

Abstract
The gut microbiota is well known to affect host metabolic phenotypes. The systemic effects of the gut microbiota on host metabolism are generally evaluated via the comparison of germfree and conventional mice, which is impossible to perform for humans. Hence, it remains difficult to determine the impact of the gut microbiota on human metabolic phenotypes. We demonstrate that a constraint-based modeling framework that simulates “germfree” and “ex-germfree” human individuals can partially fill this gap and allow for in silico predictions of systemic human-microbial co-metabolism. To this end, we constructed the first constraint-based host-microbial community model, comprising the most comprehensive model of human metabolism and 11 manually curated, validated metabolic models of commensals, probiotics, pathogens, and opportunistic pathogens. We used this host-microbiota model to predict potential metabolic host-microbe interactions under 4 in silico dietary regimes. Our model predicts that gut microbes secrete numerous health-relevant metabolites into the lumen, thereby modulating the molecular composition of the body fluid metabolome. Our key results include the following: 1. Replacing a commensal community with pathogens caused a loss of important host metabolic functions. 2. The gut microbiota can produce important precursors of host hormone synthesis and thus serves as an endocrine organ. 3. The synthesis of important neurotransmitters is elevated in the presence of the gut microbiota. 4. Gut microbes contribute essential precursors for glutathione, taurine, and leukotrienes. This computational modeling framework provides novel insight into complex metabolic host-microbiota interactions and can serve as a powerful tool with which to generate novel, non-obvious hypotheses regarding host-microbe co-metabolism.
Keywords: human gut microbiome, in silico, metabolome, metabolic modeling
Introduction
There is increasing evidence that the human gut microbiota plays a central role in human health and well-being. The gut microbiota performs essential metabolic functions for host health, such as the maturation of the host immune system 1 and protection against pathogens.2 Gut microbes produce the short-chain fatty acids acetate, propionate, and butyrate, which are utilized as carbon sources by the host.1 Furthermore, the gut microbiota synthesizes essential amino acids and vitamins,3 transforms bile acids,4 and modifies xenobiotics.5
The application of metagenomic techniques by international consortia, such as MetaHIT 6 and the Human Microbiome Project,7 has dramatically increased our understanding of the human gut microbiota and its relation to host health and disease. Species and genera over- and under-represented in disease incidence have been identified, contributing to our understanding of “who is there” in the healthy gut microbiome.7 The collective metabolic potential, rather than species composition, of the gut microbiota has been suggested to define a healthy microbiota. Losses of essential functions are associated with diseases.3 However, our understanding of the metabolic potential of the human gut microbiota and its relation with human physiology and health is limited. It remains to be discovered whether particular metabolic activities can be linked to certain key species or if multiple members are required to perform these important functions for the host.5 There is a need to systematically analyze the functions encoded in the human gut microbiome and its potential to affect host biochemistry and to identify beneficial and detrimental species and genera. Furthermore, the effects of the gut microbiota on host metabolism in body sites other than the intestine have rarely been systematically studied, and it is unclear how the gut microbiota affects the whole-body metabolism and immune homeostasis of human hosts.5
Several studies have established computational approaches with which to systematically explore the microbiota but not on an organism-resolved level.8,9 For instance, Sridharan et al. predicted and quantified products of amino acid metabolism in the mouse intestine using a supra-organism microbiota model demonstrating that computational modeling can also predict the host-microbiota co-metabolome.10 However, the supra-organism approach does not account for species boundaries and thus cannot predict host-microbe interactions on an organism-resolved level. In contrast, the constraint-based reconstruction and analysis (COBRA) approach allows for the modeling of interspecies interactions by utilizing well-curated, organism-resolved metabolic networks constructed in a bottom-up manner.11 Such metabolic reconstructions can be converted into mathematical models, which can be constrained according to environmental conditions, e.g., the availability of dietary nutrients. Subsequently, functional states can be predicted under various nutrient conditions or enzyme defects.12 Over 25 metabolic reconstructions for microbes inhabiting the human body are available.11 Using the COBRA approach, we previously demonstrated, for the first time, that the metabolite exchange between a mouse and its gut symbiont Bacteroides thetaiotaomicron could be predicted in silico.13 However, no studies have modeled the interaction between a host and more than one microbe hitherto. Here, we present a computational framework that fills this gap. The human reconstruction Recon2, accounting for most human biochemical reactions occurring in at least one human cell,14 was joined with a model community of 11 microbes, and over 2000 exchanges representing metabolic functions in humans were systematically predicted. To our knowledge, this analysis represents the first effort to predict the co-metabolism of a host and a gut microbial community using a bottom-up, organism-resolved metabolic model.
Results
We aimed to investigate the metabolic potential of a representative collection of gut microbes to affect host metabolism. Therefore, we designed a computational framework that permits us to systematically explore the effects of representative gut microbes on host metabolic functions. Using this host-microbiota model, we systematically predicted the effect of each microbe on metabolite exchanges representing host whole-body metabolic functions under 4 in silico dietary regimes. To our knowledge, this is the first constraint-based effort that integrates more than 3 metabolic models simultaneously to provide insight into the complex metabolic interactions between a host organism and its microflora.
Microbial pathway content and metabolic diversity
The human gut harbors an estimated 1000 species.6 Eleven published high-quality, manually curated gut microbe reconstructions were incorporated into our modeling framework (SI text). We evaluated the representativeness of the 11 microbe reconstructions in terms of the total metabolic capabilities of the human gut microbiota (Fig. 1). Phylum-wise, our reconstruction collection captured 3 main phyla (Bacteroidetes, Firmicutes, Proteobacteria) but lacked representatives of one major phylum (Actinobacteria) as well as minor phyla (e.g., Verrucomicrobia, Fusobacteria).15 The reconstructed organisms Bacteroides thetaiotaomicron, Faecalibacterium prausnitzii, Streptococcus thermophilus, and Escherichia coli O157:H7 were included among the 75 common species detected in the 124 human volunteers.6 The 11 microbial reconstructions included 429 of the 450 metabolic clusters of orthologous genes (COGs 16) reported to be present in the microbiome of the 124 human volunteers6 (supplemental text, Fig. S1). The remaining 662 unmapped COGs detected in the human volunteers were primarily non-metabolic and thus outside the scope of metabolic reconstructions.
Figure 1.
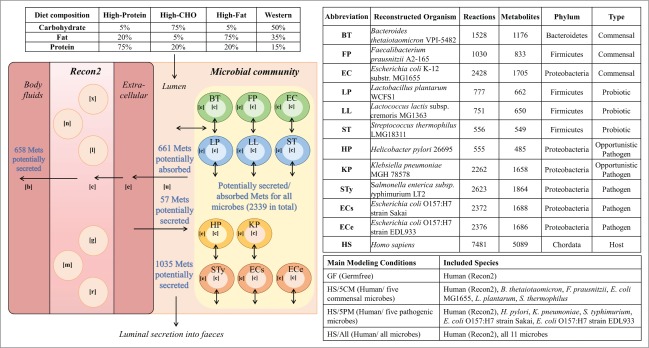
Overview of the study design, including the modeling framework, description of included organisms, main constructed host-microbe models and implemented dietary regimes. Examples of the types of exchange reactions that were optimized individually (4679 in total) are also presented. CHO = carbohydrate; Mets = metabolites; [c] = cytoplasm; [e] = extracellular space; [u] = lumen; [x] = peroxisome; [n] = nucleus; [l] = lysosome; [g] = Golgi apparatus; [m] = mitochondrion; [r] = endoplasmic reticulum.
