of neurochemistry. 2007;101(4):1002–17. doi: 10.1111/j.1471-4159.2007.04485.x. Epub 2007/04/04. [DOI] [PubMed] [Google Scholar]
- 58.Oliferenko S, Kaverina I, Small JV, Huber LA. Hyaluronic acid (HA) binding to CD44 activates Rac1 and induces lamellipodia outgrowth. The Journal of cell biology. 2000;148(6):1159–64. doi: 10.1083/jcb.148.6.1159. Epub 2000/03/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bourguignon LY, Zhu H, Shao L, Chen YW. CD44 interaction with tiam1 promotes Rac1 signaling and hyaluronic acid-mediated breast tumor cell migration. The Journal of biological chemistry. 2000;275(3):1829–38. doi: 10.1074/jbc.275.3.1829. Epub 2000/01/15. [DOI] [PubMed] [Google Scholar]
- 60.Sherman LS, Gutmann DH. Merlin: hanging tumor suppression on the Rac. Trends in cell biology. 2001;11(11):442–4. doi: 10.1016/s0962-8924(01)02128-6. Epub 2001/10/31. [DOI] [PubMed] [Google Scholar]
- 61.Kaempchen K, Mielke K, Utermark T, Langmesser S, Hanemann CO. Upregulation of the Rac1/JNK signaling pathway in primary human schwannoma cells. Human molecular genetics. 2003;12(11):1211–21. doi: 10.1093/hmg/ddg146. Epub 2003/05/23. [DOI] [PubMed] [Google Scholar]
- 62.Nakai Y, Zheng Y, MacCollin M, Ratner N. Temporal control of Rac in Schwann cell-axon interaction is disrupted in NF2-mutant schwannoma cells. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2006;26(13):3390–5. doi: 10.1523/JNEUROSCI.4865-05.2006. Epub 2006/03/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Flaiz C, Ammoun S, Biebl A, Hanemann CO. Altered adhesive structures and their relation to RhoGTPase activation in merlin-deficient Schwannoma. Brain pathology. 2009;19(1):27–38. doi: 10.1111/j.1750-3639.2008.00165.x. Epub 2008/05/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Okada T, Lopez-Lago M, Giancotti FG. Merlin/NF-2 mediates contact inhibition of growth by suppressing recruitment of Rac to the plasma membrane. The Journal of cell biology. 2005;171(2):361–71. doi: 10.1083/jcb.200503165. Epub 2005/10/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kissil JL, Wilker EW, Johnson KC, Eckman MS, Yaffe MB, Jacks T. Merlin, the product of the Nf2 tumor suppressor gene, is an inhibitor of the p21-activated kinase, Pak1. Molecular cell. 2003;12(4):841–9. doi: 10.1016/s1097-2765(03)00382-4. Epub 2003/10/29. [DOI] [PubMed] [Google Scholar]
- 66.Kissil JL, Johnson KC, Eckman MS, Jacks T. Merlin phosphorylation by p21-activated kinase 2 and effects of phosphorylation on merlin localization. The Journal of biological chemistry. 2002;277(12):10394–9. doi: 10.1074/jbc.M200083200. Epub 2002/01/10. [DOI] [PubMed] [Google Scholar]
- 67.Thaxton C, Lopera J, Bott M, Baldwin ME, Kalidas P, Fernandez-Valle C. Phosphorylation of the NF2 tumor suppressor in Schwann cells is mediated by Cdc42-Pak and requires paxillin binding. Molecular and cellular neurosciences. 2007;34(2):231–42. doi: 10.1016/j.mcn.2006.11.003. Epub 2006/12/19. [DOI] [PubMed] [Google Scholar]
- 68.Sherman LS, Rizvi TA, Karyala S, Ratner N. CD44 enhances neuregulin signaling by Schwann cells. The Journal of cell biology. 2000;150(5):1071–84. doi: 10.1083/jcb.150.5.1071. Epub 2000/09/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ahmad Z, Brown CM, Patel AK, Ryan AF, Ongkeko R, Doherty JK. Merlin knockdown in human Schwann cells: clues to vestibular schwannoma tumorigenesis. Otology & neurotology : official publication of the American Otological Society, American Neurotology Society [and] European Academy of Otology and Neurotology. 2010;31(3):460–6. doi: 10.1097/MAO.0b013e3181d2777f. Epub 2010/03/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hansen MR, Roehm PC, Chatterjee P, Green SH. Constitutive neuregulin-1/ErbB signaling contributes to human vestibular schwannoma proliferation. Glia. 2006;53(6):593–600. doi: 10.1002/glia.20316. Epub 2006/01/25. [DOI] [PubMed] [Google Scholar]
- 71.Wickremesekera A, Hovens CM, Kaye AH. Expression of ErbB-1 and 2 in vestibular schwannomas. Journal of clinical neuroscience : official journal of the Neurosurgical Society of Australasia. 2007;14(12):1199–206. doi: 10.1016/j.jocn.2007.05.009. Epub 2007/10/30. [DOI] [PubMed] [Google Scholar]
- 72.Thaxton C, Lopera J, Bott M, Fernandez-Valle C. Neuregulin and laminin stimulate phosphorylation of the NF2 tumor suppressor in Schwann cells by distinct protein kinase A and p21-activated kinase-dependent pathways. Oncogene. 2008;27(19):2705–15. doi: 10.1038/sj.onc.1210923. Epub 2007/11/14. [DOI] [PubMed] [Google Scholar]
