

Abstract
MicroRNA (miRNA) genes generally share many features common to those of protein coding genes. Various transcription factors (TFs) and co-regulators are also known to regulate miRNA genes. Here we identify novel p53 and NFκB p65/RelA responsive miRNAs and demonstrate that these 2 TFs bind to the regulatory sequences of miR-100, −146a and −150 in both mouse striatal and human cervical carcinoma cells and regulate their expression. p53 represses the miRNAs while NFκB p65/RelA induces them. Further, we provide evidence that exogenous p53 inhibits NFκB p65/RelA activity by reducing its nuclear content and competing with it for CBP binding. This suggests for the existence of a functional cross-talk between the 2 TFs in regulating miRNA expression. Moreover, promoter occupancy assay reveals that exogenous p53 excludes NFκB p65/RelA from its binding site in the upstream sequence of miR-100 gene thereby causing its repression. Thus, our work identifies novel p53 and NFκB p65/RelA responsive miRNAs in human and mouse and uncovers possible mechanisms of co-regulation of miR-100. It is to be mentioned here that cross-talks between p53 and NFκB p65/RelA have been observed to define the outcome of several biological processes and that the pro-apoptotic effect of p53 and the pro-survival functions of NFκB can be largely mediated via the biological roles of the miRNAs these TFs regulate. Our observation with cell lines thus provides an important platform upon which further work is to be done to establish the biological significance of such co-regulation of miRNAs by p53 and NFκB p65/RelA.
Keywords: microRNA, miRNA gene regulation, miR-100, miR-146a, miR-150, NFκB p65/RelA, p53, transcription factor
Abbreviations
- ChIP
Chromatin immunoprecipitation
- Co-IP
Co-immunoprecipitation
- miRNAs
microRNAs
- NFκB
nuclear factor kappa-light-chain-enhancer of activated B cells
- p53
tumor protein 53
- p65
RELA, RELA
- v-rel
avian reticuloendotheliosis viral oncogene homolog A
- RNA POL II
RNA Polymerase II
- RNA POL III
RNA Polymerase III
- RLU
Relative light unit
- RT-PCR
Reverse transcription polymerase chain reaction
- TF
Transcriptional factor
- TFBS
Transcription factor binding site
- WB
Western blot
Introduction
MicroRNAs (miRNA) are short (∼22 nucleotides) non-coding RNAs which are primarily involved in post-transcriptional regulation of gene expression. Genes coded for miRNAs are generally transcribed by RNA polymerase II (Pol II) and are often classified as Class II genes.1 However, several miRNAs have also been observed to be transcribed by RNA polymerase III2,3 and there are many intronic miRNAs that can be transcribed by both RNA polymerase II and RNA polymerase III.4 Toward the beginning of miRNA research, main focus was on studying physiological and pathological functions of miRNA genes. Considering their diverse cellular roles and their implications in major biological processes, current focus on miRNA research has widely shifted toward deciphering the molecular mechanism underlying transcriptional regulation of miRNA genes. These miRNA genes share many regulatory features common to those of RNA Pol II genes. These include regulation by core promoters and distal enhancers, 5′ capping and 3′ polyadenylation of primary transcripts.5 Similar to protein coding genes, various transcription factors (TFs) and co-regulators are known to regulate miRNA genes. It has been observed that TFs regulating miRNA transcription viz., c-myc,6,7 p538-10 NFκB p65/RelA11-13 as well as cell-type specific TFs such as MEF2, PU.114 and REST15 largely overlap with those that control protein-coding genes. Growth factor stimulation triggered by platelet-derived growth factor (PDGF), transforming growth factor-β (TGF-β) and bone-derived neurotrophic factor can also dynamically regulate miRNA gene expression.16 Besides, similar to protein coding genes, regulation of miRNA genes by TFs is a dynamic and complex process and may involve more than one TF. There have been evidences where 2 or more TFs co-regulate the expression of miRNA genes for appropriate execution of several biological events. NFI-A has been reported to repress and C/EBPα induce the expression of miR-223 associated with granulocyte differentiation.17 MyoD and Mef2 have been observed to activate miR-1, a process that contributes to enhancing myogenesis in skeletal muscles.18 The pro-apoptotic p53 and the pro-survival NFκB p65/RelA have also been known to co-regulate the expression of several miRNA genes in diverse biological processes.19-21 Since p53 and NFκB p65/RelA are mostly involved in determining cell fate, co-regulation of miRNAs by these 2 TFs, in general, may be implicated in defining final outcome of a cell undergoing cell division or apoptosis. However, till date, only in a limited number of cases the binding of the TFs to specific sites in the regulatory sequences of their target miRNA genes and the mode of such regulation have actually been experimentally determined. Hence, with a view to understanding the biological significance of combinatorial regulation of miRNAs by p53 and NFκB p65/RelA, in the present work, we first aimed at identifying novel p53 and NFκB p65/RelA responsive miRNAs which are co-regulated by these 2 TFs.
To identify miRNAs that are co-regulated by p53 and NFκB (p65/RelA), we have relied on data obtained from diverse cell types and across diverse species viz., mouse striatal STHdhQ7/HdhQ7 cells and human cervical carcinoma HeLa cells. Expression profile of 40 miRNAs in cells with over expressed p53 or NFκB p65/RelA and knocked down endogenous p53 or chemically inhibited NFκB p65/RelA was determined. The selection of 40 miRNAs was based on their possible involvement in Huntington's disease (HD)22 and several other diseases.23 Many of these miRNAs are known to be altered in cell and animal models of HD as well as in the post-mortem brains of human HD patients22,24 and also in diverse tumors originated from different tissues, cardiovascular diseases and other neurological diseases.23 Among these, we identified novel p53 and NFκB p65/RelA responsive miRNAs in both human and mouse. We observed that p53 binds to the regulatory sequences in the upstream of miR-100, −146a and −150 and represses their transcription while NFκB p65/RelA sub-unit binds to the regulatory sequences in the upstream of miR-100, −146a and −150 and induces their transcription. Although elevated NFκB p65/RelA did not affect p53 nuclear level, elevated p53 was observed to reduce NFκB p65/RelA nuclear content and activity. Thus, our results provide new data about the interplay between p53 and NFκB p65/RelA in co-regulating miRNAs which have been implicated in several diseases. The combinatorial effect of the extensive physical and functional cross-talks that exist between p53 and NFκB p65/RelA has been observed to define the outcome of several biological processes. Thus, understanding the mechanisms of regulation of these altered miRNAs by p53 and NFκB p65/RelA would likely provide an opportunity for possible therapeutic intervention in such disease processes by targeting either the regulatory pathway(s) or the miRNAs themselves.
Results
Ectopic modulation of p53 alters miRNA expression in mouse striatal ST HdhQ7/HdhQ7 cells and human cervical carcinoma HeLa cells
Exogenous expression of p53-CFP increased the expression of the protein (n = 3, p = 0.0017) 24 hours post-transfection in STHdhQ7/HdhQ7 cells (Fig. 1A). It was observed that out of 40 miRNAs whose expressions were studied, expression levels of 7 miRNAs viz., miR-145, −34a, −148a, −199a-5p, −134, −194, −182 were increased significantly (*P ≤ 0.05; ** P ≤ 0.01) and 8 miRNAs viz., miR-100, −125b, −150, −221, −146a, −138, −335 and −15b were decreased significantly (*P ≤ 0.05; ** P ≤ 0.01) in presence of exogenous p53 in STHdhQ7/HdhQ7 cells compared to control cells(Fig. 1B). Next, endogenous p53 was knocked down in the same cells with the help of p53 siRNA construct (Imgenex, USA) which down regulates the expression of p5325 72 hours post transfection (n = 3, p = 0.023) (Fig. 1C). Real time PCR analysis to detect levels of mature miRNAs from p53 siRNA transfected STHdhQ7/HdhQ7 cells showed that expressions of miR-145, −34a, −100, −125b, −146a, −199a-5p, −150, −15b and −221 were reversed in cells with knocked down p53 when compared to that with overexpressed p53 (Fig. 1D). However, expression pattern of miR-134, −148a, −182, −194, −138 and −335 were similar both in the presence of exogenous p53 as well as in cells with knocked down endogenous p53. Thus only 9 miRNAs whose expression patterns were reversed on knocking down endogenous p53 could possibly be regulated by the TF. To confirm further, exogenous p53 was expressed in p53 knocked down STHdhQ7/HdhQ7 cells and it was observed that expression of the miRNAs could be restored back to basal level (Fig. 1E). Thus, p53 regulates the expression of these 9 miRNAs in mouse STHdhQ7/HdhQ7 cells.
Figure 1.

Regulation of miRNAs by p53 in mouse striatal STHdhQ7/ HdhQ7 cells (A) (i) Western Blot showing increased p53 protein level in STHdhQ7/HdhQ7 cells transfected with p53-CFP; data are mean ± SD (n = 3); *p ≤ 0.05 compared to control. (B) Real Time PCR analysis showing changes in miRNA expression by greater than or equal to 4-fold (i.e. −ΔΔCT ≥ 2 as shown in graph) in presence of over expressed p53 in STHdhQ7/HdhQ7 cells compared with that of control; data are mean ± SD (n = 3); *P ≤ 0.05; ** P ≤ 0.01 compared to control. (C) Western Blot showing reduction in p53 protein level on knocking down endogenous p53 in STHdhQ7/HdhQ7 cells; data are mean ± SD (n = 3); *p ≤ 0.05 compared to control. (D) Real Time PCR analysis showing changes in miRNA expression in p53 knocked down STHdhQ7/HdhQ7 cells; data are mean ± SD (n = 3); *P ≤ 0.05; ** P ≤ 0.01 compared to control. (E) Real Time PCR analysis showing recovery in miRNA expression on overexpressing p53-CFP in p53 knocked down STHdhQ7/HdhQ7 cells; data are mean ± SD (n = 3); *P ≤ 0.05; ** P ≤ 0.01 compared to control STHdhQ7/HdhQ7 cells; † P ≤ 0.05; ††P ≤ 0.01 compared to p53 knocked down STHdhQ7/HdhQ7 cells (Q7 ± p53si). Error bars represent standard deviation, * or † represents statistical significance
Analysis of expression of 40 miRNAs in HeLa cells expressing exogenous p53 (Fig. 2A) revealed that expression of 11 miRNAs viz., let-7a, miR-107, −145, −182, −16, −34a, −148a, −134, −199a-5p, −154 and −194 were significantly increased (*P ≤ 0.05; ** P ≤ 0.01) and expression of 7 miRNAs viz., miR-15b, −100, −125b, −146a, −150, −205 and −221 were significantly decreased (*P ≤ 0.05; ** P ≤ 0.01) compared to control (Fig. 2B). On knocking down endogenous p53 by siRNA (Imgenex, USA) which downregulated the expression of the protein (Fig. 2C), expression pattern of all the 18 miRNAs were reversed as evident in Figure 2D. This indicated that these 18 miRNAs are regulated by p53 in HeLa cells. Comparing the expression profile of miRNAs in STHdhQ7/HdhQ7 cells and HeLa cells, it was evident that miRNA expression may be governed by the TF differently in different cell types.
