

Abstract
Background
Adolescent obesity is predictive of future weight gain, obesity, and adult-onset severe obesity (body mass index (BMI) ≥40kg/m2). Despite successful efforts to identify SNPs influencing BMI, <5% of the 40-80% heritability of the phenotype has been explained. Identification of gene-gene (G-G) interactions between known variants can help explain this hidden heritability, as well as identify potential biological mechanisms affecting weight gain during this critical developmental period.
Objective
We have recently shown distinct genetic effects on BMI across the life course, and thus it is important to examine evidence for epistasis in adolescence.
Methods
In adolescent participants of European descent (EA) from wave II of the National Longitudinal Study of Adolescent Health (Add Health, N = 5072, ages 12-21, 52.5% female), we tested 34 established BMI-related SNPs for G-G interaction effects on BMI Z-score. We used mixed-effects regression, assuming multiplicative interaction models adjusting for age, sex, and geographic region, with random effects for family and school.
Results
For 28 G-G interactions that were nominally significant (p < 0.05), we attempted to replicate our results in an adolescent sample from the Childhood European American Cohort from Philadelphia (CHOP). In the replication study, one interaction (PRKD1-FTO) was significant after correction for multiple testing.
Conclusions
Our results are suggestive of epistatic effects on BMI during adolescence and point to potentially interactive effects between genes in biological pathways important in obesity.
Keywords: genetics, BMI, obesity, epistasis, interaction, adolescence
INTRODUCTION
Adolescence is a high-risk period for weight gain, and adolescent body mass index (BMI) has been shown to influence adult obesity risk 1,2. BMI is a complex phenotype under the influence of multiple genes 3, behaviors 4-6, and environments 7. While BMI has been estimated to be 40-80% heritable 8, genetic variants identified through genome-wide association studies (GWAS) account for <5% of the variance of this adiposity measure 3. This is partly because other mechanisms may influence the heritability of complex traits such as BMI, including epigenetics, gene-environment (G-E), and gene-gene (G-G) interactions 9,10. Identifying such factors has the potential to contribute to our understanding of the heritability of complex traits, as well as understanding potential underlying biological pathways involving multiple genes 11.
Research to date on the influence of epistasis on obesity risk has been inconclusive. For obesity-related traits in adult samples, few studies have examined the effect of G-G interactions on BMI. In bulimic women, G-G interaction has been reported between an established BMI variant (BDNF) and DRD4, where subjects with both the BDNF*Met66 and DRD4*7R alleles had a significantly higher BMI than subjects lacking those variants 12. Two Chinese studies reported G-G interactions for both overall obesity risk and central adiposity 13,14. On the other hand, a genome wide association (GWA) scan for the effect of epistasis on BMI in four European cohorts, no G-G interactions reached genome-wide significance, and of the eight suggestive G-G interactions identified, none were replicated in all four cohorts, nor in the larger replication cohort (N=5173) 8. Speliotes et al. (2010) reported 33 nominally significant G-G interactions for their 32 BMI loci, but noted that none were significant after multiple-test correction.
We have recently shown distinct genetic effects on BMI in adolescence 15,16. Given that genetic effects on BMI vary with age, it is important to examine the genetic architecture of BMI (including epistasis) across the lifecourse. To our knowledge, ours is the first study to examine the effects of G-G interaction among established BMI variants on the risk of increased BMI during adolescence, a critical period for weight gain.
METHODS
Discovery Sample
National Longitudinal Study of Adolescent Health (Add Health)
Add Health is a national, prospective cohort study of adolescents representative of the U.S. school-based population in grades 7 to 12 (11-22 years of age) in 1994-95 (wave I, n=20,745) who are followed over three waves into adulthood (wave II: 1996, 12-21 years (n=14,738); wave III: 2001-2002, 18-27 years (n=15,197); wave IV: 2008-2009, 23-32 years (n=15,701). DNA was first collected from all respondents at wave IV, and consent given for banking and use in future genetic studies (n = 12,234). Add Health included a core sample plus subsamples of selected minorities, related adolescents (n = 5,524), and other groups, including well-educated African Americans, collected under protocols approved by the Institutional Review Board at the University of North Carolina at Chapel Hill. The survey design and sampling strategy have been described previously 17,18.