To estimate the metabolic closeness among the 11 microbes, the Jaccard distance between the reaction and the metabolite content as well as the subsystem coverage for each microbe reconstruction were calculated (Figs. S1 and S2). As expected, representatives of the same phyla were closest metabolically, but our in silico microbiota also exhibited considerable metabolic diversity (SI text), which has been suggested to be important for collaboration within communities.17 Large reconstructions (e.g., Escherichia coli MG1655) displayed higher subsystem coverage than smaller reconstructions (e.g., Helicobacter pylori and the probiotics) (Fig. S2, supplemental text). To further characterize the metabolic differences among the reconstructed strains, we computed the tradeoff between simultaneous host and microbe biomass production using Pareto optimality analysis.13 The majority of the microbes were capable of benefiting the host and vice versa (Fig. S3). Despite their metabolic similarity, commensal and pathogenic E. coli displayed differences in their metabolite exchange with the host (supplemental text).
Properties of the modeling framework
We developed an in silico approach to systematically study the effects of gut microbes on the human host by incorporating 11 manually curated gut microbe reconstructions and a global reconstruction of human metabolism into a constraint-based host-microbe modeling framework (Fig. 1). To elucidate the impact of various microbial groups, the host was joined with a community of either 5 commensal or 5 pathogenic species (deemed HS/5CM for human/5 commensal microbes and HS/5PM for human/5 pathogenic microbes, Fig. 1). Subsequently, the host was joined with all 11 microbes (deemed HS/All) (Fig. 1). Finally, Recon2 was joined with all 11 microbes individually as well as with pairs of 2 microbes (SI text, Table S1). The “germfree” condition (GF) included only Recon2. The required time for one FBA by the model HS/All, consisting of 20,951 reactions, was less than 2 seconds (Dell, Intel Core i5, 16GB RAM, 64 bit). Four diets, varying in carbohydrate, fat, and protein intake (Fig. 1), were simulated because dietary composition is well known to affect the gut microbiota.18
Production of mammalian-microbe co-metabolites
The gut microbiota is well known to influence the mammalian host's body fluid and tissue metabolome.19 By optimizing the fluxes through all body fluid secretion reactions individually, we systematically predicted the effects of varying the presence and the absence of the 11 microbes on the host body fluid metabolome in silico (condition GF vs. condition HS/All, Fig. 1). Of the 658 potential body fluid secretion metabolites (Fig. 1, Table S6), 342 were indeed secreted by the host under the given simulation conditions. Of these 342 non-zero metabolites, only 11 cases (butyrate, ethanol, D-ribose, D-ribitol, L-arabinose, L-homoserine, benzoic acid, and menaquinone 7, 9, 10, 11) were not secreted in the condition GF under the given dietary constraints. However, the quantitatively predicted body fluid metabolome was greatly affected by the presence of the microbes. When comparing the HS/All with the GF simulation, the secretion fluxes increased by greater than five-fold for 29 body fluid metabolites and by greater than 50-fold for an additional 23 metabolites under at least one dietary regime. Of these metabolites, 28 were derived from amino acids (Fig. 2, Table S2) and many have been detected in mammalian blood, plasma, or urine (Table S2). For instance, the fluxes of the known mammalian-microbial co-metabolites phenylacetylglutamine and 4-hydroxyphenylacetate 20 were increased by 41-fold and 90-fold, respectively, in HS/All.
Figure 2.
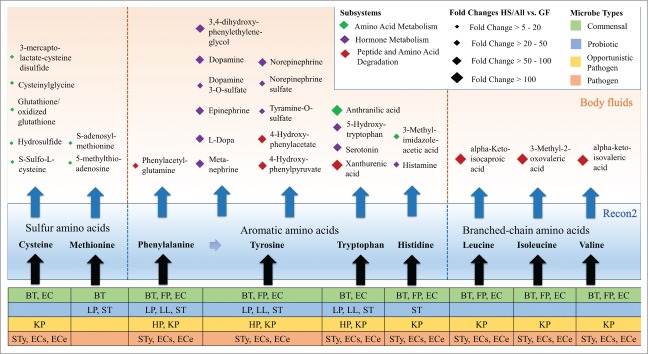
Host body-fluid secretions of amino acid-derived metabolites increased by greater than five-fold in HS/All (vs. GF) under a simulated Western diet. The diamonds represent metabolites, with subsystems indicated by color. The fold changes in secretion between the HS/All and the GF conditions are indicated by the size of a diamond. Individual microbes that contributed significantly to biosynthesis are listed for each metabolite group, with colors indicating the type of microbe. For abbreviations, refer to Figure 1.
Commensal versus pathogenic community
Dysbiosis in the gut microbiome is thought to have negative effects on host health.21 Generally, an increase in Proteobacteria associated with a loss in commensal Firmicutes is considered to be detrimental for the host. Based on this rationale, we elucidated the consequences of replacing the commensal community, including probiotic and commensal Firmicutes, with a group of pathogenic Proteobacteria by comparing the predicted body fluid metabolomes of HS/5CM and HS/5PM. Although pathogenicity cannot be directly modeled with constraint-based methods, the differences in the potentials of 2 groups to synthesize health-relevant metabolites can be readily predicted.
We quantified the global effect of the microbiota as follows: We calculated the maximum possible secretions for all 342 non-zero metabolites by optimizing the fluxes through the respective exchanges for all of the modeling conditions. The achieved percentages of the maximal flux values were compared for the condition GF and the microbe-associated conditions. The presence of the 5 commensals increased the secretions of 173 body fluid metabolites from less than 90% of the maximum possible absolute value to >99% for the simulated Western diet (saturation). In the presence of all microbes (HS/All), this number of saturated metabolites was relatively decreased to 109 (Fig. 3). This decrease was attributed to enforcing a low level of microbial growth (Methods section), which consumed resources that were consequently no longer available to maximize the metabolic objectives (Fig. 3). For 76 metabolites, the flux was constant in every model, resulting in 100% saturation under every condition (Fig. 3). The HS/5PM modeling condition, containing H. pylori and 4 Gammaproteobacteria, was unable to saturate any non-constant metabolite secretion fluxes and there were particularly low secretions of vitamins and cofactor-related metabolites into body fluids compared with HS/5CM (Fig. S5). The number of microbial reconstructions was the same under both scenarios, and the reconstructions included in HS/5CM were, on average, smaller in size and scope than for HS/5PM (Table S3). The predicted effects on host metabolism were thus a result of the metabolic capabilities represented in the reconstructions rather than the number or size of reconstructions.
Figure 3.