Figure 2.
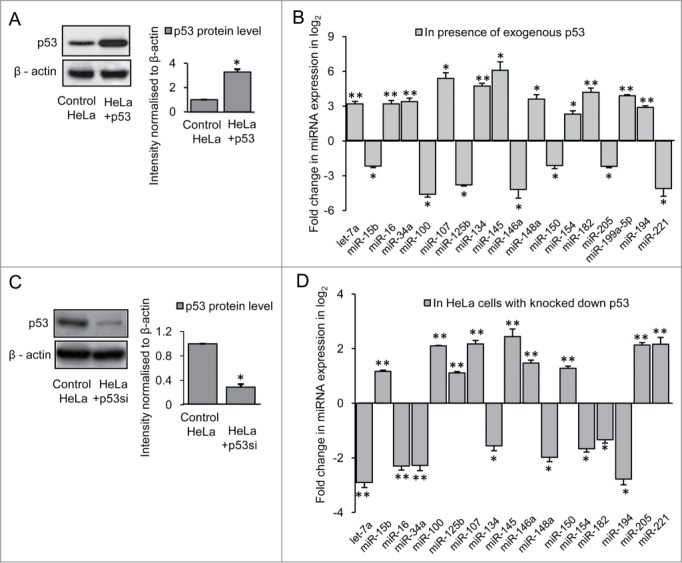
Regulation of miRNAs by p53 in HeLa cells (A) Western Blot showing increased p53 protein level in HeLa cells transfected with p53-CFP; data are mean ± SD (n = 3); *p ≤ 0.05 compared to control. (B) Real Time PCR analysis showing changes in miRNA expression by greater than or equal to 4-fold (i.e., −ΔΔCT ≥ 2 as shown in graph) in presence of over expressed p53 in HeLa cells compared with that of control; data are mean ± SD (n = 3); *P ≤ 0.05; ** P ≤ 0.01 compared to control. (C) Western Blot showing reduction in p53 protein level on knocking down endogenous p53 in HeLa cells; data are mean ± SD (n = 3); *p ≤ 0.05 compared to control. (D) Real Time PCR analysis showing changes in miRNA expression in p53 knocked down HeLa cells; data are mean ± SD (n = 3); *P ≤ 0.05; ** P ≤ 0.01 compared to control. Error bars represent standard deviation, * represents statistical significance.
Ectopic modulation of NFκB p65/RelA alters miRNA expression in mouse striatal ST HdhQ7/HdhQ7 cells and HeLa cells
In order to identify the miRNAs regulated by NFκB p65/RelA, p65 sub-unit of NFκB also known as RelA which bears the transactivation domain of the transcription factor was next over expressed in STHdhQ7/HdhQ7 cells. Twenty-four hours following transfection which increased (n = 3, p = 0.022) p65 protein level (Fig. 3A), miRNA expression profile was measured. It was observed that out of 40 miRNAs whose expressions were studied, 11 miRNAs viz., miR-15a, −34a, −146a, −107, −125b, −100, −150, −15b, −16, −221 and −135a were significantly up regulated (*P ≤ 0.05; ** P ≤ 0.01) and 6 miRNAs viz., miR-182, −214, −199a-5p, −199a-3p, −148a and −335 were decreased significantly (*P ≤ 0.05; ** P ≤ 0.01) in presence of exogenous NFκB p65/RelA in STHdhQ7/HdhQ7 cells compared to control (Fig. 3B). Endogenous NFκB p65/RelA activity was next reduced in STHdhQ7/HdhQ7 cells by treatment with aspirin26. It was observed that treating STHdhQ7/HdhQ7cells with 2.0 mM aspirin for 24 h decreased the basal p65 activity (n = 3, p = 0.0068) significantly (Fig. 3C). Under such condition, 17 miRNAs whose expressions were studied, excepting miR-107, −34a, −16, −199a-5p and −335, expression pattern of the remaining 12 miRNAs were reversed in aspirin treated cells compared to their expression pattern in presence of exogenous NFκB p65/RelA (Fig. 3D). Though NFκB p65/RelA is known to regulate miR-199a-5p27 and miR-34a28 their levels remained unaltered in the presence of aspirin.
Figure 3.

Regulation of miRNAs by NFκB p65/RelA in mouse striatal STHdhQ7/ HdhQ7 cells (A) Western Blot showing increased p65 protein level in STHdhQ7/HdhQ7 cells transfected with p65 sub-unit of NFκB; data are mean ± SD (n = 3); *P ≤ 0.05 compared to control. (B) Real Time PCR analysis showing changes in miRNA expression by greater than or equal to 4-fold (i.e. −ΔΔCT ≥ 2 as shown in graph) in presence of overexpressed p65 in STHdhQ7/HdhQ7 cells compared with that of control; data are mean ± SD (n = 3); *P ≤ 0.05; ** P ≤ 0.01 compared to control. (C) Changes in p65-RE activity in presence of exogenous p65 and different doses of Aspirin in STHdhQ7/HdhQ7 cells; data are mean ± SD (n = 3); *P ≤ 0.05; ** P ≤ 0.01 compared to control. (D) Real Time PCR analysis showing changes in miRNA expression in STHdhQ7/HdhQ7cells with reduced p65-RE activity; data are mean ± SD (n = 3); *P ≤ 0.05; ** P ≤ 0.01 compared to control. (E) Changes in p65-RE activity in presence of different doses of SN50 and its recovery following exogenous expression of p65 in STHdhQ7/HdhQ7 cells; data are mean ± SD (n = 3); ** P ≤ 0.01 compared to control; † P ≤ 0.05 compared to 15 µM SN50 treated cells (F) Real Time PCR analysis showing alteration of miRNA expression in STHdhQ7/HdhQ7cells with reduced p65-RE activity and its recovery following restoration of p65-RE activity; data are mean ± SD (n = 3); *P ≤ 0.05; ** P ≤ 0.01 compared to control STHdhQ7/HdhQ7 cells; † P ≤ 0.05; ††P ≤ 0.01 compared to STHdhQ7/HdhQ7 cells treated with 15 µM SN50. Error bars represent standard deviation, * or † represents statistical significance.
To further confirm that the expression changes of the mature miRNAs were specific due to the status of NFκB p65/RelA, endogenous NFκB p65/RelA activity was next reduced in STHdhQ7/HdhQ7 cells with the help of the peptide inhibitor SN5029. STHdhQ7/HdhQ7 cells treated with 15 µM SN50 showed decrease in basal endogenous NFκB p65/RelA activity 24 hours post treatment (n = 3, p = 0.003) (Fig. 3E). Under such condition, miRNA expression profile revealed changes in the expression of 12 miRNAs (Fig. 3F). Overexpressing NFκB p65/RelA in SN50 treated cells led to the recovery in p65-RE activity (n = 3, p = 0.03) (Fig. 3E) and also in miRNA expression compared to control (Fig. 3F). This result showed that 12 miRNAs were indeed regulated by NFκB p65/RelA in STHdhQ7/HdhQ7 cells.
Similarly, in order to find out the miRNAs which could be altered by NFκB p65/RelA in HeLa cells, we expressed exogenous NFκB p65/RelA in HeLa cells that increased p65 protein level (n = 3, p = 0.0042) compared to control cells (Fig. 4A). Under such conditions, expression of 9 miRNAs viz., miR-15a, −15b, −100, −125b, −135a, −146a, −150, −190 and −221 were significantly increased (*P ≤ 0.05; ** P ≤ 0.01) and expression of 7 miRNAs viz., miR-145, −148a, −199a-5p, −199a-3p, −214, −299 and −335 were significantly decreased (*P ≤ 0.05; ** P ≤ 0.01) in NFκB p65/RelA transfected HeLa cells compared to control (Fig. 4B). On inhibiting NFκB p65/RelA by treating HeLa cells with 2 mM aspirin which reduced basal p65 activity (n = 2, p = 0.0014) (Fig. 4C), the expression pattern of the 16 miRNAs were reversed (Fig. 4D) indicating that these miRNAs could be regulated by NFκB p65/RelA in HeLa cells. Comparing the results obtained in STHdhQ7/HdhQ7 cells and HeLa cells, it was evident that miRNA expression by NFκB p65/RelA was different in these 2 cells. These differences could be due to the differences in cell types and/or species of origin.
Figure 4.