Analytic Sample
At Wave IV, 64% (n = 6,919) of Wave I (n = 10,770) European-American (EA) respondents provided DNA samples with consent for banking and use in future genetic studies. For this study, 34 established BMI SNPs were genotyped with TaqMan (overall discordance rate across SNPs = 0.3%, average call-rate = 97.9%), using procedures described previously 15. Our eligibility criteria included each individual having at least 80% of the 34 established BMI SNPs successfully genotyped (n =6,596), and being between the ages of 12 and 21 years at either Waves II or III (n =5,285). Among the 5,285 eligible EA adolescents, we excluded the following participants: the monozygotic twin with fewer genotyped loci within each twin pair (n = 86), pregnant (n = 56) or disabled (n = 27) individuals, and those with missing data for geographic region (n = 40) or BMI (n = 1). The final analytic sample included 5,075 EA individuals.
BMI
BMI (kg/m2) was calculated from measured height and weight assessed at Waves II or III when participants were aged 12–21 years, with priority for younger age at measurement (Wave II: n = 4,812; Wave III: n = 263). In cases where respondents refused measurement or weighed more than the scale capacity, self-reported height and weight were used to estimate BMI (Wave II n = 93; Wave III n = 153). Given the heterogeneity in growth during the age range of our study (12-21 years), we used BMI Z-scores (Z-BMI) based on the CDC age- and sex-matched growth charts 19 as the outcome in all statistical analyses.
Statistical Modeling
Using a multiplicative interaction model, mixed-effects linear regression was performed to assess pairwise interaction (epistatic) effects between SNPs on BMI Z-score, adjusting for SNP main effects, sex and age with random effects for school and family, using STATA v13.1 (Stata Corp, College Station, TX). A Bonferroni significance threshold of 8.91E-05 (0.05/561) was applied to assess overall significance of individual findings.
Power
Post Hoc power calculations (Quanto v.1.2.4)20 demonstrated that, given our sample size in Add Health and assuming a minor allele frequency (MAF) of 0.5 for SNP 1 and 0.05 MAF for SNP 2, we have 14% power to detect an epistatic effect of 0.2 after correcting for multiple testing (0.05/561). As MAF for the second SNP increases, so does our power, but power is substantially decreased for smaller interaction effects. (Supplementary Table 1).
Replication Sample
CHOP cohort
We attempted to replicate G-G interactions with p<0.05 in Add Health using a sample of European-American adolescents (age 12-18) from the Childhood European American Cohort from Philadelphia study. The replication sample included 3,814 unrelated children of European ancestry with systematically recorded weight and height (52.2% female, mean age 14.74 (1.85)), excluding those with known syndromic obesity or other conditions that may influence BMI. All subjects were consecutively and randomly recruited from the greater metropolitan area of Philadelphia between 2006 and 2012 at the Children’s Hospital of Philadelphia (CHOP) (i.e., participants are not selected for any particular trait or treatment regime). The Institutional Review Board at CHOP approved the study. Parental informed consent was given for each participant for both blood collection and subsequent genotyping. SNP genotyping was performed, using the Illumina Infinium™ II HumanHap550 or Human 610 BeadChip technology (Illumina, San Diego), at the Children’s Hospital of Philadelphia’s Center for Applied Genomics, as described previously 21. Twenty-eight interactions that were nominally significant in the Add Health sample were carried forward for interrogation in the replication sample. Interactions were considered to show evidence for a nominally significant result in Add Health if the effect was directionally concordant, the p value from the replication analysis was significant after correction for multiple testing (0.05/28=1.79E-03), and there was no evidence for heterogeneity (I2>70) between samples in a meta-analysis of results between Add Health and CHOP.
RESULTS
Add Health discovery participants were a little older and heavier that the CHOP replication sample participants (Table 1). The genetic main effect of 15 of the 34 established BMI loci were replicated on BMI Z-score (p < 0.05) in the present analysis, including FTO, TMEM18, MC4R, and TFAP2B (Table 2).
Table 1.
Sample characteristics for Add Health1 and CHOP2.
| Add Health | CHOP | |||
|---|---|---|---|---|
| Mean (SD) | Males (N=2404) | Females (N=2671) | Males (N=1803) | Females (N=2011) |
| Age | 16.16 (1.83) | 16.44 (1.84) | 14.66 (1.88) | 14.82 (1.83) |
| BMI | 23.26 (4.90) | 22.84 (5.03) | 21.68 (4.59) | 22.02 (4.49) |
| Z-BMI | 0.3310 (1.16) | 0.2864 (1.04) | 0.3400 (1.12) | 0.3605 (0.98) |
Footnote: Add Health: National Longitudinal Study of Adolescent Health (sampled 1996-2002, ages 12-21, BMI 13.2-61.0);
CHOP: Childhood European American Cohort from Philadelphia (sampled 2006-2012, ages 12-18, BMI 11.1-44.2).