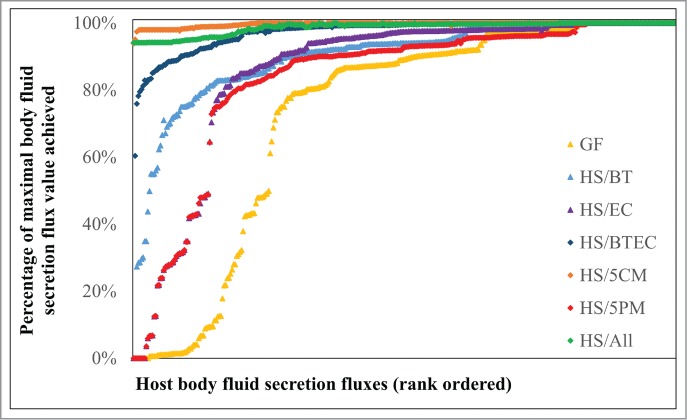
Comparison of relative amounts of host body-fluid metabolite secretion achieved by the GF condition and the host-microbe models under a Western diet (see also Fig. S4). For abbreviations, refer to Figure 1.
The metabolic distance scores for the 4 Gammaproteobacteria reconstructions included in HS/5PM were low (Fig. S1), and their subsystem coverages were similar (Fig. S2), resulting in a low combined metabolic potential. The metabolic potential of H. pylori was low because of its small genome and thus small reconstruction size (Table S3), further explaining the limited metabolic capabilities of the pathogenic community. By calculating the metabolite secretion saturation for the host linked to all 11 microbes individually or to 2 out of the 5 commensals (SI text), we determined that B. thetaiotaomicron and E. coli MG1655 affected host metabolism most strongly (Fig. 3, Fig. S5).
In total, the pathogenic community, because of its lower diversity and collective metabolic capabilities, exhibited a weaker capability to influence the predicted host body fluid metabolome than the relatively more diverse commensal community. For the high-carbohydrate, high-fat, and high-protein diets, the numbers of body fluid metabolites that increased from under 90% of the maximum possible absolute values to >99 % of the respective values were 225, 172, and 196, respectively, for the HS/5CM condition (Fig. S4), indicating that the predicted impact of the microbiota was correlated with the carbohydrate content of the simulated diet, in line with the known influence of diet on human gut microbiota.18 All of the calculated body fluid secretion flux values are presented in Table S6.
Luminal secretion
The microbiota significantly affects mammalian luminal metabolites.19 Maximizing the 1035 metabolic exchanges (Fig. 1) between the lumen compartment and “fecal” secretion individually enabled the luminal metabolome, which consisted of 266 metabolites, to be predicted. We then compared the differences in luminal secretion among the GF, HS/5CM, HS/5PM, and HS/All modeling conditions. Only 48 metabolites were secreted in the GF simulation, in line with reports that numerous metabolites detected in conventional mice are not present in germfree animals.22 The range of predicted metabolites of microbial origin was significantly higher for HS/5CM than for HS/5PM (200 compared to 99 metabolites), and numerous subsystems were saturated under the HS/5CM condition but not under HS/5PM (Fig. S6). These results demonstrate again the relatively poor metabolic potential of the pathogenic community because of its relatively lower diversity. A number of subsystems were most affected by particular species. The Gammaproteobacteria contributed the majority of inorganic iron metabolism, lipopolysaccharide biosynthesis, polyamine metabolism, and urea cycle metabolites, whereas host and plant polysaccharide degradation, the pentose phosphate pathway, cholesterol and bile acid metabolism, and vitamin and cofactor metabolism were all linked to B. thetaiotaomicron (Fig. S6). H. pylori contributed most to nitrogen metabolism but provided little other metabolic potential (Fig. S6). All of the calculated luminal secretion flux values are listed in Table S6.
The majority of the luminal metabolites were solely of microbial origin (218 of 266, 82%) (Table S4), many of which are health-relevant. For instance, the well-described microbial production of a variety of phenolic compounds from aromatic amino acids 23 was predicted (Table S4). Other modeled products derived from amino acids included sulfide and ammonia, which are toxic and disturb colonocyte energy metabolism.24 With the exception of Lactococcus lactis, the Firmicutes did not produce sulfide (Table S4). Other potentially health-relevant predicted metabolites of microbial origin included D-lactate, formate, ethanol, succinate, short-chain fatty acids, and vitamins (Table S4). As expected, only the gram-negative bacteria produced pro-inflammatory lipopolysaccharides,1 with the pathogenic Salmonella typhimurium contributing most to this subsystem (Fig. S6).
In summary, B. thetaiotaomicron, as well as the commensal and probiotic Firmicutes (F. prausnitzii and the lactic acid bacteria), produced a greater number of beneficial metabolites than the Proteobacteria. Moreover, although we could not directly model pathogenicity, we demonstrated that the community of pathogenic Proteobacteria, in particular S. typhimurium, was characterized by a greater potential to produce potentially harmful compounds (e.g., sulfide, ammonia, and lipopolysaccharides). This greater potential could not be explained entirely by their low diversity and was also attributed to their metabolic differences from the commensal Firmicutes.
It has been proposed that the gut microbiota can be considered as an endocrine organ because it produces numerous compounds that influence distal organs via the bloodstream, such as short-chain fatty acids, and regulates hormone metabolism.25 All of the reconstructed bacteria produced acetate, but propionate and butyrate production was only predicted for B. thetaiotaomicron and F. prausnitzii, respectively, (Table S4). Furthermore, we predicted a 34- to 90-fold increase in the host body fluid secretions of hormone metabolites, including epinephrine, norepinephrine, dopamine, histamine, and serotonin, on the Western diet in the presence of the 11 microbes (Fig. 2, Table S2). According to our predictions, B. thetaiotaomicron, Lactobacillus plantarum, L. lactis, and the 3 E. coli strains produced GABA and L. plantarum secreted histamine (Table S4). Both of these neurotransmitters are known to be produced by the gut microbiota.25
Host absorption
It is well known that the host absorbs and utilizes microbe-derived metabolites, such as amino acids, vitamins,3 and short-chain fatty acids.1 We examined microbe-derived metabolites that could be absorbed by the host by optimizing the fluxes through all of the 661 host absorption exchange reactions (Fig. 1, Table S6). A total of 38 dietary compounds usable by the host (e.g., simple sugars, vitamins, and amino acids) were additionally produced by the microbes (Table S4), thus increasing their respective availabilities compared with the GF condition. For instance, the microbiota produced additional aspartate, glutamate, and glutamine, which serve as energy sources to enterocytes.24 Furthermore, 60 metabolites of microbial origin that were not included in the simulated diet could be absorbed by the host, including acetate, propionate, and butyrate.
These results explain the pronounced predicted effects of the presence of gut microbes on the host body fluid metabolome described above and shown in Figure S5. We further performed a global single-gene deletion study of the human genes for all of the host-microbe pairs. Five gene deletions that were predicted to be lethal to the “germfree” host could be rescued by the presence of at least one microbe (SI text), further supporting the hypothesis that gut microbes can provide essential biomass precursors to the host. All of the calculated host absorption flux values are presented in Table S6.