- 73.Garcia C, Gutmann DH. Nf2/Merlin controls spinal cord neural progenitor function in a Rac1/ErbB2-dependent manner. PloS one. 2014;9(5):e97320. doi: 10.1371/journal.pone.0097320. Epub 2014/05/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lallemand D, Manent J, Couvelard A, Watilliaux A, Siena M, Chareyre F, et al. Merlin regulates transmembrane receptor accumulation and signaling at the plasma membrane in primary mouse Schwann cells and in human schwannomas. Oncogene. 2009;28(6):854–65. doi: 10.1038/onc.2008.427. Epub 2008/11/26. [DOI] [PubMed] [Google Scholar]
- 75.Ammoun S, Schmid MC, Ristic N, Zhou L, Hilton D, Ercolano E, et al. The role of insulin-like growth factors signaling in merlin-deficient human schwannomas. Glia. 2012;60(11):1721–33. doi: 10.1002/glia.22391. Epub 2012/07/24. [DOI] [PubMed] [Google Scholar]
- 76.Ammoun S, Flaiz C, Ristic N, Schuldt J, Hanemann CO. Dissecting and targeting the growth factor-dependent and growth factor-independent extracellular signal-regulated kinase pathway in human schwannoma. Cancer research. 2008;68(13):5236–45. doi: 10.1158/0008-5472.CAN-07-5849. Epub 2008/07/03. [DOI] [PubMed] [Google Scholar]
- 77.Ammoun S, Cunliffe CH, Allen JC, Chiriboga L, Giancotti FG, Zagzag D, et al. ErbB/HER receptor activation and preclinical efficacy of lapatinib in vestibular schwannoma. Neuro-oncology. 2010;12(8):834–43. doi: 10.1093/neuonc/noq012. Epub 2010/06/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fernandez-Valle C, Tang Y, Ricard J, Rodenas-Ruano A, Taylor A, Hackler E, et al. Paxillin binds schwannomin and regulates its density-dependent localization and effect on cell morphology. Nature genetics. 2002;31(4):354–62. doi: 10.1038/ng930. Epub 2002/07/16. [DOI] [PubMed] [Google Scholar]
- 79.Obremski VJ, Hall AM, Fernandez-Valle C. Merlin, the neurofibromatosis type 2 gene product, and beta1 integrin associate in isolated and differentiating Schwann cells. Journal of neurobiology. 1998;37(4):487–501. Epub 1998/12/19. [PubMed] [Google Scholar]
- 80.Lopez-Lago MA, Okada T, Murillo MM, Socci N, Giancotti FG. Loss of the tumor suppressor gene NF2, encoding merlin, constitutively activates integrin-dependent mTORC1 signaling. Molecular and cellular biology. 2009;29(15):4235–49. doi: 10.1128/MCB.01578-08. Epub 2009/05/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bashour AM, Meng JJ, Ip W, MacCollin M, Ratner N. The neurofibromatosis type 2 gene product, merlin, reverses the F-actin cytoskeletal defects in primary human Schwannoma cells. Molecular and cellular biology. 2002;22(4):1150–7. doi: 10.1128/MCB.22.4.1150-1157.2002. Epub 2002/01/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pelton PD, Sherman LS, Rizvi TA, Marchionni MA, Wood P, Friedman RA, et al. Ruffling membrane, stress fiber, cell spreading and proliferation abnormalities in human Schwannoma cells. Oncogene. 1998;17(17):2195–209. doi: 10.1038/sj.onc.1202141. Epub 1998/11/12. [DOI] [PubMed] [Google Scholar]
- 83.Petrilli A, Copik A, Posadas M, Chang LS, Welling DB, Giovannini M, et al. LIM domain kinases as potential therapeutic targets for neurofibromatosis type 2. Oncogene. 2014;33(27):3571–82. doi: 10.1038/onc.2013.320. Epub 2013/08/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bosco EE, Nakai Y, Hennigan RF, Ratner N, Zheng Y. NF2-deficient cells depend on the Rac1-canonical Wnt signaling pathway to promote the loss of contact inhibition of proliferation. Oncogene. 2010;29(17):2540–9. doi: 10.1038/onc.2010.20. Epub 2010/02/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhou L, Ercolano E, Ammoun S, Schmid MC, Barczyk MA, Hanemann CO. Merlin-deficient human tumors show loss of contact inhibition and activation of Wnt/beta-catenin signaling linked to the PDGFR/Src and Rac/PAK pathways. Neoplasia. 2011;13(12):1101–12. doi: 10.1593/neo.111060. Epub 2012/01/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tikoo A, Varga M, Ramesh V, Gusella J, Maruta H. An anti-Ras function of neurofibromatosis type 2 gene product (NF2/Merlin) The Journal of biological chemistry. 1994;269(38):23387–90. Epub 1994/09/23. [PubMed] [Google Scholar]
- 87.Morrison H, Sperka T, Manent J, Giovannini M, Ponta H, Herrlich P. Merlin/neurofibromatosis type 2 suppresses growth by inhibiting the activation of Ras and Rac. Cancer research. 2007;67(2):520–7. doi: 10.1158/0008-5472.CAN-06-1608. Epub 2007/01/20. [DOI] [PubMed] [Google Scholar]
- 88.Li N, Batzer A, Daly R, Yajnik V, Skolnik E, Chardin P, et al. Guanine-nucleotide-releasing factor hSos1 binds to Grb2 and links receptor tyrosine kinases to Ras signalling. Nature. 1993;363(6424):85–8. doi: 10.1038/363085a0. Epub 1993/05/06. [DOI] [PubMed] [Google Scholar]