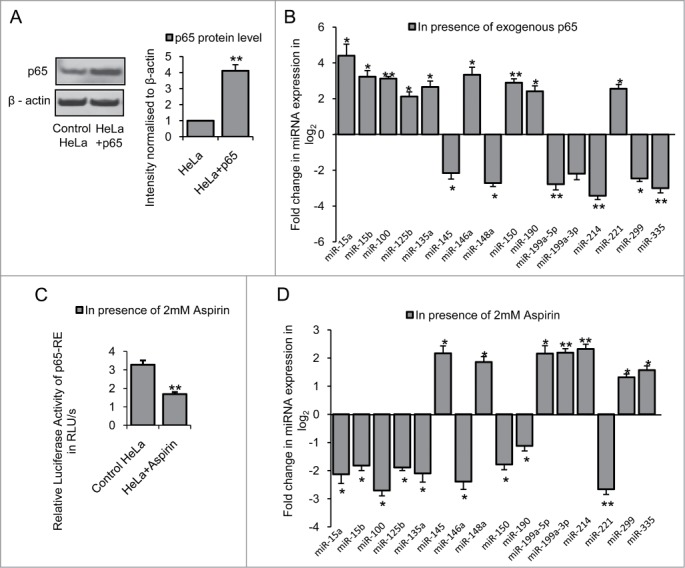
Regulation of miRNAs by NFκB p65/RelA in HeLa cells (A) Western Blot showing increased p65 protein level in HeLa cells transfected with p65 sub-unit of NFκB; data are mean ± SD (n = 3); ** P ≤ 0.01 compared to control. (B) Real Time PCR analysis showing changes in miRNA expression by greater than or equal to 4-fold (i.e., −ΔΔCT ≥ 2 as shown in graph) in presence of over expressed p65 in HeLa cells compared with that of control; data are mean ± SD (n = 3); *P ≤ 0.05; ** P ≤ 0.01 compared to control. (C) Reduction in p65-RE activity in presence of 2 mM Aspirin in HeLa cells; data are mean ± SD (n = 3); ** P ≤ 0.01 compared to control. (D) Real Time PCR analysis showing changes in miRNA expression in HeLa cells with reduced p65-RE activity; data are mean ± SD (n = 3); *P ≤ 0.05; ** P ≤ 0.01 compared to control. Error bars represent standard deviation, * represents statistical significance.
miRNAs regulated by both p53 and NFκB p65/RelA in mouse striatal STHdhQ7/HdhQ7 cells and HeLa cells
Close examination of the results obtained revealed that out of the 40 different miRNAs, p53 could increase the expression of miR-34a, −145 and −199a-5p and decrease the expression of miR-15b, −100, −125b, −146a, −150 and −221 in both mouse striatal STHdhQ7/HdhQ7 cells and human cervical carcinoma HeLa cells. NFκB p65/RelA on the other hand has been observed to increase the expression of miR-15a, −15b, −100, −125b, −135a, −146a, −150 and −221 and decrease the expression of miR-148a, −199a-3p and −214 in both mouse striatal STHdhQ7/HdhQ7 cells and human cervical carcinoma HeLa cells. Expression of 6 miRNAs viz., miR-146a, −100, −221, −15b, −150 and −125b were increased by NFκB p65/255 RelA and decreased by p53 i.e. altered by both these TFs as schematically shown in Figure 5A.
Figure 5.
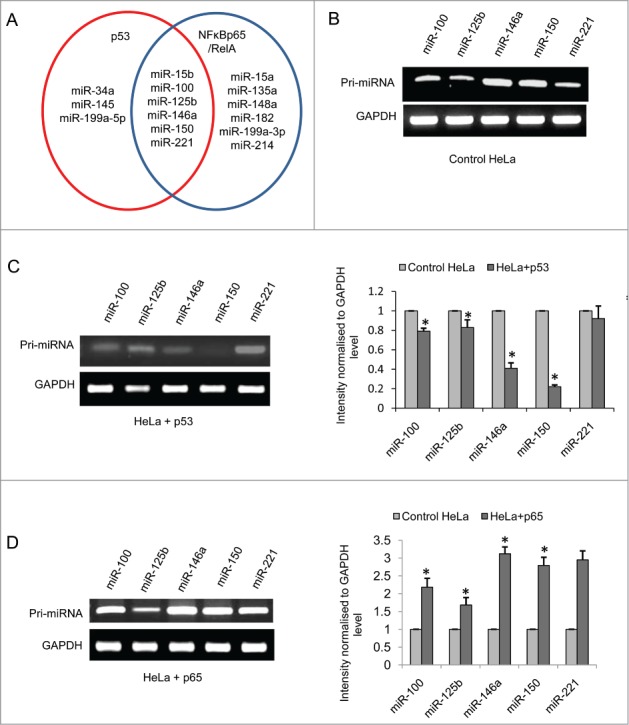
p53 and NFκB p65/RelA transcriptionally regulate the expression of miR-100, −125b, −146a, −150 and −221 (A) Schematic representation of miRNAs which are regulated by p53 only (miR-34a, −145, −199a-5p), NFκB p65/RelA only (miR-15a, −199a-3p, −148a, −135a, −182, −214) and regulated by both p53 and NFκB p65/RelA (miR-15b, −100, −125b, −146a, −150, −221) in both mouse striatal STHdhQ7/HdhQ7 cells and human cervical carcinoma HeLa cells; RT-PCR showing (B) Expression of pri-miR-15b, pri-miR-100, pri-miR-125b, pri-miR-146a, pri-miR-150 and pri-miR-221 in HeLa cells; (C) Decrease in the expression of the pri-miRNAs in presence of overexpressed p53-CFP; data are mean ± SD (n = 3); *P ≤ 0.05; ** P ≤ 0.01 compared to control. (D) Increase in the expression of the pri-miRNAs in presence of over expressed NFκB p65/RelA in HeLa cells; data are mean ± SD (n = 3); *P ≤ 0.05; ** P ≤ 0.01 compared to control. GAPDH mRNA level is taken as endogenous control in each case. Error bars represent standard deviation, * represents statistical significance.
p53 and NFκB p65/RelA alter transcription of the miRNAs in human cervical carcinoma HeLa cells
In order to examine whether miR-15b, −100, −125b, −146a, −150 and −221 are transcriptionally regulated by p53 and NFκB p65/RelA or whether their mature levels are altered by TFs affecting any of the miRNA processing events,10,30 p53 and NFκB p65/RelA were separately transfected in HeLa cells. RT-PCR carried out 24 hours following transfection revealed that p53 decreased the expression of pri-miR-100, pri-miR-125b, pri-miR-146a, pri-miR-150 and pri-miR-221 and NFκB p65/RelA increased the same at the transcriptional level (Fig. 5B-D). GAPDH transcript level was taken as endogenous control for calculating fold changes in pri-miRNA expression13. The expression level of pri-miR-15b however, remained unchanged both in presence of exogenous p53 or NFκB p65/RelA. Hence it was confirmed that 5 miRNAs viz., miR-100, −125b, −146a, −150 and −221 are regulated by both p53 and NFκB p65/RelA at the transcriptional level and not in the level of miRNA processing.
p53 modulates putative promoter activity of upstream sequence constructs of miRNA genes bearing predicted p53 binding sites in HeLa cells
In order to identify the specific binding sequences upstream of the 5 miRNA genes which were observed to be regulated by both p53 and NFκB p65/RelA, we have utilized in-house search tool www.bioinformatics.org/grn/npb3 as described earlier31 and the UCSC Genome Browser (http://genome.ucsc.edu). The details of such methods and the results obtained are shown in supplementary text. The summary of the result is shown in Table 1.
Table 1.
Summary of the transcription regulation of miRNAs by p53 and NFκB p65/RelA
| Potential p53/ NFκB p65/RelA (p65) BS |
Regulatory elements predicted based on chromatin state |
||||
|---|---|---|---|---|---|
| MiRNA genes (with chromosomal coordinates) | Region (1st nucleotide of the pre-miRNA taken as +1) | Putative p53 BS [Two (G/A)(G/A)(G/A)C(T/A)(T/A)G(C/T)(C/T)(C/T) sequences separated by 0–13 base pairs] | Putative NFκB p65/RelA BS [GG(G/A)(G/A)NN(C/T)(C/T)CC] | Functional elements predicted (UCSC Genome Browser) | Cell line where observed (Information taken from UCSC Genome Browser) |
| No. of sites | No. of sites | ||||
| hsa-miR-100 ( chr11: 122152229–122152308 [−] ) | A (−720 to −978) | p65 (2) | Strong enhancer | HSMM, NHLF | |
| B (−2486 to −2820) | p65 (1) | Active promoter | HSMM | ||
| C (−3493 to −3878) | p53 (1) | p65 (2) | Active promoter | HSMM | |
| D (−4722 to −5101) | p65 (3) | - | Active promoter | HMEC, HSMM | |
| E (−6621 to −6974) | p53 (1) | p65 (1) | Strong enhancer | HMEC, HSMM, NHEK, NHLF | |
| F (−7250 to −7628) | p65 (3) | Strong enhancer | |||
| G (−8658 to −9118) | p53 (2) | p65(1) | Strong enhancer | ||
| hsa-miR-146a ( chr5: 160485352–160485450 [+] ) | A (−19496- to 19175) | p53 (1) | p65 (2) | Weak transcription | HepG2, HMEC, NHLF |
| B1 (−17583 to −17135) | p53 (1) | p65 (5) | Active promoter | GM12878, HepG2 | |
| B2 (−14666 to −14179) | p53 (1) | p65 (3) | Active promoter | GM12878, HepG2 | |
| C (−8337 to −10136) | p65 (5) | Weak promoter | GM12878 | ||
| D (−4537 to −6336) | p53 (3) | p65 (4) | Weak promoter | GM12878 | |
| hsa-miR-150 ( chr19: 49500785–49500868 [−] ) | A (−143 to −736) | p53 (2) | p65 (1) | Weak enhancer | GM12878, K562, HepG2, HUVEC |
| B (−2097 to −2512) | p65 (4) | Weak promoter | HepG2 | ||
| C (−2688 to −3093) | p53 (1) | p65 (4) | Weak enhancer | HepG2 | |
| D (−4020 to −4258) | p53 (1) | p65 (4) | Weak enhancer | K562, HepG2 | |
| E (−4567 to −5658) | p53 (1) | p65 (5) | Weak enhancer | K562, HepG2 | |
| F (−11480 to −11640) | p53 (1) | p65 (2) | asatrong enhancer | HepG2, HUVEC, HSMM, NHLF | |
| G (−12795 to −14012) | p53 (1) | p65 (3) | Active promoter | HepG2 | |
| hsa-miR-125b ( chr11: 122099757–122099844 [−] ) | A (−1039 to −1239) | p65 (2) | Active promoter | HMEC, HSMM, NHLF | |
| B (−10601 to −10920) | p53 (1) | Weak enhancer | HUVEC | ||
| C (−125 to −414) | p53 (1) | Active promoter | HMEC, HSMM, NHLF | ||
| hsa-miR-221 ( chrX: 45746157–45746266 [−] ) | A (−4493 to −4773) | p65 (1) | Strong enhancer | HMEC, NHEK, NHLF | |
| B (−1619 to −1997) | p53 (1) | Strong enhancer | HMEC, NHEK, NHLF, HUVEC, HSMM | ||
| C (−4438 to −4458) | p53 (1) | Strong enhancer | HMEC, NHEK, NHLF | ||
The genomic co-ordinates of the miRNAs were identified from miRBase (http://www.mirbase.org/).