Glutathione, taurine, and leukotriene metabolism
We aimed to identify mechanisms by which the reconstructed gut microbes, in combination with the simulated dietary inputs, caused the predicted increases in body fluid secretions. One example of essential biomass precursors predicted to be provided by the microbiota is sulfur amino acids. Methionine is essential, and cysteine is a semi-essential amino acid for humans. Moreover, cysteine serves as a precursor for glutathione and taurine.26 To test the hypothesis that the gut microbes could provide cysteine and/or methionine, we analyzed the pathway utilization when secretions of (i) glutathione and (ii) taurine into body fluids, and the biosynthesis products requiring these compounds as precursors, were optimized for all host-microbe pairs.
The model predicted that 7 bacteria could increase glutathione biosynthesis by supplying L-cysteine, L-methionine, or L-cystine to the host (Fig. 4). Alternatively, the diet serves as sources of cysteine and methionine (Fig. 4), with the high-protein diet providing the highest inputs (Table S5). We expected a similar microbe-derived effect on the biosynthesis of leukotrienes C4, D4, E4, and F4, which are dependent on glutathione.27 In fact, their secretion was increased in the presence of all of the microbes, except the 3 probiotics and H. pylori. This effect only occurred with the high-fat diet (Table S6), indicating that a precursor other than glutathione was limiting for cysteinyl leukotriene biosynthesis. Leukotriene biosynthesis starts from arachidonic acid, which is not predicted to be synthesized by any of the microbes included in this study (Table S6). Consequently, the supply of arachidonic acid was dependent entirely on the dietary inputs under the given simulation conditions. Hence, the secretion of leukotriene into body fluids could only be increased by the presence of the microbes in combination with the high-fat diet (Fig. 4). Thus, the effects of the microbe- and the diet-supplied inputs of biosynthesis precursor were complementary.
Figure 4.

Schematic depiction of glutathione and leukotriene biosynthesis in Recon2 and the entry points of microbe- and diet-derived metabolites with increased fluxes through the pathway and increased secretion into body fluids. For abbreviations, refer to Figure 1.
Similarly, B. thetaiotaomicron and F. prausnitzii increased the secretion flux of taurine by supplying L-cysteine, L-methionine (B. thetaiotaomicron), or L-cystine (F. prausnitzii) (Fig. 5). The main taurine biosynthesis pathway starts from L-cysteine, with L-cysteinesulfinic acid and hypotaurine as intermediates (Fig. 5). In the case of F. prausnitzii, this effect was limited by the supply of L-serine in the diet, which this microbe requires for L-cysteine/L-cystine biosynthesis. Furthermore, the model predicted an alternate pathway for taurine biosynthesis that starts from L-serine and sulfate via L-cysteic acid (Fig. 5), which is a minor pathway for taurine biosynthesis.28 B. thetaiotaomicron was able to increase the flux of taurine synthesized via this pathway by providing sulfate. As a result, the secretions of taurocholic and taurodeoxycholic acid into body fluids were also increased by the high-protein diet and/or in the presence of B. thetaiotaomicron or F. prausnitzii (Fig. 5). In summary, similar to glutathione biosynthesis, the presence of gut microbes, in addition to the supply of diet-derived biosynthesis precursors, increased taurine biosynthesis.
Figure 5.
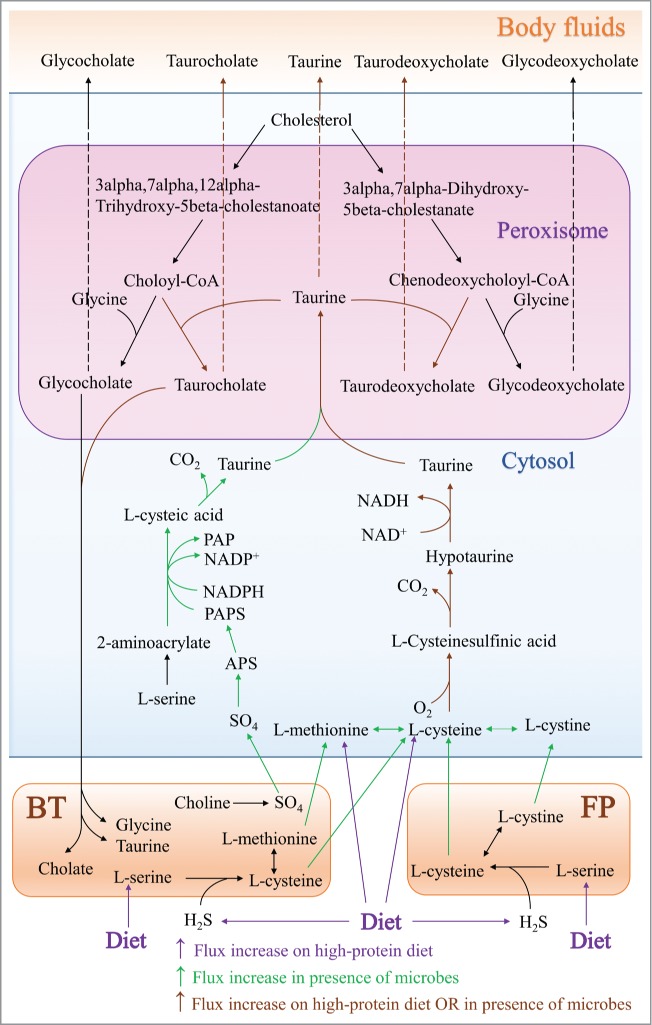
Schematic depiction of taurine and bile acid biosynthesis in Recon2 and the entry points of microbe- and diet-derived metabolites with increased fluxes through the pathway and increased secretion into body fluids. The transformation of bile acid by B. thetaiotaomicron is also shown. For abbreviations, refer to Figure 1.
Discussion
In this study, we present a framework designed to model the metabolic interactions between the human host and its microbiota. For the first time, a constraint-based model of a host and as many as 11 microbes was constructed. We systematically explored the effects of metabolites produced by commensal probiotics, opportunistic pathogens, and pathogens on human metabolic functions. A community of 5 commensals consistently provided a higher metabolic potential than a community of 5 pathogens. The gut has the metabolic potential to serve as an endocrine organ for the host. The synthesis of important neurotransmitters is elevated in the presence of the gut microbiota. Gut microbes contribute essential precursors for glutathione, taurine, and leukotrienes. This computational modeling framework provides novel insight into the complex metabolic interactions between a host and its microflora, and it can serve as a powerful tool with which to generate novel, non-obvious hypotheses regarding host-microbe co-metabolism.
Bottom-up systems biology permits us to evaluate, at the species level, the individual and combined metabolic potentials of microorganisms. We demonstrated that our constraint-based modeling framework (Fig. 1) was able to accurately predict the metabolic potential of the included 11 microbes as well as their global effects on host metabolism. As a result of the increased availability of automated reconstruction tools, such as Model SEED,29 and the catalog of reference genomes established by MetaHIT 6 and the Human Microbiome Project,7 a greater number of metabolic reconstructions for important species inhabiting the gut, including low-abundance and/or understudied species, should become available in the near future. Important metabolic functions may be performed by low-abundance microbes (the “rare biosphere”).3 Using our framework, the metabolic potential of such “keystone” species could be elucidated and placed in context with host metabolism without a need to cultivate the microbe.