- 89.Geissler KJ, Jung MJ, Riecken LB, Sperka T, Cui Y, Schacke S, et al. Regulation of Son of sevenless by the membrane-actin linker protein ezrin. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(51):20587–92. doi: 10.1073/pnas.1222078110. Epub 2013/12/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ammoun S, Provenzano L, Zhou L, Barczyk M, Evans K, Hilton DA, et al. Axl/Gas6/NFkappaB signalling in schwannoma pathological proliferation, adhesion and survival. Oncogene. 2014;33(3):336–46. doi: 10.1038/onc.2012.587. Epub 2013/01/16. [DOI] [PubMed] [Google Scholar]
- 91.Houshmandi SS, Emnett RJ, Giovannini M, Gutmann DH. The neurofibromatosis 2 protein, merlin, regulates glial cell growth in an ErbB2- and Src-dependent manner. Molecular and cellular biology. 2009;29(6):1472–86. doi: 10.1128/MCB.01392-08. Epub 2008/12/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ammoun S, Schmid MC, Zhou L, Hilton DA, Barczyk M, Hanemann CO. The p53/mouse double minute 2 homolog complex deregulation in merlin-deficient tumours. Molecular oncology. 2015;9(1):236–48. doi: 10.1016/j.molonc.2014.08.005. Epub 2014/09/14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hilton DA, Ristic N, Hanemann CO. Activation of ERK, AKT and JNK signalling pathways in human schwannomas in situ. Histopathology. 2009;55(6):744–9. doi: 10.1111/j.1365-2559.2009.03440.x. Epub 2009/11/19. [DOI] [PubMed] [Google Scholar]
- 94.Rong R, Tang X, Gutmann DH, Ye K. Neurofibromatosis 2 (NF2) tumor suppressor merlin inhibits phosphatidylinositol 3-kinase through binding to PIKE-L. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(52):18200–5. doi: 10.1073/pnas.0405971102. Epub 2004/12/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jacob A, Lee TX, Neff BA, Miller S, Welling B, Chang LS. Phosphatidylinositol 3-kinase/AKT pathway activation in human vestibular schwannoma. Otology & neurotology : official publication of the American Otological Society, American Neurotology Society [and] European Academy of Otology and Neurotology. 2008;29(1):58–68. doi: 10.1097/mao.0b013e31816021f7. Epub 2008/01/18. [DOI] [PubMed] [Google Scholar]
- 96.Mawrin C, Sasse T, Kirches E, Kropf S, Schneider T, Grimm C, et al. Different activation of mitogen-activated protein kinase and Akt signaling is associated with aggressive phenotype of human meningiomas. Clinical cancer research : an official journal of the American Association for Cancer Research. 2005;11(11):4074–82. doi: 10.1158/1078-0432.CCR-04-2550. Epub 2005/06/03. [DOI] [PubMed] [Google Scholar]
- 97.Ye K. Phosphorylation of merlin regulates its stability and tumor suppressive activity. Cell adhesion & migration. 2007;1(4):196–8. doi: 10.4161/cam.1.4.5192. Epub 2007/10/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li Y, Tennekoon GI, Birnbaum M, Marchionni MA, Rutkowski JL. Neuregulin signaling through a PI3K/Akt/Bad pathway in Schwann cell survival. Molecular and cellular neurosciences. 2001;17(4):761–7. doi: 10.1006/mcne.2000.0967. Epub 2001/04/21. [DOI] [PubMed] [Google Scholar]
- 99.Petrilli AM, Fuse MA, Donnan MS, Bott M, Sparrow NA, Tondera D, et al. A chemical biology approach identified PI3K as a potential therapeutic target for neurofibromatosis type 2. American journal of translational research. 2014;6(5):471–93. Epub 2014/11/02. [PMC free article] [PubMed] [Google Scholar]
- 100.Lee TX, Packer MD, Huang J, Akhmametyeva EM, Kulp SK, Chen CS, et al. Growth inhibitory and anti-tumour activities of OSU-03012, a novel PDK-1 inhibitor, on vestibular schwannoma and malignant schwannoma cells. European journal of cancer. 2009;45(9):1709–20. doi: 10.1016/j.ejca.2009.03.013. Epub 2009/04/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Johnson MD, Okedli E, Woodard A, Toms SA, Allen GS. Evidence for phosphatidylinositol 3-kinase-Akt-p7S6K pathway activation and transduction of mitogenic signals by platelet-derived growth factor in meningioma cells. Journal of neurosurgery. 2002;97(3):668–75. doi: 10.3171/jns.2002.97.3.0668. Epub 2002/09/26. [DOI] [PubMed] [Google Scholar]
- 102.Bush ML, Oblinger J, Brendel V, Santarelli G, Huang J, Akhmametyeva EM, et al. AR42, a novel histone deacetylase inhibitor, as a potential therapy for vestibular schwannomas and meningiomas. Neuro-oncology. 2011;13(9):983–99. doi: 10.1093/neuonc/nor072. Epub 2011/07/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen CS, Weng SC, Tseng PH, Lin HP, Chen CS. Histone acetylation-independent effect of histone deacetylase inhibitors on Akt through the reshuffling of protein phosphatase 1 complexes. The Journal of biological chemistry. 2005;280(46):38879–87. doi: 10.1074/jbc.M505733200. Epub 2005/09/28. [DOI] [PubMed] [Google Scholar]