20 kb upstream sequences of the pre-miRNAs (1st nucleotide of the pre-miRNA taken as +1) were downloaded from UCSC Genome Browser (http://genome.ucsc.edu) for both human and mouse.
The putative p53 and NFκB p65/RelA binding sites (BS) at miRNA upstream sequences were analyzed by using the in-house search tool www.bioinformatics.org/grn/npb3.
The p53 binding site is a dimer, comprising of 2 monomers, each 10 nucleotides long, with a variable spacer that can range between 0 to 13 nucleotides. The consensus sequence of the monomer is RRRCWWGYYY (R = G or A, W = T or A, Y = C or T).The NFκB p65/RelA binding site is a monomer consisting of 10 nucleotides73 and the consensus sequence of the monomer is GGGRNNYYCC.
Presence of predicted regulatory regions based on ENCODE annotation data in the upstream sequences of the human miRNA genes were also evident from UCSC Genome Browser.
For any given miRNA the region in the 20 kb upstream with the highest score was considered. The 20 kb upstream sequence may have more than one putative p53 or p65 binding site with identical or lesser score other than the ones mentioned in the table.
Abbreviations used: GM12878: B-lymphocyte; HepG2: Hepatocellular carcinoma; HMEC: Mammary epithelial cells; HSMM: Skeletal muscle myoblasts; HUVEC: Umbilical vein endothelial cells; K562: Leukemia; NHEK: Epidermal keratinocytes; NHLF: Lung fibroblasts.
Region C in the upstream of miR-100 (Fig. S1A), region B1 in the upstream of miR-146a (Fig. S1B), region C in the upstream of miR-150 (Fig. S1C), region B in the upstream of miR-125b (Fig. S1D) and region B in the upstream of miR-221 (Fig. S1E) were PCR amplified from human genomic DNA and cloned into pGL3 basic vector and co-transfected in HeLa cells in presence of exogenous p53. It was observed that ectopic expression of p53 significantly reduced the basal luciferase activities of the constructs bearing segments of region C in the upstream of miR-100 (n = 3, p = 0.032), region B1 in the upstream of miR-146a (n = 3, p = 0.025) and region C in the upstream of miR-150 (n = 3, p = 0.019) respectively in HeLa cells (Fig. 6A). Conversely, knocking down endogenous p53 in HeLa cells by p53 siRNA markedly increased the luciferase activities of these constructs bearing the upstream sequences of miR-100 (n = 3, p = 0.0035), miR-146a (n = 3, p = 0.021) and miR-150 (n = 3, p = 0.029) respectively (Fig. 6A). However, although hsa-miR-125b and hsa-miR-221 are already known to be repressed by p53 in vivo,8,9 no direct evidence of p53 regulation at the putative binding sites i.e. at region B in the upstream of hsa-miR-125b and region B in the upstream of hsa-miR-221 were observed either in presence of exogenous p53 or by knocking down endogenous p53 in HeLa cells. Thus, it is likely that p53 might exert its suppressive effect on miR-125b and −221 through other regulatory sites. These results indicate that p53 represses the expression of miR-100, −146a and −150 in HeLa cells by inhibiting transcriptional activities at these sites in the regulatory sequences of the miRNA genes.
Figure 6.

p53 binds to the regulatory sequences in the upstream of miR-100, −146a and −150 genes in both human and mouse and represses their transcription (A) Changes in relative luciferase activity of upstream sequence constructs of miR-100, −146a, −150, −125b and −221 bearing putative p53 binding sites in presence of both exogenous p53 and on knocking down basal endogenous p53 by siRNA mediated gene silencing in human cervical carcinoma HeLa cells; data are mean ± SD (n = 3); *P ≤ 0.05; ** P ≤ 0.01 compared to control. Chromatin immunoprecipitation (ChIP) assay showing binding of ectopic p53 to its respective binding sites within the regulatory elements in the upstream of (B) miR-100, (C) miR-146a and (D) miR-150 in HeLa cells, (n = 3). (ChIP) assay showing binding of ectopic p53 as well as endogenous p53 to corresponding conserved regions bearing putative p53 binding sites in the upstream of (E) miR-100, (F) miR-146a and (G) miR-150 in mouse striatal STHdhQ7/HdhQ7 cells; (n = 3). Error bars represent standard deviation, * represents statistical significance. Solid lines represent putative NFκB p65/RelA binding sites and solid triangles represent putative p53 binding sites in the regulatory sequences of the miRNA genes.
Binding of p53 at regulatory sequences causes transcriptional inhibition of miR-100, −146a and −150 genes in HeLa and ST HdhQ7/HdhQ7 cells
Finally, p53 was over expressed in HeLa cells and ChIP assays were performed to detect its direct interaction if any with these putative cis-regulatory elements. Thus, isolated chromatin from p53 transfected HeLa cells were immunoprecipitated by anti-p53 antibody, followed by PCR analysis with primers targeted to sequences in region C in the upstream of miR-100, region B1 in the upstream of miR-146a and region C in the upstream of miR-150 respectively in HeLa cells. PCR analysis with primers targeted to a region of GAPDH promoter32 was taken as negative control in ChIP performed with anti p53 antibody. It was observed that p53 physically binds to regulatory sequences in the upstream of miR-100 (Fig. 6B), −146a (Fig. 6C) and −150 (Fig. 6D) in HeLa cells. Thus, these results demonstrate for the first time that p53 could directly bind to its response elements in the upstream sequence of miR-100, −146a and −150 and repress their transcription in human cervical carcinoma cell line viz., HeLa cells.
In order to see whether such regulation by p53 is also conserved in mouse as predicted earlier by analyzing miRNA profiling data in STHdhQ7/HdhQ7 cells in presence of exogenous p53, ChIP assays were carried out by immunoprecipitating isolated chromatin from both wild type STHdhQ7/HdhQ7 cells as well as from p53 transfected STHdhQ7/HdhQ7 cells with anti-p53 antibody. This was followed by PCR analysis with primers targeted to corresponding conserved region C' in the upstream of miR-100, region B1' in the upstream of miR-146a and region C' in the upstream of miR-150 respectively. It was observed that both endogenous p53 as well as ectopic p53 could physically bind to regulatory sequences in the upstream of miR-100 (Fig. 6E), −146a (Fig. 6F) and −150 (Fig. 6G) in mouse striatal cells as well. These results demonstrate for the first time that p53 binds to regulatory sequences in the upstream of miR-100, −146a and −150 in both human and mouse and represses transcription of these miRNA genes.
NFκB p65/RelA modulates putative promoter activity of upstream sequence constructs of miRNA genes bearing predicted NFκB p65/RelA binding sites in HeLa cells
In order to investigate the role of NFκB p65/RelA and to test the putative promoter activity of the upstream sequences of miRNA genes bearing putative NFκB p65/RelA binding sites region A, B and C in the upstream of miR-100 (Fig. S1A), region B1 in the upstream of miR-146a (Fig. S1B), region B and C in the upstream of miR-150 (Fig. S1C), region A in the upstream of miR-125b (Fig. S1D) and region A in the upstream of miR-221 (Fig. S1E) were PCR amplified from human genomic DNA, cloned into pGL3 basic vector and co-transfected in HeLa cells in presence of exogenous NFκB p65/RelA. It was observed that ectopic expression of NFκB p65/RelA significantly increased the basal luciferase activities of the constructs bearing segments of region B (n = 3, p = 0.0019) and C (n = 3, p = 0.023) in the upstream of miR-100 (Fig. 7A(i)), region B1 in the upstream of miR-146a (n = 3, p = 0.037) (Fig. 7A(ii)), region B (n = 3, p = 0.0028) and C (n = 3, p = 0.0034) in the upstream of miR-150 (Fig. 7A(iii)), region A in the upstream of miR-125b (n = 3, p = 0.0015) (Fig. 7A(iv)) and region A in the upstream of miR-221 (n = 3, p = 0.0026) (Fig. 7A(v)) respectively in HeLa cells. Conversely, inhibiting basal endogenous NFκB p65/RelA activity by treating HeLa cells with 2mM Aspirin markedly repressed the luciferase activities of these constructs bearing the upstream sequences of miR-100 (region B, n = 3, p = 0.021; region C, n = 3, p = 0.014) (Fig. 7A(i)), miR-146a (region B1, n = 3, p = 0.025) (Fig. 7A(ii)), miR-150 (region B, n = 3, p = 0.0027; region C, n = 3, p = 0.0023) (Fig. 7A(iii)), miR-125b (region A, n = 3, p = 0.013) (Fig. 7A(iv)) and miR-221 (region A, n = 3, p = 0.0045) (Fig. 7A(v)) respectively. These results confirm that NFκB p65/RelA regulates the expression of miR-146a,11 −125b12 and −22133 and demonstrate for the first time that NFκB p65/RelA induces the expression of miR-100 and −150 in HeLa cells by enhancing transcriptional activities at regulatory sites in the upstream sequences of the miRNA genes. It had already been reported that NFκB p65/RelA induces the expression of miR-146a by binding to and activating transcription at sites −1121 and −3432 in the upstream sequence of miR-146a gene.11 This report identifies a novel NFκB p65/RelA binding site at −17,290 which also contributes to miR-146a regulation by the TF.
Figure 7.