Such host-microbiota modeling may reveal the mechanisms underlying correlations among various taxa and particular mammalian-microbial co-metabolites. For example, phenylacetylglutamine and 4-hydroxyphenylacetate, which were predicted to be produced by the host and to be affected by the presence of the microbes, have been positively correlated with Subdoligranulum variable, and additionally, phenylacetylglutamine has been positively correlated with Bifidobacterium pseudocatenulatum.20 Our modeling framework can be expanded by the inclusion of any number of representative, well-curated gut microbe reconstructions, which will further elucidate the impacts and the importance of the gut ecosystem on human health. Notably, the human reconstruction currently lacks biosynthesis pathways for certain important mammalian-microbial co-metabolites (e.g., hippurate and 4-cresyl sulfate 1), which will be included in future revisions of Recon2.
We have provided a comprehensive chart that details the metabolic potential of each gut microbial species (Table S4). Overall, we predict that a diverse commensal community spanning 3 phyla will have a higher global metabolic potential than a less diverse community of 5 pathogenic Proteobacteria (Fig. 3), leading to the production of a wider range of microbe-derived compounds to be absorbed by the host. Species considered to be beneficial, e.g., F. prausnitzii, produce more beneficial metabolites (e.g., butyrate 30), whereas harmful or pro-inflammatory compounds, such as sulfide, ammonia 24 and lipopolysaccharides,1 are more highly associated with bacteria considered to be detrimental (e.g., Proteobacteria) (Table S4, Fig. S6). Based on these results, a loss of beneficial Firmicutes and gain in Proteobacteria would result in a depletion of beneficial metabolites (e.g., butyrate) but not harmful or pro-inflammatory microbial products (e.g., lipopolysaccharides, ammonium ions). Indeed, changes in the gut microbiome, such as an increase in Proteobacteria, have been associated with detrimental effects on host health.21 It has to be noted that we predict the theoretical maximal metabolite secretion potential of each species. In a natural community, each species would most likely optimize their own biomass production resulting in lower secretion fluxes than predicted as well as the secretion of waste products rather than valuable compounds. Moreover, certain metabolites would only be synthesized and secreted in certain environmental conditions. Simulating a more realistic community behavior in silico would require a well-defined objective function enabling each species to strive for optimal biomass production. Such modeling approaches will be addressed in future efforts.
Low bacterial gene richness has been associated with adiposity and a higher inflammatory status,31 which may suggest that an increase in gram-negative bacteria, such as Proteobacteria, and corresponding loss in gram-positive Firmicutes are correlated with a loss of richness in encoded functions. Similarly, a variety of metabolic subsystems have been shown to be affected by antibiotic perturbations of the gut microbiota.32 We predicted the production of aromatic amino acid-derived phenolic metabolites by gut microbes (Table S4), which is supported by experimental data and may have implications for vascular health.23 In our model, cadaverine was only produced by the 5 Gammaproteobacteria (Table S4). This compound is elevated in the fecal extracts of ulcerative colitis (UC) patients.33 Such findings can link microbial groups that are over- or under-represented in certain disease conditions with alterations in metabolomes. Indeed, numerous studies have reported increases in Enterobacteria, such as E. coli, in inflammatory bowel disease patients,34 whereas butyrate-producing Firmicutes, which have a protective role in the intestine, were decreased.35
It has been proposed that the gut microbiota serves as an additional endocrine organ.25 Accordingly, we predicted that the secretions of hormones and hormone precursors, including serotonin and its precursors tryptophan and 5-hydroxytryptophan, would increase by over 34-fold in the presence of the 11 microbes (Fig. 2, Table S2). Supporting this hypothesis, consistently higher tryptophan levels in urine were reported during the establishment of a gut microbiota compared with germfree animals 36 though another study reported lower serum tryptophan levels in conventional mice compared to germfree mice.37 Interestingly, ex-germfree mice also displayed higher levels of phenylalanine, tyrosine, and tryptophan in the cerebral metabolome compared with germfree mice.38 There is evidence that the gut microbiota influences the brain and behavior by regulating tryptophan availability as well as by synthesising and degrading tryptophan.25 All of the microbes, except F. prausnitzii, were predicted to synthesize tryptophan. B. thetaiotaomicron and the Gammaproteobacteria degraded tryptophan to skatole and indole, respectively (Table S4). Host enzymes convert indole to indoxyl sulfate, which is a uremic toxin and has been directly linked to vascular disease and mortality in chronic kidney disease patients.39 Indole is thus another example of a detrimental metabolic product specifically predicted for the mostly pathogenic Proteobacteria. This finding again highlights that metabolic modeling is a useful tool for the prediction of detrimental metabolic profiles associated with a dysbiotic microbiota.
We propose that serotonin levels may be affected by the gut microbial potential to produce and degrade its precursor tryptophan. Based on our predictions, the dopamine secretion into body fluids is increased for individuals on a Western diet in the presence of the 11 microbes (Fig. 4, Table S2). Consistently, conventional mice were reported to exhibit a greater than 1.5-fold increase in dopamine levels in serum compared to germfree mice.37 Importantly, we predicted that all of the hormones and hormone precursors could be produced by the “germfree” host, but their secretion levels were drastically increased in the presence of the 11 microbes. These predictions are supported by experimental observations that germfree animals have a functional hormonal repertoire but exhibit an altered behavior, likely because of the lack of microbial influence on their hormone metabolism.25
Over 10% of detected blood metabolites differ by at least 50% between germfree and conventional mice.22 Accordingly, we predicted a global effect of microbial presence on human body-fluid metabolites (Fig. 2, Table S2). For instance, the microbes were predicted to provide essential sulfur amino acids, thereby increasing the host's potential to synthesize glutathione, leukotrienes, and taurine-conjugated bile acids (Figs. 4 and 5). Consistently, up to 20% of the amounts of circulating lysine, threonine, and leucine have been synthesized by microbes,40 supporting our hypothesis that microbes also provide essential methionine and cysteine. Luminal glutathione performs important functions, including detoxification and the maintenance of mucosal integrity.41 The availability of cysteine is limiting for glutathione biosynthesis,26 and germfree mice colonized with human baby flora display impaired metabolism, including glutathione depletion,42 supporting a link between the gut microbiota and glutathione levels. Based on our predictions, we propose a relationship between the gut microbiota and the redox state in the intestine. In support, B. thetaiotaomicron has been suggested to generate energy via bile acid dehydrogenation,4 forming a feedback loop of increased taurocholic acid secretion by B. thetaiotaomicron, which would benefit itself.