Table 2.
Main effects of risk alleles on BMI Z-score in Add Health.
| SNP | In/Near | Risk Allele | FEA | Main effect βhat(SE) | p |
|---|---|---|---|---|---|
| rs2444217 | ADCY9 | A | 0.57 | −0.02 (0.02) | 0.257 |
| rs10767664 | BDNF | A | 0.79 | 0.05 (0.03) | 0.041 |
| rs13078807 | CADM2 | G | 0.20 | 0.06 (0.03) | 0.042 |
| rs7647305 | ETV5 | C | 0.79 | 0.03 (0.03) | 0.279 |
| rs7138803 | FAIM2 | A | 0.38 | 0.02 (0.02) | 0.315 |
| rs887912 | FANCL | T | 0.28 | 0.06 (0.02) | 0.020 |
| rs2112347 | FLJ35779 | T | 0.63 | 0.02 (0.02) | 0.377 |
| rs9939609 | FTO | A | 0.39 | 0.16 (0.02) | 2.53E-13 |
| rs10938397 | GNPDA2 | G | 0.43 | 0.05 (0.02) | 0.041 |
| rs12444979 | GPRC5B | C | 0.86 | 0.06 (0.03) | 0.045 |
| rs29941 | KCTD15 | G | 0.68 | 0.03 (0.02) | 0.150 |
| rs867559 | LMX1B | G | 0.19 | 0.02 (0.03) | 0.366 |
| rs2890652 | LRP1B | C | 0.16 | 0.03 (0.03) | 0.241 |
| rs10968576 | LRRN6C | G | 0.31 | 0.01 (0.02) | 0.650 |
| rs543874 | LZTR2 | G | 0.20 | 0.08 (0.03) | 0.002 |
| rs2241423 | MAP2K5 | G | 0.77 | 0.06 (0.03) | 0.026 |
| rs571312 | MC4R | A | 0.23 | 0.10 (0.03) | 7.78E-05 |
| rs3817334 | MTCH2 | T | 0.40 | 0.03 (0.02) | 0.137 |
| rs4771122 | MTIF3 | G | 0.22 | 3.90E-04 (0.03) | 0.988 |
| rs1077393 | NCR3_BAT2 | G | 0.49 | −5.73E-04(0.02) | 0.979 |
| rs2568958 | NEGR1 | A | 0.63 | 0.04 (0.02) | 0.081 |
| rs10146997 | NRXN3 | G | 0.21 | −0.03 (0.03) | 0.219 |
| rs206936 | NUDT3 | G | 0.21 | 0.04 (0.03) | 0.099 |
| rs713586 | POMC | C | 0.48 | 0.06 (0.02) | 0.003 |
| rs11847697 | PRKD1 | T | 0.05 | 0.14 (0.05) | 0.004 |
| rs1555543 | PTBP2 | C | 0.59 | 0.05 (0.02) | 0.029 |
| rs2287019 | QPCTL | C | 0.81 | 0.03 (0.03) | 0.280 |
| rs4929949 | RPL27A | C | 0.51 | −0.02 (0.02) | 0.450 |
| rs4788102 | SH2B1 | A | 0.39 | 0.01 (0.02) | 0.615 |
| rs13107325 | SLC39A8 | T | 0.08 | 0.04 (0.04) | 0.328 |
| rs987237 | TFAP2B | G | 0.18 | 0.10 (0.03) | 2.44E-04 |
| rs6548238 | TMEM18 | C | 0.83 | 0.15 (0.03) | 1.12E-07 |
| rs1514175 | TNNI3K | A | 0.44 | 0.07 (0.02) | 0.002 |
| rs3810291 | ZC3H4 | A | 0.67 | 0.04 (0.02) | 0.123 |
|
| |||||
| FEA = Frequency of effect (risk) allele | |||||
Twenty-eight interactions were nominally significant in Add Health, which is what would be expected by chance (561*0.05=28.05) (Table 3). Only one of these interactions, LMX1B/MTIF3, remained significant after correction for multiple testing in the discovery sample (beta = -0.18 (0.04), p = 2.88E-05). However, this interaction was not significant in CHOP (beta = 1.49E-03 (0.06), p=0.979). Seventeen of the 28 nominally significant interactions in Add Health were directionally consistent in the CHOP replication sample (binomial one-sided p = 0.08). Of the 28 interactions examined in the meta-analysis, seven had I2 values >70 – indicative of heterogeneity of effects between studies, including the strongest result in the Add Health cohort (LMX1B-MTIF3: I2=85.8). However, one interaction was significant in the replication analysis after correction for multiple testing and had I2 <30 in the meta-analysis (PRKD1-FTO: beta =-0.20 (0.05), p = 1.27E-04, I2=27.5).