- 104.Jacob A, Oblinger J, Bush ML, Brendel V, Santarelli G, Chaudhury AR, et al. Preclinical validation of AR42, a novel histone deacetylase inhibitor, as treatment for vestibular schwannomas. The Laryngoscope. 2012;122(1):174–89. doi: 10.1002/lary.22392. Epub 2011/11/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Burns SS, Akhmametyeva EM, Oblinger JL, Bush ML, Huang J, Senner V, et al. Histone deacetylase inhibitor AR-42 differentially affects cell-cycle transit in meningeal and meningioma cells, potently inhibiting NF2-deficient meningioma growth. Cancer research. 2013;73(2):792–803. doi: 10.1158/0008-5472.CAN-12-1888. Epub 2012/11/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Agnihotri S, Gugel I, Remke M, Bornemann A, Pantazis G, Mack SC, et al. Gene-expression profiling elucidates molecular signaling networks that can be therapeutically targeted in vestibular schwannoma. Journal of neurosurgery. 2014:1–100. doi: 10.3171/2014.6.JNS131433. Epub 2014/09/24. [DOI] [PubMed] [Google Scholar]
- 107.James MF, Han S, Polizzano C, Plotkin SR, Manning BD, Stemmer-Rachamimov AO, et al. NF2/merlin is a novel negative regulator of mTOR complex 1, and activation of mTORC1 is associated with meningioma and schwannoma growth. Molecular and cellular biology. 2009;29(15):4250–61. doi: 10.1128/MCB.01581-08. Epub 2009/05/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Giovannini M, Bonne NX, Vitte J, Chareyre F, Tanaka K, Adams R, et al. mTORC1 inhibition delays growth of neurofibromatosis type 2 schwannoma. Neuro-oncology. 2014;16(4):493–504. doi: 10.1093/neuonc/not242. Epub 2014/01/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer research. 2006;66(3):1500–8. doi: 10.1158/0008-5472.CAN-05-2925. Epub 2006/02/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cloughesy TF, Yoshimoto K, Nghiemphu P, Brown K, Dang J, Zhu S, et al. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS medicine. 2008;5(1):e8. doi: 10.1371/journal.pmed.0050008. Epub 2008/01/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.James MF, Stivison E, Beauchamp R, Han S, Li H, Wallace MR, et al. Regulation of mTOR complex 2 signaling in neurofibromatosis 2-deficient target cell types. Molecular cancer research : MCR. 2012;10(5):649–59. doi: 10.1158/1541-7786.MCR-11-0425-T. Epub 2012/03/20. [DOI] [PubMed] [Google Scholar]
- 112.Petrilli A, Bott M, Fernandez-Valle C. Inhibition of SIRT2 in merlin/NF2-mutant Schwann cells triggers necrosis. Oncotarget. 2013;4(12):2354–65. doi: 10.18632/oncotarget.1422. Epub 2013/11/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Muranen T, Gronholm M, Lampin A, Lallemand D, Zhao F, Giovannini M, et al. The tumor suppressor merlin interacts with microtubules and modulates Schwann cell microtubule cytoskeleton. Human molecular genetics. 2007;16(14):1742–51. doi: 10.1093/hmg/ddm122. Epub 2007/06/15. [DOI] [PubMed] [Google Scholar]
- 114.Bensenor LB, Barlan K, Rice SE, Fehon RG, Gelfand VI. Microtubule-mediated transport of the tumor-suppressor protein Merlin and its mutants. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(16):7311–6. doi: 10.1073/pnas.0907389107. Epub 2010/04/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pan D. The hippo signaling pathway in development and cancer. Developmental cell. 2010;19(4):491–505. doi: 10.1016/j.devcel.2010.09.011. Epub 2010/10/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Piccolo S, Dupont S, Cordenonsi M. The Biology of YAP/TAZ: Hippo Signaling and Beyond. Physiological reviews. 2014;94(4):1287–312. doi: 10.1152/physrev.00005.2014. Epub 2014/10/08. [DOI] [PubMed] [Google Scholar]
- 117.Kodaka M, Hata Y. The mammalian Hippo pathway: regulation and function of YAP1 and TAZ. Cellular and molecular life sciences : CMLS. 2014 doi: 10.1007/s00018-014-1742-9. Epub 2014/10/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Barron DA, Kagey JD. The role of the Hippo pathway in human disease and tumorigenesis. Clinical and translational medicine. 2014;3:25. doi: 10.1186/2001-1326-3-25. Epub 2014/08/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Boin A, Couvelard A, Couderc C, Brito I, Filipescu D, Kalamarides M, et al. Proteomic screening identifies a YAP-driven signaling network linked to tumor cell proliferation in human schwannomas. Neuro-oncology. 