In summary, we developed a computational framework that allows for the generation of novel testable hypotheses regarding the identity and impacts of influential gut microbiota within the gut microbial community and the mechanisms by which gut microbiota influence host metabolism. Although it is imperative that these hypotheses be subjected to experimental validation, we highlight that computational approaches, such as the one presented here, are required to assess the complexity of the interaction network between the host and its associated microbiota. The presented computational modeling framework has the potential to serve as an additional, complementary tool to existing cellular and animal models for the study of human-microbe co-metabolism.
Materials and Methods
Construction of host-microbe models
We retrieved 11 manually constructed and validated reconstructions of human gut microbes 13,43-50 and an extensive, high-quality reconstruction of human metabolism.14 The 11 microbes included commensal, probiotic and pathogenic species (Fig. 1). Quality assurance and, if possible, expansion of each reconstruction content was performed. If necessary, additional reactions/genes were included and/or the reconstruction structure was revised (details in SI text). An in silico framework combining host and microbes that enables host-microbe interactions through an in silico compartment representing the intestinal lumen, deemed [u], was constructed. The host and the microbes each had their own extracellular spaces [e] through which they could exchange metabolites with the lumen compartment and, via the lumen, with each other (Fig. 1). The lumen compartment provided an outlet for simulated luminal secretion into feces. The host could secrete metabolites into a separate outlet representing host secretion into body fluids (i.e., blood and urine) (Fig. 1). To model the effects of the presence and absence of certain microbes, a total of 25 models containing the host and zero to 11 microbes were constructed. Briefly, the host was combined with each microbe separately, with 2 commensal microbes, with communities of 5 microbes and with all 11 microbes. The unassociated host represented a “germfree” human (SI text, Table S1). The 25 models are described in spreadsheet format in Table S7 and are available in COBRA format at http://thielelab.eu.
Comparison of microbial reconstruction content with a human microbiome gene catalog
A human gene catalog constructed from the fecal samples of 124 human subjects, assembled by Qin et al.,6 consisting of 1112 clusters of orthologous genes (COGs 16) was downloaded. The metabolic functions represented in the COGs were manually mapped to the metabolic reactions contained in the 11 microbe reconstructions. Non-metabolic COGs were not translated, as they are outside the scope of metabolic reconstructions. Moreover, where possible, the unmapped reactions contained in the 11 reconstructions were translated into additional COGs not reported in the Qin et al. dataset. The translations were performed using Enzyme Commission (EC) numbers and KEGG Orthologies (KOs).51
Definition of diets
We defined the following diets: Western diet, which approximates the amounts of protein, carbohydrate, and fat consumed by a typical Western citizen (http://www.ars.usda.gov/); high-carbohydrate diet; high-fat diet; and high-protein diet (Fig. 1). All diets contained the same simulated nutrients but with varying uptake rates (Table S5).
Quantification of fluxes
The fluxes through biomass objective functions and the exchange reactions of the 25 models were predicted using flux balance analysis (FBA). Briefly, FBA calculates the flow of metabolites through a metabolic network that results in an optimal solution for a given objective function while assuming steady state.12 By definition, the uptake flux of a metabolite through an exchange reaction is negative, whereas the secretion flux is positive. Alternate solutions for optimizing for biomass production were predicted using flux variability analysis (FVA). This analysis identifies the allowed minimal and maximal flux spans for each reaction that occur when a certain percentage of the maximal flux through the objective function is enforced.52 All of the simulations were performed using the COBRA Toolbox 53 in the MATLAB (Mathworks, Inc., Natick, MA, USA) programming environment. We used Tomopt (Tomlab, Inc., Seattle, WA, USA) and ILOG CPLEX (IBM, Armonk, NY, USA) as the linear programming solvers for the flux balance analysis 12 and for the flux variability analysis,52 respectively.
Pareto optimality analysis
Pareto optimality analysis was performed for all host-microbe pairs as previously described.13 Briefly, the fluxes through the host and microbe biomass objective functions were fixed at multiple intervals while optimizing the flux through the other respective biomass objective function, resulting in the prediction of the tradeoff between host and microbe biomass production. This analysis was conducted for the 4 diets defined above.
Systematic analysis of host metabolic functions
The effect of microbial presence on host metabolism was examined systematically as follows: using FBA, the fluxes through all of the exchange reactions in the joint models were optimized individually. These exchange reactions included (i) the absorption/secretion between the host and the lumen as well as between the microbes and the lumen, (ii) luminal secretion into feces, and (iii) host secretion into body fluids (Fig. 1). Minimal biomass growth was enforced for the host and the included microbes by setting the lower bounds on the respective biomass objective functions to 0.01 hr−1, representing the low growth rates (ca. 0.02–0.2 hr−1) measured for the intestinal microflora of mammals.54 In total, 4679 exchanges representing metabolite secretion/absorption were set as the objective function, resulting in 4679 simulations (Fig. 1, Table S6). The analysis was performed for all 25 models (Table S1) for each of the 4 defined diets by optimizing the flux through all 4679 exchanges individually. In total, the approach resulted in the systematic quantification of (i) the potential of Recon2 to secrete body fluid metabolites, (ii) the potential of Recon2 to absorb metabolites of dietary and microbial origin, (iii) the fecal secretion of metabolites of human and microbial origin and (iv) the absorption and secretion of metabolites of dietary, human and microbial origin by each microbe (Fig. 1). See Table S6 for all of the optimized metabolic functions and computed fluxes.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Funding
The authors thank Mr. Eugen Bauer for help with calculating the reaction and metabolite distance scores. This work was funded by an ATTRACT program grant from the Luxembourg National Research Fund (FNR) to I.T. (FNR/A12/01).