Table 3.
Interaction effects1 for nominally significant G-G interactions on BMI Z-score in Add Health, CHOP, and meta-analysis.
| SNP1/SNP2 | Add Health (N=5075) | CHOP (N=3814) | Meta-analysis (N= 8889) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Beta (SE) | p | Beta (SE) | p | Beta (SE) | p | Direction | Het ISq | HetPVal | |
| LMX1B/MTIF3 | −0.18 (0.04) | 2.88E-05 | 1.49E-03 (0.06) | 0.979 | −0.12 (0.03) | 2.82E-04 | −+ | 85.8 | 0.008 |
| TNNI3K/LRP1B | −0.12 (0.04) | 0.005 | −0.04 (0.05) | 0.459 | −0.09 (0.03) | 0.005 | − − | 42.6 | 0.187 |
| SLC39A8/FTO | 0.15 (0.06) | 0.007 | −0.05 (0.06) | 0.417 | 0.05 (0.04) | 0.257 | +− | 82.2 | 0.018 |
| POMC/LMX1B | −0.10 (0.04) | 0.008 | −0.07 (0.04) | 0.119 | −0.09 (0.03) | 0.004 | − − | 0 | 0.617 |
| LZTR2/KCTD15 | 0.10 (0.04) | 0.009 | 0.04 (0.05) | 0.422 | 0.07 (0.03) | 0.015 | ++ | 0 | 0.325 |
| CADM2/NRXN3 | −0.11 (0.05) | 0.018 | 0.01 (0.05) | 0.905 | −0.06 (0.04) | 0.123 | −+ | 59.7 | 0.115 |
| LRP1B/NRXN3 | −0.12 (0.05) | 0.019 | −0.13 (0.06) | 0.037 | −0.13 (0.04) | 1.48E-03 | − − | 0 | 0.866 |
| TFAP2B/MAP2K5 | 0.11 (0.05) | 0.020 | 0.02 (0.05) | 0.740 | 0.06 (0.04) | 0.072 | ++ | 41.5 | 0.191 |
| ETV5/LRRN6C | 0.09 (0.04) | 0.027 | −0.04 (0.05) | 0.345 | 0.03 (0.03) | 0.271 | +− | 78.9 | 0.029 |
| FANCL/MTCH2 | −0.07 (0.03) | 0.028 | 0.01 (0.04) | 0.844 | −0.04 (0.02) | 0.087 | −+ | 60.8 | 0.110 |
| PRKD1/FTO | −0.14 (0.07) | 0.029 | −0.27 (0.08) | 0.001 | −0.19 (0.05) | 2.81E-04 | − − | 27.5 | 0.240 |
| MTCH2/MC4R | 0.08 (0.04) | 0.030 | 3.05E-03 (0.04) | 0.941 | 0.04 (0.03) | 0.136 | ++ | 44.1 | 0.181 |
| LZTR2/MAP2K5 | 0.09 (0.04) | 0.030 | −0.04 (0.05) | 0.484 | 0.04 (0.03) | 0.172 | +− | 72.9 | 0.055 |
| TNNI3K/TFAP2B | 0.08 (0.04) | 0.031 | 0.07 (0.04) | 0.087 | 0.08 (0.03) | 0.009 | ++ | 0 | 0.906 |
| GNPDA2/KCTD15 | −0.07 (0.03) | 0.031 | −0.01 (0.04) | 0.822 | −0.05 (0.02) | 0.051 | − − | 40.4 | 0.195 |
| NEGR1/KCTD15 | −0.07 (0.03) | 0.032 | 0.02 (0.04) | 0.671 | −0.04 (0.02) | 0.124 | −+ | 69.3 | 0.071 |
| NEGR1/POMC | 0.07 (0.03) | 0.033 | 0.01 (0.04) | 0.776 | 0.04 (0.02) | 0.050 | ++ | 40.4 | 0.195 |
| FANCL/MTIF3 | −0.08 (0.04) | 0.035 | 0.07 (0.05) | 0.134 | −0.02 (0.03) | 0.589 | −+ | 83.2 | 0.015 |