2014;16(9):1196–209. doi: 10.1093/neuonc/nou020. Epub 2014/02/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Striedinger K, VandenBerg SR, Baia GS, McDermott MW, Gutmann DH, Lal A. The neurofibromatosis 2 tumor suppressor gene product, merlin, regulates human meningioma cell growth by signaling through YAP. Neoplasia. 2008;10(11):1204–12. doi: 10.1593/neo.08642. Epub 2008/10/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Li W, You L, Cooper J, Schiavon G, Pepe-Caprio A, Zhou L, et al. Merlin/NF2 suppresses tumorigenesis by inhibiting the E3 ubiquitin ligase CRL4(DCAF1) in the nucleus. Cell. 2010;140(4):477–90. doi: 10.1016/j.cell.2010.01.029. Epub 2010/02/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mori T, Gotoh S, Shirakawa M, Hakoshima T. Structural basis of DDB1-and-Cullin 4-associated Factor 1 (DCAF1) recognition by merlin/NF2 and its implication in tumorigenesis by CD44-mediated inhibition of merlin suppression of DCAF1 function. Genes to cells : devoted to molecular & cellular mechanisms. 2014;19(8):603–19. doi: 10.1111/gtc.12161. Epub 2014/06/11. [DOI] [PubMed] [Google Scholar]
- 123.Cooper J, Li W, You L, Schiavon G, Pepe-Caprio A, Zhou L, et al. Merlin/NF2 functions upstream of the nuclear E3 ubiquitin ligase CRL4DCAF1 to suppress oncogenic gene expression. Science signaling. 2011;4(188):pt6. doi: 10.1126/scisignal.2002314. Epub 2011/09/01. [DOI] [PubMed] [Google Scholar]
- 124.Li W, Cooper J, Zhou L, Yang C, Erdjument-Bromage H, Zagzag D, et al. Merlin/NF2 loss-driven tumorigenesis linked to CRL4(DCAF1)-mediated inhibition of the hippo pathway kinases Lats1 and 2 in the nucleus. Cancer cell. 2014;26(1):48–60. doi: 10.1016/j.ccr.2014.05.001. Epub 2014/07/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yin F, Yu J, Zheng Y, Chen Q, Zhang N, Pan D. Spatial organization of Hippo signaling at the plasma membrane mediated by the tumor suppressor Merlin/NF2. Cell. 2013;154(6):1342–55. doi: 10.1016/j.cell.2013.08.025. Epub 2013/09/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Flaiz C, Utermark T, Parkinson DB, Poetsch A, Hanemann CO. Impaired intercellular adhesion and immature adherens junctions in merlin-deficient human primary schwannoma cells. Glia. 2008;56(5):506–15. doi: 10.1002/glia.20629. Epub 2008/02/02. [DOI] [PubMed] [Google Scholar]
- 127.Low BC, Pan CQ, Shivashankar GV, Bershadsky A, Sudol M, Sheetz M. YAP/TAZ as mechanosensors and mechanotransducers in regulating organ size and tumor growth. FEBS letters. 2014;588(16):2663–70. doi: 10.1016/j.febslet.2014.04.012. Epub 2014/04/22. [DOI] [PubMed] [Google Scholar]
- 128.Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes & development. 2007;21(21):2747–61. doi: 10.1101/gad.1602907. Epub 2007/11/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474(7350):179–83. doi: 10.1038/nature10137. Epub 2011/06/10. [DOI] [PubMed] [Google Scholar]
- 130.Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N, et al. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell. 2013;154(5):1047–59. doi: 10.1016/j.cell.2013.07.042. Epub 2013/08/21. [DOI] [PubMed] [Google Scholar]
- 131.Wada K, Itoga K, Okano T, Yonemura S, Sasaki H. Hippo pathway regulation by cell morphology and stress fibers. Development. 2011;138(18):3907–14. doi: 10.1242/dev.070987. Epub 2011/08/13. [DOI] [PubMed] [Google Scholar]
- 132.Boggiano JC, Fehon RG. Growth control by committee: intercellular junctions, cell polarity, and the cytoskeleton regulate Hippo signaling. Developmental cell. 2012;22(4):695–702. doi: 10.1016/j.devcel.2012.03.013. Epub 2012/04/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Robinson BS, Moberg KH. Cell-cell junctions: alpha-catenin and E-cadherin help fence in Yap1. Current biology : CB. 2011;21(21):R890–2. doi: 10.1016/j.cub.2011.09.019. Epub 2011/11/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Schroeder MC, Halder G. Regulation of the Hippo pathway by cell architecture and mechanical signals. Seminars in cell & developmental biology. 2012;23(7):803–11. doi: 10.1016/j.semcdb.2012.06.001. Epub 2012/07/04. [DOI] [PubMed] [Google Scholar]
- 135.Yi C, Shen Z, Stemmer-Rachamimov A, Dawany N, Troutman S, Showe LC, et al. The p130 isoform of angiomotin is required for Yap-mediated hepatic epithelial cell proliferation and tumorigenesis. Science signaling. 2013;6(291):ra77. doi: 10.1126/scisignal.2004060. Epub 2013/09/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Yi C, Troutman S, Fera D, Stemmer-Rachamimov A, Avila JL, Christian N, et al. A tight junction-associated Merlin-angiomotin complex mediates Merlin’s regulation of mitogenic signaling and tumor suppressive functions. Cancer cell. 2011;19(4):527–40. doi: 10.1016/j.ccr.2011.02.017. Epub 2011/04/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Pass HI, Vogelzang N, Hahn S, Carbone M. Malignant pleural mesothelioma. Current problems in cancer. 2004;28(3):93–174. doi: 10.1016/j.currproblcancer.2004.04.001. Epub 2004/06/16. [DOI] [PubMed] [Google Scholar]