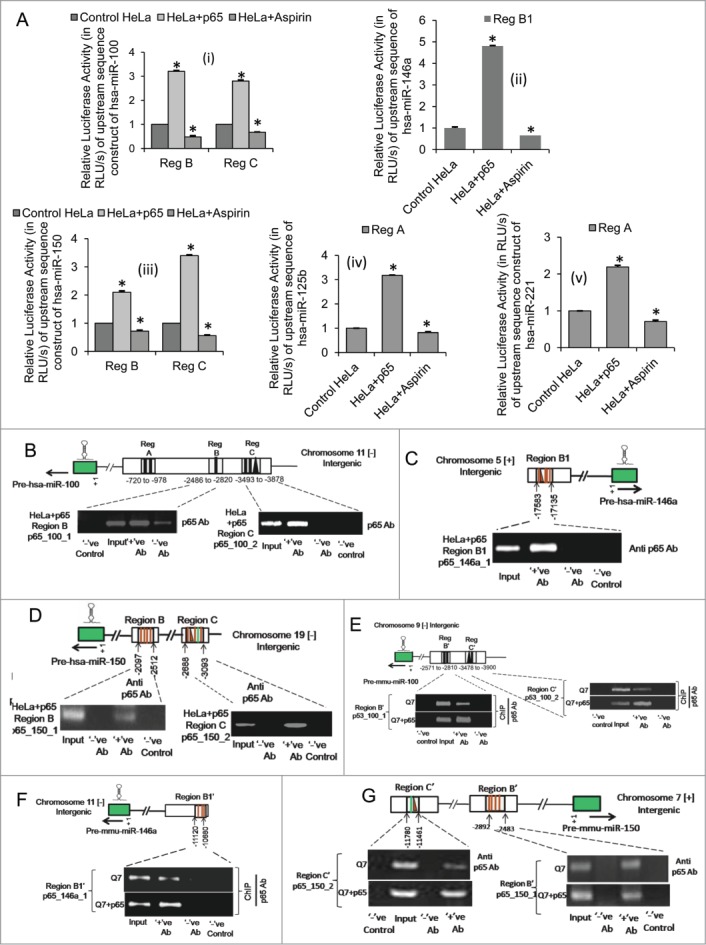
NFκB p65/RelA binds to the regulatory sequences in the upstream of miR-100, −146a and −150 genes in both human and mouse and activates their transcription (A) Changes in relative luciferase activity of upstream sequence constructs of (i) miR-100, (ii) miR-146a, (iii) miR-150, (iv) miR-125b and (v) miR-221 bearing putative NFκB p65/RelA binding sites in presence of both exogenous NFκB p65/RelA and on inhibition of basal endogenous NFκB p65/RelA activity in human cervical carcinoma HeLa cells; data are mean ± SD (n = 3); *P ≤ 0.05; ** P ≤ 0.01 compared to control. Chromatin immunoprecipitation (ChIP) assay showing binding of ectopic NFκB p65/RelA to its respective binding sites within the regulatory elements in the upstream of (B) miR-100, (C) miR-146a and (D) miR-150 in HeLa cells, (n = 3). (ChIP) assay showing binding of ectopic NFκB p65/RelA as well as endogenous NFκB p65/RelA to corresponding conserved regions bearing putative NFκB p65/RelA binding sites in the upstream of (E) miR-100, (F) miR-146a and (G) miR-150 in mouse striatal STHdhQ7/HdhQ7 cells; (n = 3). Error bars represent standard deviation, * represents statistical significance. Solid lines represent putative NFκB p65/RelA binding sites and solid triangles represent putative p53 binding sites in the regulatory sequences of the miRNA genes.
Promoter binding of NFκB p65/RelA sub-unit is required for the transcription of NFκB dependant miR-100, -146a and -150 genes in HeLa and STHdhQ7/HdhQ7 cells
Finally, NFκB p65/RelA was overexpressed in HeLa cells and ChIP assays were performed to detect its direct interaction if any with these putative cis-regulatory elements. Thus, isolated chromatin from NFκB p65/RelA transfected HeLa cells were immunoprecipitated by anti-p65 antibody, followed by PCR analysis with primers targeted to sequences in region B and C in the upstream of miR-100, region B1 in the upstream of miR-146a and region B and C in the upstream of miR-150 respectively in HeLa cells. PCR analysis with primers targeted to a region on chromosome 1 (Chr1: 204,366,822–204,366,872)6 was taken as negative control in ChIP peformed with anti-p65 antibody. It was observed that NFκB p65/RelA physically binds to regulatory sequences in the upstream of miR-100 (Fig. 7B), miR-146a (Fig. 7C) and miR-150 (Fig. 7D) in HeLa cells. Thus, these results identify a novel NFκB p65/RelA regulatory site in the upstream of miR-146a and demonstrate for the first time that NFκB p65/RelA could directly bind to its response elements in the upstream sequence of miR-100 and −150 and activate their transcription in human cervical carcinoma cell line viz., HeLa cells.
In order to see whether such regulation by NFκB p65/RelA is also conserved in mouse as predicted earlier by analyzing miRNA profiling data in STHdhQ7/HdhQ7 cells in presence of exogenous NFκB p65/RelA, ChIP assays were carried out by immunoprecipitating isolated chromatin from both wild type STHdhQ7/HdhQ7 cells and from p65 sub-unit transfected STHdhQ7/HdhQ7 cells with anti-p65 antibody. This was followed by PCR analysis with primers targeted to corresponding conserved regions B' and C' in the upstream of miR-100, region B1' in the upstream of miR-146a and region B' and C' in the upstream of miR-150 respectively. It was observed that both endogenous NFκB p65/RelA as well as ectopic NFκB p65/RelA could physically bind to regulatory sequences in the upstream of miR-100 (Fig. 7E), miR-146a (Fig. 7F) and miR-150 (Fig. 7G) in mouse striatal cells as well. These results demonstrate for the first time that NFκB p65/RelA binds to regulatory sequences in the upstream of miR-100, −146a and −150 in both human and mouse and induces transcription of these miRNA genes.
Functional cross-talk between p53 and p65 (NFκB/RelA) governs NFκB p65/RelA activity in mouse STHdhQ7/HdhQ7 cells
These functional p53 and NFκB p65/RelA binding sites in the upstream regulatory sequences lie in close proximity (Figs. S1A, B, C) and the fact that p53 binds and represses transcription and NFκB p65/RelA binds and activates transcription of these miRNAs at these binding sites suggest for the possibility of a functional cross-talk between p53 and NFκB p65/RelA in co-regulating miRNA expression. In order to validate our hypothesis of the existence of a possible cross-talk between p53 and NFκB p65/RelA, p53 was over expressed in STHdhQ7/HdhQ7 cells. Ectopic expression of p53 in STHdhQ7/HdhQ7 cells decreased NFκB p65/RelA response element (p65-RE) activity (n = 3, p = 0.027) 48 hours post transfection and reduction in endogenous p53 level via p53 siRNA mediated gene silencing led to an increase (n = 3, p = 0.045) in the same (Fig. 8A). This observation is supported by immunoblot analysis with nuclear and cytoplasmic extract from p53 transfected STHdhQ7/HdhQ7 cells which showed reduced (n = 3, p = 0.018) nuclear content of NFκB p65/RelA in presence of overexpressed p53 (Fig. 8B). Similarly, expression of NFκB p65/RelA responsive chemokine CCL534 was significantly decreased (n = 3, p = 0.021) in STHdhQ7/HdhQ7 cells in presence of ectopic p53 (Fig. 8C). This showed that ectopic p53 inhibited NFκB p65/RelA in STHdhQ7/HdhQ7 cells. Conversely, over expression of NFκB p65/RelA in STHdhQ7/HdhQ7 cells did not alter p53 nuclear content (Fig. 8D) or the expression of p53 responsive CASP1 gene (Fig. 8E) indicating that NFκB p65/RelA did not influence p53 activity in mouse striatal cells.
Figure 8.
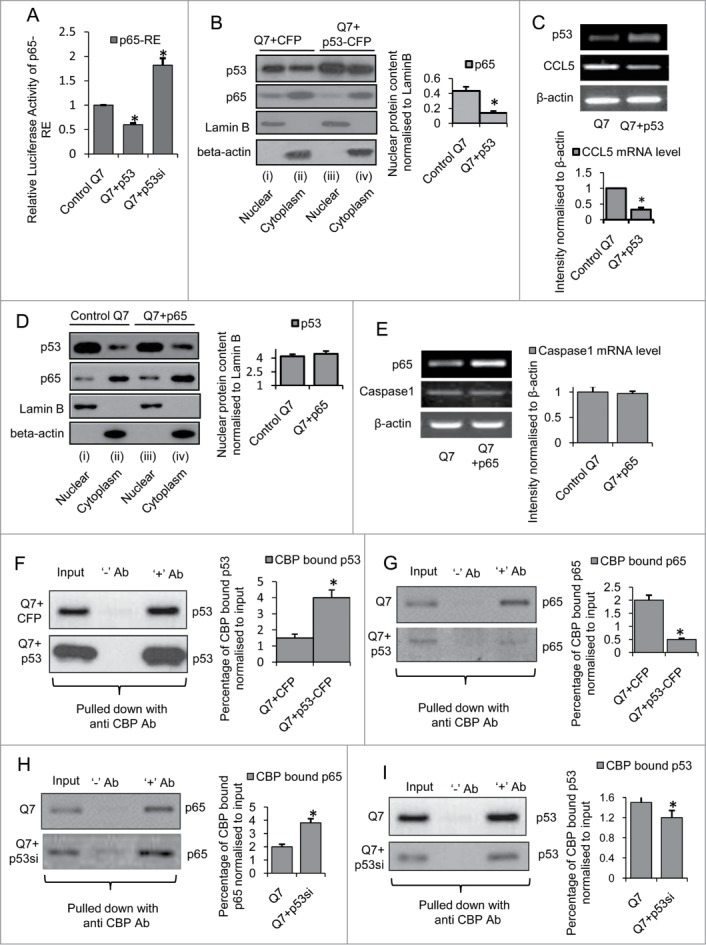
Ectopic expression of p53 in mouse striatal STHdhQ7/HdhQ7 cells competes with NFκB p65/RelA for CBP binding and inhibits NFκB p65/RelA activity (A) Luciferase assay showing reduced NFκB p65/RelA response element (p65-RE) activity in presence of overexpressed p53 in STHdhQ7/HdhQ7 cells and increased p65-RE activity in STHdhQ7/HdhQ7 cells with knocked down p53; data are mean ± SD (n = 3); *P ≤ 0.05; ** P ≤ 0.01 compared to control. (B) Western Blot showing increased p53 nuclear content and decreased p65 nuclear content and (C) RT-PCR showing reduction in CCL5 expression in STHdhQ7/HdhQ7 in presence of exogenous p53-CFP; data are mean ± SD (n = 3); *P ≤ 0.05; ** P ≤ 0.01 compared to control. (D) Western Blot showing no change in p53 nuclear content on exogenous expression of NFκB p65/RelA in STHdhQ7/HdhQ7 cells, n = 3. (E) RT-PCR showing no change in Caspase1 mRNA level on ectopic expression of NFκB p65/RelA in STHdhQ7/HdhQ7 cells, n = 3. Co-immunoprecipitation assay showing (F) increased p53 binding and (G) decreased p65 binding to CBP in p53-CFP transfected STHdhQ7/HdhQ7 cells compared to wild type STHdhQ7/HdhQ7 cells; data are mean ± SD (n = 3); *P ≤ 0.05; ** P ≤ 0.01 compared to control. Co-immunoprecipitation assay showing (H) increased p65 binding and (I) decreased p53 binding to CBP on knocking down endogenous p53 in STHdhQ7/HdhQ7 cells; data are mean ± SD (n = 3); *P ≤ 0.05; ** P ≤ 0.01 compared to control. Error bars represent standard deviation, * represents statistical significance.