| NUDT3/MAP2K5 | 0.09 (0.04) | 0.036 | 3.59E-04 (0.05) | 0.994 | 0.05 (0.03) | 0.081 | ++ | 50.3 | 0.156 |
| LZTR2/MC4R | 0.09 (0.04) | 0.039 | −0.02 (0.05) | 0.683 | 0.05 (0.03) | 0.120 | +− | 64.5 | 0.093 |
| GNPDA2/NRXN3 | −0.08 (0.04) | 0.039 | 0.02 (0.04) | 0.734 | −0.04 (0.03) | 0.210 | −+ | 60.7 | 0.111 |
| NCR3_BAT2/QPCTL | 0.08 (0.04) | 0.040 | 0.03 (0.04) | 0.430 | 0.06 (0.03) | 0.046 | ++ | 0 | 0.429 |
| ETV5/NUDT3 | −0.09 (0.05) | 0.041 | 0.10 (0.05) | 0.047 | 2.80E-03 (0.04) | 0.938 | −+ | 86 | 0.007 |
| NUDT3/KCTD15 | −0.08 (0.04) | 0.045 | −0.02 (0.05) | 0.681 | −0.05 (0.03) | 0.074 | − − | 0 | 0.322 |
| FANCL/ETV5 | 0.08 (0.04) | 0.048 | 0.01 (0.05) | 0.908 | 0.05 (0.03) | 0.110 | ++ | 31.6 | 0.227 |
| POMC/GNPDA2 | −0.06 (0.03) | 0.048 | 0.04 (0.04) | 0.302 | −0.02 (0.02) | 0.407 | −+ | 77.2 | 0.036 |
| CADM2/MC4R | −0.09 (0.05) | 0.049 | 9.85E-04 (0.05) | 0.984 | −0.04 (0.04) | 0.209 | −+ | 39.8 | 0.197 |
| MTCH2/ZC3H4 | −0.06 (0.03) | 0.050 | −0.04 (0.04) | 0.332 | −0.05 (0.02) | 0.030 | − − | 0 | 0.647 |
Footnote: Mixed effects model, , Betas shown in table refer to βSNP1×SNP2.
The interaction for PRKD1-FTO relative to BMI Z-score is shown in Figure 1. The FTO risk allele (A) by itself is positively associated with Z-BMI, with each additional copy increasing Z-BMI by 0.175 units. The addition of a single PRKD1 risk allele to this model attenuates the influence of the FTO risk allele, so that each additional copy increases Z-BMI by only 0.031 units. The addition of two PRKD1 risk alleles has a more dramatic impact, so that each additional copy of the FTO risk allele decreases Z-BMI by 0.114 units.
Figure 1.
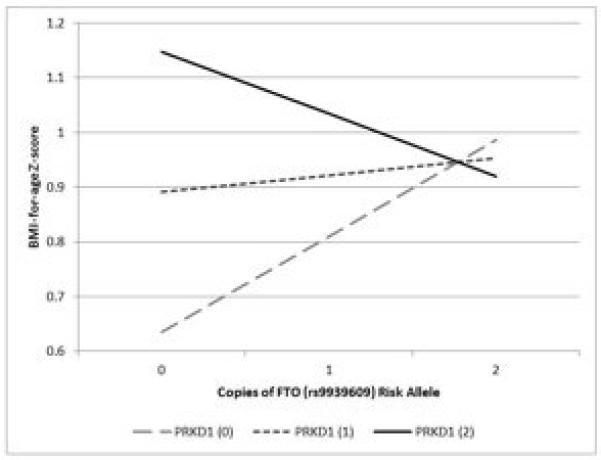
Effect of interaction1 between PRKD1 and FTO2 on BMI-for-age Z-score in adolescence.
Footnote: 1Mixed effects model, BMI = β + βSNP1xSNP2 + βSNP1 + βSNP2 + βage + βsex + f + s + ε
2PRKD1 (rs11847697) risk allele (T), FTO (rs9939609) risk allele (A) – Dashed grey line: Main effect of FTO risk allele on Z-BMI with no influence from PRKD1 risk allele. Dotted dark grey line: Effect of 1 PRKD1 risk allele on the relationship between FTO and Z-BMI. Solid black line: Effect of 2 PRKD1 risk alleles on the FTO/Z-BMI association.