References
- 1. Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, Pettersson S. Host-gut microbiota metabolic interactions. Science 2012; 336:1262-7; PMID:22674330; http://dx.doi.org/ 10.1126/science.1223813 [DOI] [PubMed] [Google Scholar]
- 2. Lawley TD, Walker AW. Intestinal colonization resistance. Immunology 2013; 138:1-11; PMID:23240815; http://dx.doi.org/ 10.1111/j.1365-2567.2012.03616.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shafquat A, Joice R, Simmons SL, Huttenhower C. Functional and phylogenetic assembly of microbial communities in the human microbiome. Trends Microbiol 2014; 22:261-6; PMID:24618403; http://dx.doi.org/ 10.1016/j.tim.2014.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res 2006; 47:241-59; PMID:16299351; http://dx.doi.org/ 10.1194/jlr.R500013-JLR200 [DOI] [PubMed] [Google Scholar]
- 5. Maurice CF, Haiser HJ, Turnbaugh PJ. Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell 2013; 152:39-50; PMID:23332745; http://dx.doi.org/10.1016%2Fj.cell.2012.10.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010; 464:59-65; PMID:20203603; http://dx.doi.org/ 10.1038/nature08821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Human Microbiome Project C . Structure, function and diversity of the healthy human microbiome. Nature 2012; 486:207-14; PMID:22699609; http://dx.doi.org/ 10.1038/nature11234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Greenblum S, Turnbaugh PJ, Borenstein E. Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proc Natl Acad Sci U S A 2012; 109:594-9; PMID:22184244; http://dx.doi.org/ 10.1073/pnas.1116053109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Abubucker S, Segata N, Goll J, Schubert AM, Izard J, Cantarel BL, Rodriguez-Mueller B, Zucker J, Thiagarajan M, Henrissat B, et al. Metabolic reconstruction for metagenomic data and its application to the human microbiome. Plos Comput Biol 2012; 8:e1002358; PMID:22719234; http://dx.doi.org/ 10.1371/journal.pcbi.1002358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sridharan GV, Choi K, Klemashevich C, Wu C, Prabakaran D, Pan LB, Steinmeyer S, Mueller C, Yousofshahi M, Alaniz RC, et al. Prediction and quantification of bioactive microbiota metabolites in the mouse gut. Nat Commun 2014; 5:5492; PMID:25411059; http://dx.doi.org/ 10.1038/ncomms6492 [DOI] [PubMed] [Google Scholar]
- 11. Thiele I, Heinken A, Fleming RM. A systems biology approach to studying the role of microbes in human health. Curr Opin Biotechnol 2013; 24:4-12; PMID:23102866; http://dx.doi.org/ 10.1016/j.copbio.2012.10.001 [DOI] [PubMed] [Google Scholar]
- 12. Orth JD, Thiele I, Palsson BO. What is flux balance analysis? Nat Biotechnol 2010; 28:245-8; PMID:20212490; http://dx.doi.org/ 10.1038/nbt.1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Heinken A, Sahoo S, Fleming RM, Thiele I. Systems-level characterization of a host-microbe metabolic symbiosis in the mammalian gut. Gut Microbes 2013; 4:28-40; PMID:23022739; http://dx.doi.org/ 10.4161/gmic.22370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Thiele I, Swainston N, Fleming RM, Hoppe A, Sahoo S, Aurich MK, Haraldsdottir H, Mo ML, Rolfsson O, Stobbe MD, et al. A community-driven global reconstruction of human metabolism. Nat Biotechnol 2013; 31:419-25; PMID:23455439; http://dx.doi.org/ 10.1038/nbt.2488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto JM, et al. Enterotypes of the human gut microbiome. Nature 2011; 473:174-80; PMID:21508958; http://dx.doi.org/ 10.1038/nature09944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics 2003; 4:41; PMID:12969510; http://dx.doi.org/ 10.1186/1471-2105-4-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mazumdar V, Amar S, Segre D. Metabolic proximity in the order of colonization of a microbial community. PloS One 2013; 8:e77617; PMID:24204896; http://dx.doi.org/ 10.1371/journal.pone.0077617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Scott KP, Gratz SW, Sheridan PO, Flint HJ, Duncan SH. The influence of diet on the gut microbiota. Pharmacol Res: Off J Ital Pharmacol Soc 2013; 69:52-60; PMID:23147033; http://dx.doi.org/ 10.1016/j.phrs.2012.10.020 [DOI] [PubMed] [Google Scholar]
- 19. Claus SP, Tsang TM, Wang Y, Cloarec O, Skordi E, Martin FP, Rezzi S, Ross A, Kochhar S, Holmes E, et al. Systemic multicompartmental effects of the gut microbiome on mouse metabolic phenotypes. Mol Syst Biol 2008; 4:219; PMID:18854818; http://dx.doi.org/ 10.1038/msb.2008.56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li M, Wang B, Zhang M, Rantalainen M, Wang S, Zhou H, Zhang Y, Shen J, Pang X, Zhang M, et al. Symbiotic gut microbes modulate human metabolic phenotypes. Proc Natl Acad Sci U S A 2008; 105:2117-22; PMID:18252821; http://dx.doi.org/ 10.1073/pnas.0712038105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guinane CM, Cotter PD. Role of the gut microbiota in health and chronic gastrointestinal disease: understanding a hidden metabolic organ. Therap Adv Gastroenterol 2013; 6:295-308; PMID:23814609; http://dx.doi.org/ 10.1177/1756283X13482996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, Siuzdak G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci U S A 2009; 106:3698-703; PMID:19234110; http://dx.doi.org/ 10.1073/pnas.0812874106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Russell WR, Duncan SH, Scobbie L, Duncan G, Cantlay L, Calder AG, Anderson SE, Flint HJ. Major phenylpropanoid-derived metabolites in the human gut can arise from microbial fermentation of protein. Mol Nutr Food Res 2013; 57:523-35; PMID:23349065; http://dx.doi.org/ 10.1002/mnfr.201200594 [DOI] [PubMed] [Google Scholar]
- 24. Davila AM, Blachier F, Gotteland M, Andriamihaja M, Benetti PH, Sanz Y, Tomé D. Re-print of "Intestinal luminal nitrogen metabolism: role of the gut microbiota and consequences for the host". Pharmacol Res: Off J Ital Pharmacol Soc 2013; 69:114-26; PMID:23318949; http://dx.doi.org/ 10.1016/j.phrs.2013.01.003 [DOI] [PubMed] [Google Scholar]
- 25. Clarke G, Stilling RM, Kennedy PJ, Stanton C, Cryan JF, Dinan TG. Gut microbiota: the neglected endocrine organ. Mol Endocrinol 2014; 28:1221-38; me20141108; PMID:24892638; http://dx.doi.org/ 10.1210/me.2014-1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Brosnan JT, Brosnan ME. The sulfur-containing amino acids: an overview. J Nutr 2006; 136:1636S-40S; PMID:16702333 [DOI] [PubMed] [Google Scholar]
- 27. Savari S, Vinnakota K, Zhang Y, Sjolander A. Cysteinyl leukotrienes and their receptors: bridging inflammation and colorectal cancer. World J Gastroenterol: WJG 2014; 20:968-77; PMID:24574769; http://dx.doi.org/ 10.3748/wjg.v20.i4.968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jacobsen JG, Smith LH. Biochemistry and physiology of taurine and taurine derivatives. Physiol Rev 1968; 48:424-511; PMID:4297098 [DOI] [PubMed] [Google Scholar]
- 29. Henry CS, DeJongh M, Best AA, Frybarger PM, Linsay B, Stevens RL. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat Biotechnol 2010; 28:977-82; PMID:20802497; http://dx.doi.org/ 10.1038/nbt.1672 [DOI] [PubMed] [Google Scholar]