Moreover, elevated p53 was also observed to compete with NFκB p65/RelA for CBP binding. In a series of co-immunoprecipitation experiments using cell extracts from both control STHdhQ7/HdhQ7 cells and STHdhQ7/HdhQ7 cells transfected with exogenous p53 pulled down with anti-CBP antibody it was revealed that p53 could bind more strongly with CBP (p = 0.024) in p53 transfected STHdhQ7/HdhQ7 cells compared to control STHdhQ7/HdhQ7 cells (Fig. 8F) and NFκB p65/RelA could bind less strongly with CBP (p = 0.013) under such conditions (Fig. 8G). In order to examine whether this reduced binding of NFκB p65/RelA with CBP in p53 transfected STHdhQ7/HdhQ7 cells was due to competition by elevated nuclear p53 for CBP, endogenous p53 was knocked down in STHdhQ7/HdhQ7 cells and immunoprecipitation with cell extracts prepared 72 hours post transfection (which significantly decreased nuclear p53 content, p = 0.035) showed an increased binding of NFκB p65/RelA with CBP (p = 0.0041) (Fig 8H) and a reduced binding of p53 with CBP (Fig. 8I) compared to control STHdhQ7/HdhQ7 cells. Thus, the results obtained indicate that sequestration of transcriptional co-activator p300/CBP by elevated p53 in p53 transfected STHdhQ7/HdhQ7 cells lead to reduced NFκB p65/RelA binding with p300/CBP which possibly accounts for the observed p53 mediated repression of endogenous NFκB p65/RelA activity in these cells. Similar nuclear competition between p53 and NFκB p65/RelA for a limiting amount of CBP were also reported earlier.36–38 Thus, these observations suggest for the co-regulation of miRNAs by p53 and NFκB p65/RelA where NFκB p65/RelA induces transcription of miR-100, −146a and −150 and p53 represses the same by inhibiting endogenous NFκB p65/RelA activity.
Co-regulation of miR-100, −146a and −150 by p53 and NFκB p65/RelA in mouse striatal STHdhQ7/HdhQ7 cells
In order to validate our hypothesis, upstream sequence constructs bearing functional p53 and NFκB p65/RelA binding sites in close proximity viz., region C in the upstream of miR-100 (Fig. S1A), region B1 in the upstream of miR-146a (Fig. S1B) and region C in the upstream of miR-150 (Fig. S1C) were co-transfected in mouse striatal STHdhQ7/HdhQ7 cells along with p53 and/or NFκB p65/RelA. Luciferase assay with region C bearing both functional p53 and NFκB p65/RelA binding sites in the upstream of miR-100 revealed that ectopic NFκB p65/RelA could increase the luciferase activity of the construct by over 7-fold (n = 3, p = 0.042, * represents statistical significance where * P ≤ 0.05; ** P ≤ 0.01) of the basal value. However, such increase was significantly reduced in presence of ectopic p53 (n = 3, p = 0.029, † represents statistical significance where † P ≤ 0.05; †† P ≤ 0.01) (Fig 9A). Interestingly, on overexpressing NFκB p65/RelA along with p53, the luciferase activity of the construct could not revive and was significantly less (n = 3, p = 0.011, † represents statistical significance where † P ≤ 0.05; †† P ≤ 0.01) than that observed in presence of NFκB p65/RelA alone (Fig. 9A). These results indicate that apart from repressing miR-100 expression via inhibiting NFκB p65/RelA activity, p53 here could also decrease miR-100 expression directly by binding to its putative binding site in the upstream sequence of miR-100.
Figure 9.

Functional cross-talk between p53 and NFκB p65/RelA regulate miR-100, −146a and −150 expressions in mouse striatal STHdhQ7/HdhQ7 cells Changes in relative luciferase activity of (A) miR-100 upstream sequence construct (B) miR-146a upstream sequence construct and (C) miR-150 upstream sequence construct in presence of exogenous p53-CFP and NFκB p65/RelA in mouse striatal STHdhQ7/HdhQ7 cells. In presence of exogenous NFκB p65/RelA luciferase activities of the constructs were increased whereas in presence of exogenous p53-CFP the luciferase activities of the constructs were significantly reduced; data are mean ± SD (n = 3); *P ≤ 0.05; ** P ≤ 0.01 compared to control. (D) Western blot showing increase in p65 protein level by more than 3 fold (*p≤ 0.05) and increase in p53 protein level by more than two fold († p≤ 0.05) when 2.5 μg each of p65 and p53 plasmid clones was transfected either in isolation or in combination in STHdhQ7/HdhQ7 cells compared to control STHdhQ7/HdhQ7 cells; (n = 3). (E) Chromatin immunoprecipitation assay showing decreased NFκB p65/RelA occupancy at its binding site in miR-100 upstream sequence (Reg C) in p53-CFP transfected STHdhQ7/HdhQ7 cells compared to control STHdhQ7/HdhQ7 cells, data are mean ± SD (n = 3); *P ≤ 0.05; ** P ≤ 0.01 compared to control. This indicates p53 driven exclusion of NFκB p65/RelA from its binding site that partially contributes to miR-100 repression. Error bars represent standard deviation, * represents statistical significance.
In order to investigate the role of p53 and NFκB p65/RelA in co-regulating miR-146a expression, promoter luciferase assay with the upstream construct encompassing region B1 with both functional p53 and NFκB p65/RelA binding sites was performed in presence of overexpressed p53 and NFκB p65/RelA. The results obtained revealed that ectopic NFκB p65/RelA could increase the expression of miR-146a by 6.4-fold of the basal level (n = 3, p = 0.039, * represents statistical significance where * P ≤ 0.05; †† P ≤ 0.01) (Fig. 9B) which was significantly reduced in presence of exogenous p53 (n = 3, p = 0.0085, † represents statistical significance where † P ≤ 0.05; †† P ≤ 0.01). However, on addition of NFκB p65/RelA along with p53 luciferase activity of the construct could not revive and was significantly less (n = 3, p = 0.0085, † represents statistical significance where † P ≤ 0.05; †† P ≤ 0.01) than that observed in presence of NFκB p65/RelA alone (Fig. 9B). These indicate inhibition of NFκB p65/RelA activity by p53. In order to investigate the role of p53 and NFκB p65/RelA in co-regulating miR-150 expression, promoter luciferase assay with the upstream construct containing region C with both functional p53 and NFκB p65/RelA binding sites revealed that ectopic NFκB p65/RelA could increase the luciferase activity of the construct by over 2-fold (n = 3, p = 0.0033, * represents statistical significance where * P ≤ 0.05; ** P ≤ 0.01) (Fig. 9C) which was reduced to the basal level in presence of p53) (n = 3, p = 0.021, † represents statistical significance where † P ≤ 0.05; †† P ≤ 0.01). On overexpressing NFκB p65/RelA along with p53, the luciferase activity of the construct could not revive and was significantly less (n = 3, p = 0.0039, † represents statistical significance where † P ≤ 0.05; †† P ≤ 0.01) than that observed in presence of NFκB p65/RelA alone (Fig. 9C). This indicated that p53 could strongly inhibit miR-150 at this upstream region.