DISCUSSION
Previous work by our team 15,16 demonstrates that variants associated with adult BMI, also influence body mass index earlier in the lifecourse. In the present study, main effects were generally directionally consistent with published literature 3,22, with the exception of SNPs in/near NRXN3, RPL27A, NCR3_BAT2, and ADCY9, which showed generally null effects on Z-BMI in our sample. G-G interactions are expected to play a role in the genetic architecture of common, complex diseases, including obesity. Such interactions could perhaps account for some of the missing heritability in these traits, and point to potential underlying biological pathways and mechanisms influencing overall obesity risk. The purpose of our study was to examine the estimated effect of G-G interaction of established BMI variants on risk of increased BMI Z-score in adolescence.
Seven out of 28 of the nominally significant interactions in Add Health showed evidence of heterogeneity in the meta-analysis (I2>70), suggesting that these interactions are either null (high I2 due to “winner’s curse” of pairing results from CHOP to only the nominally significant results in Add Health) or strongly influenced by differences in sampling strategy and/or age between the discovery (nationally-representative; mean age ~16 years) and replication samples (geographically restricted; mean age ~14.5 years). However, the G-G interaction between PRKD1 and FTO (shown in Figure 1) was statistically significant after correction for multiple testing in CHOP and had consistent direction of effect between the discovery and replication samples. Given the result did not reach the threshold of statistical significance in the discovery study (Add Health), a future study from a similar population of adolescents should be performed to replicate the interaction between these two loci.
The epistatic interaction between PRKD1 and FTO reduced the additive effects of having one or more risk alleles at both these BMI risk variants. While Speliotes et al. (2010) reported no significant GG interactions influencing BMI after correction for multiple testing, and none of the epistatic effects identified in the present study were found in their list of nominally significant G-G interactions, this lack of replication is not surprising given that Speliotes et al. (2010) use a different outcome (inverse normally transformed BMI) and an older cohort (average age 54.3). Both PRKD1 and FTO have been implicated in epistatic interactions in other European cohorts 8. Our results suggest that several known BMI variants may warrant further examination with regards to epistatic effects in adolescence.
Both of the loci indicating suggestive evidence for G-G interaction influencing BMI Z-score in our study are have mRNA transcripts expressed in the brain and adipose tissue23. FTO is also expressed in the arcurate nucleus of the hypothalamus, a region responsible for feeding behavior, and has been positively associated with both emotional and uncontrolled eating24. In addition to being associated with BMI, FTO variants have been associated with glucose intolerance25, with increased FTO mRNA expression in subcutaneous fat tissue of insulin resistant individuals 26. PRKD1 also plays a role in glucose regulation by influencing insulin signaling 27, suggesting a potential biological pathway for G-G interaction.
A strength of our study was the selection of well-established loci associated with BMI among EA adults for genotyping in Add Health EA adolescents, a reasonable strategy given our previous research showing that many of these variants have a stronger estimated effect in adolescence 16. We also have a unique sample from a sensitive developmental period, when risk of obesity is high, and we conducted the analysis in an independent cohort to assess whether results replicated.
Studies like ours can be underpowered for detecting G-G interactions. One way to increase power and reduce our multiple testing burden would be to focus our analysis on only those SNPs with a known functional role on BMI. However, given the complex nature of BMI, few causal SNPs have been identified to date, necessitating a broader interrogation. In addition, we have limited our analysis to two-locus interactions due to sample size, though we acknowledge that three- or more locus interactions are certainly plausible for this complex phenotype 28. Finally, due to the comparatively small Add Health African American, Hispanic, and Asian sample sizes, and the lack of appropriate replication samples, we restricted our analysis to adolescents of European descent. This strategy, while reasonable considering these loci were discovered in EA adults, limits the generalizability of our study.
In conclusion, the reported statistically significant interaction in European descent adolescents warrant further follow up in additional cohorts. Our results are suggestive of possible epistatic effects on BMI during adolescence and may point to potential underlying biological pathways important in the development of obesity.
Supplementary Material
Supplementary Table 1. Power calculations for G-G interactions in Add Health.
What is already known about this subject
- BMI is estimated to be 40-80% heritable
- GWAS have identified numerous genetic loci that affect BMI in adults of European descent
- Adolescent obesity is predictive of future weight gain, obesity, and adult-onset severe obesity
What this study adds
- An investigation of the influence of epistasis (gene-gene interaction) between 34 established BMI SNPs with body mass index (BMI) Z-score in a nationally representative sample of European American adolescents.