- 138.Sekido Y, Pass HI, Bader S, Mew DJ, Christman MF, Gazdar AF, et al. Neurofibromatosis type 2 (NF2) gene is somatically mutated in mesothelioma but not in lung cancer. Cancer research. 1995;55(6):1227–31. Epub 1995/03/15. [PubMed] [Google Scholar]
- 139.Cheng JQ, Lee WC, Klein MA, Cheng GZ, Jhanwar SC, Testa JR. Frequent mutations of NF2 and allelic loss from chromosome band 22q12 in malignant mesothelioma: evidence for a two-hit mechanism of NF2 inactivation. Genes, chromosomes & cancer. 1999;24(3):238–42. Epub 1999/08/19. [PubMed] [Google Scholar]
- 140.Baser ME, De Rienzo A, Altomare D, Balsara BR, Hedrick NM, Gutmann DH, et al. Neurofibromatosis 2 and malignant mesothelioma. Neurology. 2002;59(2):290–1. doi: 10.1212/wnl.59.2.290. Epub 2002/07/24. [DOI] [PubMed] [Google Scholar]
- 141.Thurneysen C, Opitz I, Kurtz S, Weder W, Stahel RA, Felley-Bosco E. Functional inactivation of NF2/merlin in human mesothelioma. Lung cancer. 2009;64(2):140–7. doi: 10.1016/j.lungcan.2008.08.014. Epub 2008/10/07. [DOI] [PubMed] [Google Scholar]
- 142.Fleury-Feith J, Lecomte C, Renier A, Matrat M, Kheuang L, Abramowski V, et al. Hemizygosity of Nf2 is associated with increased susceptibility to asbestos-induced peritoneal tumours. Oncogene. 2003;22(24):3799–805. doi: 10.1038/sj.onc.1206593. Epub 2003/06/13. [DOI] [PubMed] [Google Scholar]
- 143.Altomare DA, Vaslet CA, Skele KL, De Rienzo A, Devarajan K, Jhanwar SC, et al. A mouse model recapitulating molecular features of human mesothelioma. Cancer research. 2005;65(18):8090–5. doi: 10.1158/0008-5472.CAN-05-2312. Epub 2005/09/17. [DOI] [PubMed] [Google Scholar]
- 144.Xiao GH, Gallagher R, Shetler J, Skele K, Altomare DA, Pestell RG, et al. The NF2 tumor suppressor gene product, merlin, inhibits cell proliferation and cell cycle progression by repressing cyclin D1 expression. Molecular and cellular biology. 2005;25(6):2384–94. doi: 10.1128/MCB.25.6.2384-2394.2005. Epub 2005/03/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Altomare DA, You H, Xiao GH, Ramos-Nino ME, Skele KL, De Rienzo A, et al. Human and mouse mesotheliomas exhibit elevated AKT/PKB activity, which can be targeted pharmacologically to inhibit tumor cell growth. Oncogene. 2005;24(40):6080–9. doi: 10.1038/sj.onc.1208744. Epub 2005/05/18. [DOI] [PubMed] [Google Scholar]
- 146.Lallemand D, Curto M, Saotome I, Giovannini M, McClatchey AI. NF2 deficiency promotes tumorigenesis and metastasis by destabilizing adherens junctions. Genes & development. 2003;17(9):1090–100. doi: 10.1101/gad.1054603. Epub 2003/04/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yokoyama T, Osada H, Murakami H, Tatematsu Y, Taniguchi T, Kondo Y, et al. YAP1 is involved in mesothelioma development and negatively regulated by Merlin through phosphorylation. Carcinogenesis. 2008;29(11):2139–46. doi: 10.1093/carcin/bgn200. Epub 2008/08/30. [DOI] [PubMed] [Google Scholar]
- 148.Murakami H, Mizuno T, Taniguchi T, Fujii M, Ishiguro F, Fukui T, et al. LATS2 is a tumor suppressor gene of malignant mesothelioma. Cancer research. 2011;71(3):873–83. doi: 10.1158/0008-5472.CAN-10-2164. Epub 2011/01/20. [DOI] [PubMed] [Google Scholar]
- 149.Smole Z, Thoma CR, Applegate KT, Duda M, Gutbrodt KL, Danuser G, et al. Tumor suppressor NF2/Merlin is a microtubule stabilizer. Cancer research. 2014;74(1):353–62. doi: 10.1158/0008-5472.CAN-13-1334. Epub 2013/11/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Poulikakos PI, Xiao GH, Gallagher R, Jablonski S, Jhanwar SC, Testa JR. Re-expression of the tumor suppressor NF2/merlin inhibits invasiveness in mesothelioma cells and negatively regulates FAK. Oncogene. 2006;25(44):5960–8. doi: 10.1038/sj.onc.1209587. Epub 2006/05/03. [DOI] [PubMed] [Google Scholar]
- 151.McClatchey AI, Saotome I, Mercer K, Crowley D, Gusella JF, Bronson RT, et al. Mice heterozygous for a mutation at the Nf2 tumor suppressor locus develop a range of highly metastatic tumors. Genes & development. 1998;12(8):1121–33. doi: 10.1101/gad.12.8.1121. Epub 1998/05/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bianchi AB, Hara T, Ramesh V, Gao J, Klein-Szanto AJ, Morin F, et al. Mutations in transcript isoforms of the neurofibromatosis 2 gene in multiple human tumour types. Nature genetics. 1994;6(2):185–92. doi: 10.1038/ng0294-185. Epub 1994/02/01. [DOI] [PubMed] [Google Scholar]