It is to be mentioned here that under identical experimental conditions with those maintained in the luciferase assays, western blot analysis revealed an increase in NFκB p65/RelA protein level by more than 3-fold when 2.5 μg of the plasmid was transfected both in isolation (n = 3, p = 0.029, * represents statistical significance where * P ≤ 0.05; ** P ≤ 0.01) and in combination with equal amount of p53-CFP (n = 3, p = 0.036, * represents statistical significance where * P ≤ 0.05; ** P ≤ 0.01) in STHdhQ7/HdhQ7 cells (Fig. 9D). Similarly, ectopic expression of p53-CFP in isolation (n = 3, p = 0.021, † represents statistical significance where † P ≤ 0.05; †† P ≤ 0.01) as well as in combination with equal amount of NFκB p65/RelA (n = 3, p = 0.018, † represents statistical significance where † P ≤ 0.05; †† P ≤ 0.01) demonstrate a 2-fold increase in p53 protein level in both the cases (Fig. 9D). Thus, NFκB p65/RelA protein level remains the same when expressed isolated and in combination with p53-CFP in STHdhQ7/HdhQ7 cells. Therefore the partial recovery in luciferase activities of the constructs as observed in Figure 9A-C in presence of both NFκB p65/RelA and p53 as compared to those in presence of NFκB p65/RelA alone is not due to reduced NFκB p65/RelA protein level but due to inhibition of NFκB p65/RelA activity by p53 or due to other p53 mediated repression mechanism(s).
p53 binds to region C in the upstream of miR-100 and displaces NFκB p65/RelA from its functional binding site thereby repressing transcription of miR-100 gene
The co-regulation of miR-100 by p53 and NFκB p65/RelA as observed in the present work could be explained based on prediction in the context of nuclear cross-talk or functional interaction between p53 and NFκB p65/RelA by models previously described by Schneider et al.39 The co-regulation of miR-100 at region C (−3493 to −3878) (Fig. 9A) by p53 and NFκB p65/RelA can be explained by Exclusion model where elevated p53 in p53 transfected STHdhQ7/HdhQ7 cells might have prevented binding of endogenous NFκB p65/RelA at the closely placed or overlapping p65 binding site thereby reducing miR-100 transcription. In order to validate our hypothesis, chromatin immunoprecipitation was done using anti-p65 antibody both in control STHdhQ7/HdhQ7 cells and in STHdhQ7/HdhQ7 cells transfected with exogenous p53. Over expression of p53-CFP in wild type STHdhQ7/HdhQ7 cells resulted in decreased p65 occupancy (p = 0.032) at region C in the upstream of miR-100 (Fig. 9E) 48 hours post transfection compared to control STHdhQ7/HdhQ7 cells transfected with empty vector CFP-C1. These results demonstrate that ectopic p53 in STHdhQ7/HdhQ7 cells binds to miR-100 upstream sequence and displaces NFκB p65/RelA from its nearby binding site. This may partially account for the p53 mediated repression of miR-100 expression in STHdhQ7/HdhQ7 cells. However, since p53 mediated suppression of miRNA expression could not be entirely retrieved by overexpressing NFκB p65/RelA, therefore co-regulation of miR-100 by p53 and NFκB p65/RelA cannot be solely explained by the Exclusion Model. Presently we are investigating additional mechanisms of co-regulation of miR-100 by p53 and NFκB p65/RelA.
Discussion
Like protein-coding genes, most miRNA genes are initially transcribed by RNA Polymerase II in the nucleus as primary transcripts1 and can be regulated by transcription factors (TFs).5 In this context, good many number of papers have been published describing transcription factors that bind to promoter regions of miRNA genes and regulate their expression either in physiological or pathological conditions.13 In this work, it was observed that p53 could increase the expression of 3 miRNAs viz., miR-145, −34a, −199a-5p and decrease the expression of 6 miRNAs viz., miR-15b, −100, −125b, −146a, −150 and −221 (Figs. 1 and 2) and NFκB p65/RelA could increase the expression of 8 miRNAs viz., miR-15a, −15b, −100, −125b, −135a, −146a, −150 and −221 and decrease the expression of 4 miRNAs viz., miR-214, −199a-3p, −148a and −182 in mouse striatal STHdhQ7/HdhQ7 cells and human cervical carcinoma HeLa cells (Figs. 3 and 4). However, miR-145, −34a, −15b, −100, −125b and −221 were already known to be regulated by p53.8,40–42 Similarly, miR-146a, −221, −15a, −34a, −214 and −125b were already known to be regulated by NFκB p65/RelA.11,12,28,27,33 In our earlier work, we had reported that miR-146a could be regulated by p53.43 Thus, this report demonstrates for the first time that -146a, −150 and −199a-5p could be regulated by p53 and that NFκB p65/RelA increases the expression of miR-100, −150 and −135a and decreases the expression of miR-199a-3p and −148a in both mouse and human and across diverse cell lines. Of these miRNAs, miR-100, −125b, −146a, −150 and −221 have been observed to be transcriptionally repressed by p53 and induced by NFκB p65/RelA (Fig. 5). 20 kb upstream sequences of these miRNA genes also harboured putative p53 and NFκB p65/RelA binding sites (Table 1). Promoter luciferase assays and chromatin immunoprecipitation (ChIP) demonstrated p53 and NFκB p65/RelA to bind to their respective binding sites in the upstream sequences of miR-100, −146a and −150 and regulate their expression both in HeLa and STHdhQ7/HdhQ7 cells (Figs. 6 and 7). Regulation of miRNA genes by direct physical binding of p53 has been previously shown for several miRNAs viz., miR-34a,40 miR-34b, −34c,44 miR-192/194/215,45 miR-17–92 cluster,32 miR-107,46 miR-200c/141,47 miR-149*,48 miR-605,49 miR-124650 and miR-1204.51 Similarly, NFκB p65/RelA has been earlier reported to bind with upstream sequence and regulate the expression of miR-146a,11 miR-9,52 miR-125b-1, −21, −23b, −30b,12,13 miR-17–92, −30a, −130a,13 miR-301,53 miR-221,33 miR-34a.28 In the present work, it has been observed for the first time that p53 can bind with the upstream sequence and suppress the expression of miR-100, −146a and −150 and that NFκB p65/RelA can bind with the upstream sequence and induce the expression of miR-100 and −150 in both human and mouse. A novel NFκB p65/RelA binding site in the upstream sequence of miR-146a has also been identified in the present work.
These functional p53 and NFκB p65/RelA binding sites in the upstream regulatory sequences lie in close proximity (Fig. S1A, B, C) and the fact that p53 binds and represses transcription and NFκB p65/RelA binds and activates transcription of these miRNAs at these binding sites suggest for the possibility of a functional cross-talk between p53 and NFκB p65/RelA in co-regulating miRNA expression. Interestingly, elevated p53 in STHdhQ7/HdhQ7 cells was observed to inhibit NFκB p65/RelA nuclear content and activity and repress NFκB p65/RelA dependant CCL5 expression (Fig. 8A–C). However, ectopic NFκB p65/RelA did not alter p53 nuclear content or p53 mediated CASP1 gene expression (Fig. 8D & E). Moreover, elevated p53 was also observed to compete with NFκB p65/RelA for CBP binding (Fig. 8F-I). Since nuclear cross-talk between 2 transcription factors (TFs) can occur at several levels where any one TF can influence the activity of the other to define the outcome of a process,39 therefore observations in the present work support for the existence of a cross-talk between these 2 TFs in co-regulating miRNA expression. In fact, luciferase assays demonstrated p53 and NFκB p65/RelA to co-regulate miR-100, −146a and −150 expression where p53 mediated inhibition of NFκB p65/RelA activity possibly led to miRNA repression (Fig. 9A–D). In such case, ectopic expression of p53 in STHdhQ7/HdhQ7 cells was observed to reduce NFκB p65/RelA occupancy at Region C in the upstream of miR-100 (Fig. 9E). This suggests one of the possible mechanisms by which p53 represses miR-100 expression by inhibiting NFκB p65/RelA mediated activation. However, since such p53 mediated suppression of miRNA expression could not be entirely retrieved by over expressing NFκB p65/RelA as suggested by luciferase assays (Fig. 9A–C), this indicates the involvement of other mechanisms in the process.
Such prediction in the context of transcriptional cross-talk between p53 and NFκB p65/RelA by models were previously described by Schneider et al.39 and illustrated in Figure 10. The upstream region bearing both p53 and NFκB p65/RelA binding sites through which the miRNAs can be co-regulated are indicated by region C (−3493 to −3878) in the upstream of miR-100 (Fig. S1 A), region B1 (−17583 to −17135) in the upstream of miR-146a (Fig. S1 B) and region C (−2688 to −3093) in the upstream of miR-150 (Fig. S1 C). p53 mediated repression of miR-100, −146a and −150 could be due to binding of p53 at these sites which could (i) lead to exclusion of NFκB p65/RelA from the nearly overlapping NFκB p65/RelA binding site (Exclusion model) or (ii) competition with NFκB p65/RelA for limited pool of co-activator CBP (Competition model) thereby causing such repression or (iii) facilitate recruitment of other factors necessary for repression (Cis-repression Model) or (iv) by more than one of these mechanisms.
Figure 10.
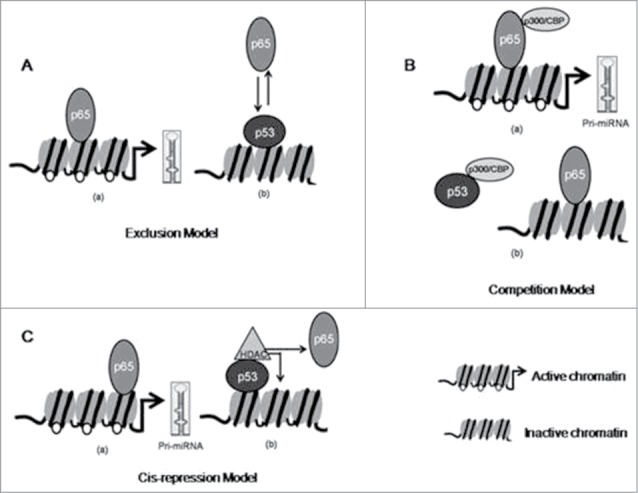
Probable mechanism of cross-talk between p53 and NFκB p65/RelA (p65) in regulating miRNA expression Cross-talk between p53 and p65 in regulating miRNA expression where p53 represses p65 transactivation by (A) Exclusion Model in which elevated p63 displaces p65 from overlapping or nearby DNA binding sites; (B) Competition Model in which elevated p53 competes with p65 for p300/CBP binding; (C) Cis-repression Model in which p53 binds with DNA and recruits HDAC and suppresses expression of genes.