- Twenty-eight nominally significant G-G interactions (p<0.05) in Add Health
- Replication of one G-G interactions in the CHOP sample
ACKNOWLEDGEMENT
We thank Amy Perou of the BioSpecimen Processing facility and Jason Luo of the Mammalian Genotyping Core at the University of North Carolina at Chapel Hill. This work was funded by National Institutes of Health grants R01HD057194 for Add Health and R01HD056465 for CHOP. K.E.N, E.L., and K.L.Y. contributed to study design, K.L.Y. and A.S.R to data analysis, and K.L.Y., K.E.N., and P.G.L. contributed to writing the manuscript, all other authors provided critical evaluation of the manuscript. K.L.Y., K.E.N., and P.G.L. had full access to all data in the study and take responsibility for data integrity and analysis accuracy. This research uses data from Add Health, a program project directed by Kathleen Mullan Harris and designed by J. Richard Udry, Peter S. Bearman, and Kathleen Mullan Harris at the University of North Carolina at Chapel Hill, and funded by grant P01-HD31921 from the Eunice Kennedy Shriver National Institute of Child Health and Human Development, with cooperative funding from 23 other federal agencies and foundations. Special acknowledgement is due to Ronald R. Rindfuss and Barbara Entwistle for assistance in the original design. Information on how to obtain Add Health data files is available on the Add Health website (http://www.cpc.unc.edu/addhealth). No direct support was received from grant P01-HD31921 for this analysis. We are grateful to the Carolina Population Center for general support.
ABBREVIATIONS
- G-E
Gene-Environment
- G-G
Gene-Gene
- GWA
Genome-Wide Association
- EA
non-Hispanic European American
- CHOP
Childhood European American Cohort from Philadelphia
Footnotes
CONFLICT OF INTEREST
There were no potential or real conflicts of financial or personal interest with the financial sponsors of the research project.
References
- 1.Cuypers KF, Loos RJF, Kvaløy K, Kulle B, Romundstad P, Holmen TL. Obesity-Susceptibility Loci and Their Influence on Adiposity-Related Traits in Transition from Adolescence to Adulthood - The HUNT Study. PLoS ONE. 2012;7(10):e46912. doi: 10.1371/journal.pone.0046912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.The NS, Suchindran C, North KE, Popkin BM, Gordon-Larsen P. Association of adolescent obesity with risk of severe obesity in adulthood. JAMA. 2010;304(18):2042–2047. doi: 10.1001/jama.2010.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Speliotes EK, Willer CJ, Berndt SI, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nature Genetics. 2010;42(11):937–948. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gordon-Larsen P, Nelson MC, Beam K. Associations among active transportation, physical activity, and weight status in young adults. Obesity Research. 2005;13(5):868–875. doi: 10.1038/oby.2005.100. [DOI] [PubMed] [Google Scholar]
- 5.Richardson AS, North KE, Graff M, et al. Moderate to vigorous physical activity interactions with genetic variants and body mass index in a large US ethnically diverse cohort. Pediatric Obesity. 2014;9(2):e35–46. doi: 10.1111/j.2047-6310.2013.00152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Graff M, North KE, Richardson AS, et al. Screen time behaviours may interact with obesity genes, independent of physical activity, to influence adolescent BMI in an ethnically diverse cohort. Pediatric Obesity. 2013;8(6):e74–9. doi: 10.1111/j.2047-6310.2013.00195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gordon-Larsen P, Nelson MC, Page P, Popkin BM. Inequality in the built environment underlies key health disparities in physical activity and obesity. Pediatrics. 2006;117(2):417–424. doi: 10.1542/peds.2005-0058. [DOI] [PubMed] [Google Scholar]
- 8.Wei W-H, Hemani G, Gyenesei A, et al. Genome-wide analysis of epistasis in body mass index using multiple human populations. European Journal of Human Genetics. 2012;20(8):857–862. doi: 10.1038/ejhg.2012.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gibson G. Hints of hidden heritability in GWAS. Nature Genetics. 2010;42(7):558–560. doi: 10.1038/ng0710-558. [DOI] [PubMed] [Google Scholar]