- 153.Yoo NJ, Park SW, Lee SH. Mutational analysis of tumour suppressor gene NF2 in common solid cancers and acute leukaemias. Pathology. 2012;44(1):29–32. doi: 10.1097/PAT.0b013e32834c3599. Epub 2011/11/15. [DOI] [PubMed] [Google Scholar]
- 154.Baylin SB, Herman JG, Graff JR, Vertino PM, Issa JP. Alterations in DNA methylation: a fundamental aspect of neoplasia. Advances in cancer research. 1998;72:141–96. Epub 1997/10/24. [PubMed] [Google Scholar]
- 155.Esteller M. CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene. 2002;21(35):5427–40. doi: 10.1038/sj.onc.1205600. Epub 2002/08/03. [DOI] [PubMed] [Google Scholar]
- 156.Larsson J, Ohishi M, Garrison B, Aspling M, Janzen V, Adams GB, et al. Nf2/merlin regulates hematopoietic stem cell behavior by altering microenvironmental architecture. Cell stem cell. 2008;3(2):221–7. doi: 10.1016/j.stem.2008.06.005. Epub 2008/08/07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ramalho-Santos M, Yoon S, Matsuzaki Y, Mulligan RC, Melton DA. “Stemness”: transcriptional profiling of embryonic and adult stem cells. Science. 2002;298(5593):597–600. doi: 10.1126/science.1072530. Epub 2002/09/14. [DOI] [PubMed] [Google Scholar]
- 158.Hiemer SE, Varelas X. Stem cell regulation by the Hippo pathway. Biochimica et biophysica acta. 2013;1830(2):2323–34. doi: 10.1016/j.bbagen.2012.07.005. Epub 2012/07/25. [DOI] [PubMed] [Google Scholar]
- 159.Ramos A, Camargo FD. The Hippo signaling pathway and stem cell biology. Trends in cell biology. 2012;22(7):339–46. doi: 10.1016/j.tcb.2012.04.006. Epub 2012/06/05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zhao B, Tumaneng K, Guan KL. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nature cell biology. 2011;13(8):877–83. doi: 10.1038/ncb2303. Epub 2011/08/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Yaegashi S, Sachse R, Ohuchi N, Mori S, Sekiya T. Low incidence of a nucleotide sequence alteration of the neurofibromatosis 2 gene in human breast cancers. Japanese journal of cancer research : Gann. 1995;86(10):929–33. doi: 10.1111/j.1349-7006.1995.tb03003.x. Epub 1995/10/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Allison KH, Sledge GW., Jr Heterogeneity and Cancer. Oncology. 2014;28(9) Epub 2014/09/17. [PubMed] [Google Scholar]
- 163.Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314(5797):268–74. doi: 10.1126/science.1133427. Epub 2006/09/09. [DOI] [PubMed] [Google Scholar]
- 164.Shrestha Y, Schafer EJ, Boehm JS, Thomas SR, He F, Du J, et al. PAK1 is a breast cancer oncogene that coordinately activates MAPK and MET signaling. Oncogene. 2012;31(29):3397–408. doi: 10.1038/onc.2011.515. Epub 2011/11/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Arakawa H, Hayashi N, Nagase H, Ogawa M, Nakamura Y. Alternative splicing of the NF2 gene and its mutation analysis of breast and colorectal cancers. Human molecular genetics. 1994;3(4):565–8. doi: 10.1093/hmg/3.4.565. Epub 1994/04/01. [DOI] [PubMed] [Google Scholar]
- 166.Rustgi AK, Xu L, Pinney D, Sterner C, Beauchamp R, Schmidt S, et al. Neurofibromatosis 2 gene in human colorectal cancer. Cancer genetics and cytogenetics. 1995;84(1):24–6. doi: 10.1016/0165-4608(95)00059-3. Epub 1995/10/01. [DOI] [PubMed] [Google Scholar]
- 167.Cacev T, Aralica G, Loncar B, Kapitanovic S. Loss of NF2/Merlin expression in advanced sporadic colorectal cancer. Cellular oncology. 2014;37(1):69–77. doi: 10.1007/s13402-013-0164-2. Epub 2013/12/11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Pineau P, Marchio A, Nagamori S, Seki S, Tiollais P, Dejean A. Homozygous deletion scanning in hepatobiliary tumor cell lines reveals alternative pathways for liver carcinogenesis. Hepatology. 2003;37(4):852–61. doi: 10.1053/jhep.2003.50138. Epub 2003/04/02. [DOI] [PubMed] [Google Scholar]
- 169.Benhamouche S, Curto M, Saotome I, Gladden AB, Liu CH, Giovannini M, et al. Nf2/Merlin controls progenitor homeostasis and tumorigenesis in the liver. Genes & development. 2010;24(16):1718–30. doi: 10.1101/gad.1938710. Epub 2010/08/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, et al. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer cell. 2009;16(5):425–38. doi: 10.1016/j.ccr.2009.09.026. Epub 2009/11/03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Song H, Mak KK, Topol L, Yun K, Hu J, Garrett L, et al. Mammalian Mst1 and Mst2 kinases play essential roles in organ size control and tumor suppression. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(4):1431–6. doi: 10.1073/pnas.0911409107. Epub 2010/01/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lu L, Li Y, Kim SM, Bossuyt W, Liu P, Qiu Q, et al. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(4):1437–42. doi: 10.1073/pnas.0911427107. Epub 2010/01/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Zhang N, Bai H, David KK, Dong J, Zheng Y, Cai J, et al. The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Developmental cell. 2010;19(1):27–38. doi: 10.1016/j.devcel.2010.06.015. Epub 2010/07/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Kawana Y, Ichikawa T, Suzuki H, Ueda T, Komiya A, Ichikawa Y, et al. Loss of heterozygosity at 7q31.1 and 12p13-12 in advanced prostate cancer. The Prostate. 2002;53(1):60–4. doi: 10.1002/pros.10131. Epub 2002/09/05. [DOI] [PubMed] [Google Scholar]
- 175.Malhotra A, Shibata Y, Hall IM, Dutta A. Chromosomal structural variations during progression of a prostate epithelial cell line to a malignant metastatic state inactivate the NF2, NIPSNAP1, UGT2B17, and LPIN2 genes. Cancer biology & therapy. 2013;14(9):840–52. doi: 10.4161/cbt.25329. Epub 2013/06/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Horiguchi A, Zheng R, Shen R, Nanus DM. Inactivation of the NF2 tumor suppressor protein merlin in DU145 prostate cancer cells. The Prostate. 2008;68(9):975–84. doi: 10.1002/pros.20760. Epub 2008/03/26. [DOI] [PubMed] [Google Scholar]
- 177.Lau YK, Murray LB, Houshmandi SS, Xu Y, Gutmann DH, Yu Q. Merlin is a potent inhibitor of glioma growth. Cancer research. 2008;68(14):5733–42. doi: 10.1158/0008-5472.CAN-08-0190. Epub 2008/07/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Morales FC, Molina JR, Hayashi Y, Georgescu MM. Overexpression of ezrin inactivates NF2 tumor suppressor in glioblastoma. Neuro-oncology. 2010;12(6):528–39. doi: 10.1093/neuonc/nop060. Epub 2010/02/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Guerrero PA, Yin W, Camacho L, Marchetti D. Oncogenic role of Merlin/NF2 in glioblastoma. Oncogene. 2014 doi: 10.1038/onc.2014.185. Epub 2014/07/22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Sheikh HA, Tometsko M, Niehouse L, Aldeeb D, Swalsky P, Finkelstein S, et al. Molecular genotyping of medullary thyroid carcinoma can predict tumor recurrence. The American journal of surgical pathology. 2004;28(1):101–6. doi: 10.1097/00000478-200401000-00012. Epub 2004/01/07. [DOI] [PubMed] [Google Scholar]
- 181.Murray LB, Lau YK, Yu Q. Merlin is a negative regulator of human melanoma growth. PloS one. 2012;7(8):e43295. doi: 10.1371/journal.pone.0043295. Epub 2012/08/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Frontzek F, Nitzlaff S, Horstmann M, Schwab A, Stock C. Functional interdependence of NHE1 and merlin in human melanoma cells. Biochemistry and cell biology = Biochimie et biologie cellulaire. 2014:1–11. doi: 10.1139/bcb-2014-0041. Epub 2014/10/03. [DOI] [PubMed] [Google Scholar]
- 183.Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343(6166):84–7. doi: 10.1126/science.1247005. Epub 2013/12/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Reinhold WC, Varma S, Sousa F, Sunshine M, Abaan OD, Davis SR, et al. NCI-60 whole exome sequencing and pharmacological CellMiner analyses. PloS one. 2014;9(7):e101670. doi: 10.1371/journal.pone.0101670. Epub 2014/07/18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483(7391):570–5. doi: 10.1038/nature11005. Epub 2012/03/31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic acids research. 2013;41(Database issue):D955–61. doi: 10.1093/nar/gks1111. Epub 2012/11/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468(7326):973–7. doi: 10.1038/nature09626. Epub 2010/11/26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Sabha N, Au K, Agnihotri S, Singh S, Mangat R, Guha A, et al. Investigation of the in vitro therapeutic efficacy of nilotinib in immortalized human NF2-null vestibular schwannoma cells. PloS one. 2012;7(6):e39412. doi: 10.1371/journal.pone.0039412. Epub 2012/06/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Mukherjee J, Kamnasaran D, Balasubramaniam A, Radovanovic I, Zadeh G, Kiehl TR, et al. Human schwannomas express activated platelet-derived growth factor receptors and c-kit and are growth inhibited by Gleevec (Imatinib Mesylate) Cancer research. 2009;69(12):5099–107. doi: 10.1158/0008-5472.CAN-08-4475. Epub 2009/06/11. [DOI] [PubMed] [Google Scholar]
- 190.Ammoun S, Schmid MC, Triner J, Manley P, Hanemann CO. Nilotinib alone or in combination with selumetinib is a drug candidate for neurofibromatosis type 2. Neuro-oncology. 2011;13(7):759–66. doi: 10.1093/neuonc/nor056. Epub 2011/07/06. [DOI] [PMC free article] [PubMed] [Google Scholar]