- 30. Russell WR, Hoyles L, Flint HJ, Dumas ME. Colonic bacterial metabolites and human health. Curr Opin Microbiol 2013; 16:246-54; PMID:23880135; http://dx.doi.org/ 10.1016/j.mib.2013.07.002 [DOI] [PubMed] [Google Scholar]
- 31. Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, Almeida M, Arumugam M, Batto JM, Kennedy S, et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013; 500:541-6; PMID:23985870; http://dx.doi.org/ 10.1038/nature12506 [DOI] [PubMed] [Google Scholar]
- 32. Zheng X, Xie G, Zhao A, Zhao L, Yao C, Chiu NH, Zhou Z, Bao Y, Jia W, Nicholson JK, et al. The footprints of gut microbial-mammalian co-metabolism. J Proteome Res 2011; 10:5512-22; PMID:21970572; http://dx.doi.org/ 10.1021/pr2007945 [DOI] [PubMed] [Google Scholar]
- 33. Le Gall G, Noor SO, Ridgway K, Scovell L, Jamieson C, Johnson IT, Colquhoun IJ, Kemsley EK, Narbad A. Metabolomics of fecal extracts detects altered metabolic activity of gut microbiota in ulcerative colitis and irritable bowel syndrome. J Proteome Res 2011; 10:4208-18; PMID:21761941; http://dx.doi.org/ 10.1021/pr2003598 [DOI] [PubMed] [Google Scholar]
- 34. Loh G, Blaut M. Role of commensal gut bacteria in inflammatory bowel diseases. Gut Microbes 2012; 3:544-55; PMID:23060017; http://dx.doi.org/ 10.4161/gmic.22156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. PNAS 2007; 104:13780-5; PMID:17699621; http://dx.doi.org/ 10.1073/pnas.0706625104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. El Aidy S, Derrien M, Merrifield CA, Levenez F, Dore J, Boekschoten MV, Dekker J, Holmes E, Zoetendal EG, van Baarlen P, et al. Gut bacteria-host metabolic interplay during conventionalisation of the mouse germfree colon. Isme J 2013; 7:743-55; PMID:23178667; http://dx.doi.org/ 10.1038/ismej.2012.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Velagapudi VR, Hezaveh R, Reigstad CS, Gopalacharyulu P, Yetukuri L, Islam S, Felin J, Perkins R, Borén J, Oresic M, et al. The gut microbiota modulates host energy and lipid metabolism in mice. J Lipid Res 2010; 51:1101-12; PMID:20040631; http://dx.doi.org/ 10.1194/jlr.M002774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Matsumoto M, Kibe R, Ooga T, Aiba Y, Sawaki E, Koga Y, Benno Y. Cerebral low-molecular metabolites influenced by intestinal microbiota: a pilot study. Front Syst Neurosci 2013; 7:9; PMID:23630473; http://dx.doi.org/ 10.3389/fnsys.2013.00009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Barreto FC, Barreto DV, Liabeuf S, Meert N, Glorieux G, Temmar M, Choukroun G, Vanholder R, Massy ZA, European Uremic Toxin Work Group (EUTox) . Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin J Am Soc Nephrol: CJASN 2009; 4:1551-8; PMID:19696217; http://dx.doi.org/ 10.2215/CJN.03980609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Morowitz MJ, Carlisle EM, Alverdy JC. Contributions of intestinal bacteria to nutrition and metabolism in the critically ill. Surg Clin N Am 2011; 91:771-85, viii; PMID:21787967; http://dx.doi.org/ 10.1016/j.suc.2011.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Circu ML, Aw TY. Intestinal redox biology and oxidative stress. Semin Cell Dev Biol 2012; 23:729-37; PMID:22484611; http://dx.doi.org/ 10.1016/j.semcdb.2012.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Martin FP, Dumas ME, Wang Y, Legido-Quigley C, Yap IK, Tang H, Zirah S, Murphy GM, Cloarec O, Lindon JC, et al. A top-down systems biology view of microbiome-mammalian metabolic interactions in a mouse model. Mol Syst Biol 2007; 3:112; PMID:17515922; http://dx.doi.org/ 10.1038/msb4100153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pastink MI, Teusink B, Hols P, Visser S, de Vos WM, Hugenholtz J. Genome-scale model of Streptococcus thermophilus LMG18311 for metabolic comparison of lactic acid bacteria. Appl Environ Microbiol 2009; 75:3627-33; PMID:19346354; http://dx.doi.org/ 10.1128/AEM.00138-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Thiele I, Hyduke DR, Steeb B, Fankam G, Allen DK, Bazzani S, Charusanti P, Chen FC, Fleming RM, Hsiung CA, et al. A community effort towards a knowledge-base and mathematical model of the human pathogen Salmonella Typhimurium LT2. BMC Syst Biol 2011; 5:8; PMID:21244678; http://dx.doi.org/ 10.1186/1752-0509-5-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Thiele I, Vo TD, Price ND, Palsson BO. Expanded metabolic reconstruction of Helicobacter pylori (iIT341 GSM/GPR): an in silico genome-scale characterization of single- and double-deletion mutants. J Bacteriol 2005; 187:5818-30; PMID:16077130; http://dx.doi.org/ 10.1128/JB.187.16.5818-5830.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Teusink B, Wiersma A, Molenaar D, Francke C, de Vos WM, Siezen RJ, Smid EJ. Analysis of growth of Lactobacillus plantarum WCFS1 on a complex medium using a genome-scale metabolic model. J Biol Chem 2006; 281:40041-8; PMID:17062565; http://dx.doi.org/ 10.1074/jbc.M606263200 [DOI] [PubMed] [Google Scholar]
- 47. Flahaut NA, Wiersma A, van de Bunt B, Martens DE, Schaap PJ, Sijtsma L, Dos Santos VA, de Vos WM. Genome-scale metabolic model for Lactococcus lactis MG1363 and its application to the analysis of flavor formation. Appl Microbiol Biotechnol 2013; 97:8729-39; PMID:23974365; http://dx.doi.org/ 10.1007/s00253-013-5140-2 [DOI] [PubMed] [Google Scholar]
- 48. Heinken A, Khan MT, Paglia G, Rodionov DA, Harmsen HJ, Thiele I. A functional metabolic map of Faecalibacterium prausnitzii, a beneficial human gut microbe. J Bacteriol 2014; 6:3289-302; PMID:25002542; http://dx.doi.org/ 10.1128/JB.01780-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liao YC, Huang TW, Chen FC, Charusanti P, Hong JS, Chang HY, Tsai SF, Palsson BO, Hsiung CA. An experimentally validated genome-scale metabolic reconstruction of Klebsiella pneumoniae MGH 78578, iYL1228. J Bacteriol 2011; 193:1710-7; PMID:21296962; http://dx.doi.org/ 10.1128/JB.01218-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Baumler DJ, Peplinski RG, Reed JL, Glasner JD, Perna NT. The evolution of metabolic networks of E. coli. BMC Syst Biol 2011; 5:182; PMID:22044664; http://dx.doi.org/ 10.1186/1752-0509-5-182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res 2008; 36:D480-4; PMID:18077471; http://dx.doi.org/ 10.1093/nar/gkm882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gudmundsson S, Thiele I. Computationally efficient flux variability analysis. BMC Bioinformatics 2010; 11:489; PMID:20920235; http://dx.doi.org/ 10.1186/1471-2105-11-489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schellenberger J, Que R, Fleming RM, Thiele I, Orth JD, Feist AM, Zielinski DC, Bordbar A, Lewis NE, Rahmanian S, et al. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nat Protoc 2011; 6:1290-307; PMID:21886097; http://dx.doi.org/ 10.1038/nprot.2011.308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gibbons RJ, Kapsimalis B. Estimates of the overall rate of growth of the intestinal microflora of hamsters, guinea pigs, and mice. J Bacteriol 1967; 93:510-2; PMID:6020422 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.