- 10.Wang X, Elston RC, Zhu X. The meaning of interaction. Human Heredity. 2010;70(4):269–277. doi: 10.1159/000321967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rajapakse I, Perlman MD, Martin PJ, Hansen JA, Kooperberg C. Multivariate detection of gene-gene interactions. Genetic Epidemiology. 2012;36(6):622–30. doi: 10.1002/gepi.21656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaplan AS, Levitan RD, Yilmaz Z, Davis C, Tharmalingam S, Kennedy JL. A DRD4/BDNF gene–gene interaction associated with maximum BMI in women with bulimia nervosa. International Journal of Eating Disorders. 2008;41(1):22–28. doi: 10.1002/eat.20474. [DOI] [PubMed] [Google Scholar]
- 13.Ding Y, Guo Z-R, Wu M, Chen Q, Yu H, Luo W-S. Gene-Gene interaction between PPARδ and PPARγ is associated with abdominal obesity in a Chinese population. Journal of Genetics and Genomics. 2012;39(12):625–631. doi: 10.1016/j.jgg.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 14.Luo W, Guo Z, Wu M, et al. Association of Peroxisome Proliferator-Activated Receptor α/γ/δ; with obesity, and gene-gene interaction, in the Chinese Han population. Journal of Epidemiology. 2013;23(3):187–194. doi: 10.2188/jea.JE20120110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graff M, North KE, Mohlke KL, et al. Estimation of genetic effects on BMI during adolescence in an ethnically diverse cohort: The National Longitudinal Study of Adolescent Health. Nutrition and Diabetes. 2012;2(9):e47–8. doi: 10.1038/nutd.2012.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Graff M, Ngwa JS, Workalemahu T, et al. Genome-wide analysis of BMI in adolescents and young adults reveals additional insight into the effects of genetic loci over the life course. Human Molecular Genetics. 2013;22(17):3597–607. doi: 10.1093/hmg/ddt205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Resnick MD, Bearman PS, Blum RW, et al. Protecting adolescents from harm. Findings from the National Longitudinal Study on Adolescent Health. JAMA. 1997;278(10):823–832. doi: 10.1001/jama.278.10.823. [DOI] [PubMed] [Google Scholar]
- 18.Harris KM. An integrative approach to health. Demography. 2010;47(1):1–22. doi: 10.1353/dem.0.0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ogden CL, Kuczmarski RJ, Flegal KM, et al. Centers for Disease Control and Prevention 2000 growth charts for the United States: improvements to the 1977 National Center for Health Statistics version. Pediatrics. 2002;109(1):45–60. doi: 10.1542/peds.109.1.45. [DOI] [PubMed] [Google Scholar]
- 20.Gauderman WJ. Sample size requirements for association studies of gene-gene interaction. American Journal of Epidemiology. 2002;155:478–484. doi: 10.1093/aje/155.5.478. [DOI] [PubMed] [Google Scholar]
- 21.Hakonarson H, Grant SFA, Bradfield JP, et al. A genome-wide association study identifies KIAA0350 as a type 1 diabetes gene. Nature. 2007;448(7153):591–594. doi: 10.1038/nature06010. [DOI] [PubMed] [Google Scholar]
- 22.Willer CJ, Speliotes EK, Loos RJF, et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nature Genetics. 2008;41(1):25–34. doi: 10.1038/ng.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uhlen M, Oksvold P, Fagerberg L, et al. Towards a knowledge-based Human Protein Atlas. Nature Biotechnology. 2010;28(12):1248–1250. doi: 10.1038/nbt1210-1248. [DOI] [PubMed] [Google Scholar]
- 24.Cornelis MC, Rimm EB, Curhan GC, et al. Obesity susceptibility loci and uncontrolled eating, emotional eating and cognitive restraint behaviors in men and women. Obesity. 2013;22(5):E135–41. doi: 10.1002/oby.20592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garver WS, Newman SB, Gonzales-Pacheco DM, et al. The genetics of childhood obesity and interaction with dietary macronutrients. Genes & nutrition. 2013;8(3):271–287. doi: 10.1007/s12263-013-0339-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bravard A, Veilleux A, Disse E, et al. The expression of FTO in human adipose tissue is influenced by fat depot, adiposity, and insulin sensitivity. Obesity. 2013;21(6):1165–1173. doi: 10.1002/oby.20110. [DOI] [PubMed] [Google Scholar]
- 27.Williams MJ, Almén MS, Fredriksson R, Schiöth HB. What model organisms and interactomics can reveal about the genetics of human obesity. Cellular and Molecular Life Sciences. 2012;69(22):3819–3834. doi: 10.1007/s00018-012-1022-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fang G, Haznadar M, Wang W, et al. High-order SNP combinations associated with complex diseases: efficient discovery, statistical power and functional interactions. PLoS ONE. 2012;7(4):e33531. doi: 10.1371/journal.pone.0033531. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Power calculations for G-G interactions in Add Health.