Thus, the co-regulation of miR-100 at region C (Fig. 9A) by p53 and NFκB p65/RelA can be partly explained by Exclusion model (Fig. 10A) where elevated p53 in p53 transfected cells might have prevented binding of low endogenous p65 at the closely placed or overlapping site thereby reducing miR-100 transcription as evident in Figure 9D. Besides, co-regulation of miR-100 at region C by p53 and NFκB p65/RelA can also be explained by Cis-repression model (Fig. 10C) where acetylated Histone H3 at the regulatory region C (Fig. S1A) might be reduced by the recruitment of HDAC by p53. Moreover, NFκB p65/RelA itself functions as a transcriptional activator or as a repressor by interacting with HAT and HDAC respectively.54 Hence increased NFκB p65/RelA association with HDAC-containing co-repressor complexes might have also contributed to reduced miR-100 expression. Presently, we are investigating whether such Cis-repression Model may also explain the mechanism of co-regulation of miR-100 by p53 and NFκB p65/RelA. Similarly, the co-regulation of miR-146a at region B1 (Fig. 9B) can be explained by the Exclusion model or by Cis-repression model as evident in Supplementary Fig. 1B. Additionally, the presence of p300/CBP binding site at region E in the upstream of miR-100 (Fig. S1A) and near Region B1 in the upstream of miR-146a (Fig. S1B) may include the possibility of co-regulation of miR-100 and −146a by Competition model (Fig. 10B) as well. Similarly, the co-regulation of miR-150 at region C (Fig. 9C) by p53 and NFκB p65/RelA could be explained by Cis-repression model (Fig. 10) as evident in Supplementary Figure 1C. Although region A in the upstream sequence of miR-125b did not bear a putative p53 binding site (Fig. S1D) yet miR-125b could be regulated by p53 at this site through non-specific DNA binding following Cis-repression Model or by inhibiting NFκB p65/RelA transactivation following Competition Model. Although not verified by us, the presence of putative p53 binding site in close proximity with functional NFκB p65/RelA binding site immediately adjacent to Region A of miR-221 upstream sequence (Fig. S1E) may also explain for the observed repression in miR-221 expression by p53 following the Exclusion Model discussed or it may also occur following Cis-repression model at region A. Further extensive studies would be required to validate our prediction.
Thus results obtained in the present work demonstrate the existence of a functional cross-talk between p53 and NFκB p65/RelA in co-regulating miRNA expression. In fact, several instances of cross-talks between p53 and NFκB p65/RelA had been previously reported where one TF was observed to enhance or repress the activity of the other. Depending upon cellular needs, p53 may either repress transcription from NFκB p65/RelA gene promoter,3 inhibit NFκB p65/RelA activity,36–38,55,56 or may enhance the transcriptional activity of p65.57,58 NFκB p65/RelA on the other hand may either increase the expression of p5359,60 or may inhibit p53 transactivation.37,61,62 There are even instances of transcriptional co-operation between these 2 TFs in the co-regulation of miR-224 expression in mouse ovarian granulosa cells.20 Besides, there has been a report where p53 has been observed to act as a co-factor binding to RelA/p65 and inducing miR-21 expression in human heart failure.19 Very recently p53 and NFκB p65/RelA have been observed to play significant role in head and neck squamous cell carcinoma (HNSCC) by regulating the expression of miR-21 and −34a/c thereby modulating downstream target genes.21 This discrepancy in the functional relationship between p53 and NFκB p65/RelA may be attributed to different cell types and methods for activating/inactivating p53 and NFκB p65/RelA used by different investigators in their studies.
The importance of the present findings lies in the fact that the pro-apoptotic effect of p53 and the pro-survival functions of NFκB can be largely mediated via the biological roles of the miRNAs these TFs regulate. In general, p53 induced miRNAs viz., miR−34, −200, −15/16 and −192/194/215 families, −145 and −107 are mediators of tumor suppression and stress responses thereby executing these well characterized functions of p53.30 Upon induction, these miRNAs target genes affecting important biological processes such as metabolism, cell cycle progression, migration, epithelial–mesenchymal transition, stemness, differentiation and cell survival. NFκB p65/RelA regulated miRNAs on the other hand generally target genes critical to innate and adaptive immunity, cell proliferation, inflammation, and tumor development thereby largely mediating the biological functions of the TF.63 NFκB p65/RelA directly binds and induces the transcription of miR-146a, −125b, −9, −155, −21, −221, −222 among others while suppresses the expression of miR-199a/214 cluster.27 The 2 TFs p53, NFκB p65/RelA and miRNAs are involved in several TF-miRNA loops. The two master TFs p53 and NFκB p65/RelA are also known to influence the transcriptional activities of one another depending on cellular context. The combinatorial effect of the extensive physical and functional cross-talks that exist between p53 and NFκB p65/RelA has been observed to define the outcome of several biological processes. This work reports for the first time the existence of a functional cross-talk between p53 and NFκB p65/RelA that resulted in regulation of miR-100, −146a and −150 expressions in diverse cell types and diverse species.
Materials and Methods
Mouse striatal STHdhQ7/HdhQ7 cells and human cervical carcinoma HeLa cells
Immortalized striatal STHdhQ7/HdhQ7 cells were established from wild type (Q7/7) Hdh knock-in mice which express full-length Huntingtin gene (HTT) with 7 Glu (Q) residues.64 These STHdhQ7/HdhQ7 cells are primarily used as controls in experiments where STHdhQ111/HdhQ111 cells expressing endogenous full-length mutant HTT gene with 111 Glu (Q) residues are extensively used as cell model of Huntington's disease (HD) for identifying molecular alterations in the disease pathogenesis. Prof. Marcy E. MacDonald of Massachusetts General Hospital, USA kindly donated us these cell lines. The human cervical carcinoma cell line i.e., HeLa cells were obtained from National Cell Science Center, Pune, India.
Cell culture and transfection
Immortalized striatal STHdhQ7/HdhQ7 cells were cultured in DMEM (Cat No. AT006, HiMedia) supplemented with 10% (v/v) heat inactivated FBS (Cat No. S1810–500, Biowest), antibiotics penicillin/streptomycin PS 1% (v/v) and 400 μg/ml G418 (Cat No. sc-29065A, Santa Cruz Biotechnology) at 33°C in humidified condition and 5% CO2. Human cervical carcinoma cells (HeLa) were cultured in MEM (Cat No. AT020, HiMedia), 10% (v/v) FBS (Cat No. S1810–500, Biowest), 1% (v/v) PS at 37°C in humidified condition and 5% CO2.
All transfections were carried on 70–80% confluent cells using Lipofectamine 2000 (Cat No. 11668–019, Invitrogen) as per manufacturer's protocol. Unless otherwise mentioned, for single transfection experiment 1 µg (30 mm plate), 2.5 μg (60 mm plate) or 5 μg (100 mm plate) i.e. amounting to 0.25 nM of plasmid DNA constructs as well as 5 μl, 10 μl or 15 µl of Lipofectamine 2000 respectively were used. Transfection efficiency was normalized by co-transfecting cells with GFP-C1 and counting and determining the percentage of GFP positive cells under the microscope.22 For knocking down p53, cells were transfected with 2.5 μg (60 mm plate) of p53 siRNA construct (Cat No. IMG-701, Imgenex).25 For inhibiting basal NFκB p65/RelA activity, cells were treated with 2 mM aspirin (Cat No. 027039, Central Drug House Laboratory Reagent)26 or with 15 µM SN50 (Cat No. 481480–1MG, Millipore).29 The transfected cells were harvested at definite time intervals and treated in accordance with experimental requirement.
Identification of putative p53 and NFκB p65/RelA (p65) binding site(s) in the upstream sequences of miRNA genes
The genomic co-ordinates of the miRNAs were identified from miRBase (http://www.mirbase.org/).65 Next, 20 kb upstream sequences of the pre-miRNAs (1st nucleotide of the pre-miRNA taken as +1) were downloaded from UCSC Genome Browser (http://genome.ucsc.edu)66 for both human and mouse. The putative p53 and NFκB p65/RelA binding sites (BS) at miRNA upstream sequences were analyzed by using the in-house search tool www.bioinformatics.org/grn/npb3 as described earlier.31 The detailed procedure has been described in Supplementary Text and the results obtained in Supplementary Table S1. Presence of predicted regulatory regions based on ENCODE annotation data in the upstream sequences of the human miRNA genes were also evident from UCSC Genome Browser.67 The corresponding conserved regions in upstream of mouse miRNA genes were identified by NCBI BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
DNA constructs
For overexpression studies, full-length human p53 cDNA was obtained by PCR from human brain cDNA library and cloned into CFP vector using BamH1 (Cat No. R0136L, NEB) and Sal1 sites (Cat No. R3138L, NEB). The primers used for cloning were p53_CFP_F: 5′-ACGCGTCGACGTGGAGCCGCAGTCAGATCCTA-3′ and p53_CFP_R: 5′-CGCGGATCCCAGTCTGAGTCAGGCCCTTC-3′. Full-length p65 subunit of NFkB (RelA) i.e., NFκB p65/RelA cloned into pLG3 vector was obtained as a kind gift from Dr. Susanta Roychoudhury, IICB, Kolkata. For knocking down endogenous p53, pSuppressorNeo p53 plasmid DNA containing p53 siRNA construct (Cat No. IMG-701, Imgenex) was used and described earlier.25
For functional assay of NFκB p65/RelA promoter, the plasmid NFκB luciferase (p65-RE), containing multiple copies of NFκB response elements in pGL3 vector was obtained as a kind gift from Prof. Susanta Roychoudhury, IICB, Kolkata. For promoter luciferase assays of the regulatory sequences of miRNA genes, we cloned upstream sequence regions of miR-100, −125b, −146a, −150 and −221 bearing one or more putative p53 and NFκB p65/RelA binding sites into pGL3 Basic vector (Cat No. E1751, Promega). These regions were amplified by PCR from human genomic DNA and cloned in vector using the Mlu I (Cat No. R0198L, NEB) and Hind III (Cat No. R0104L, NEB) sites. The constructs were named accordingly to indicate different regions of the upstream sequences of the different miRNA genes they harbor. The primers used to generate the upstream sequence constructs were as follow:
RegA_100_F:CGACGCGTCATGTGTGTTTCCCAGCA-TC and RegA_100_R: CCCAAGCTT CCCCGCAATACT-GTTTGAA; RegB_100_F: CGACGCGTTGTGTTCTGAATTCCTGGAA-GA and RegB_100_R: CCCAAGCTTGAATC-CGTGGGCTCTCTTAG; RegC_100_F: CGAC-GCGTAGC-CCTATCAGCCGAGAGAA and RegC_100_R: CCCAAGCTTGAAAA ACATC -GGGAACCATT; RegB1_146a_F: CGA-CGCGTAAAAGCCAACAGGCTCATTG and RegB1_ 146a_R: CCCAAGCTTGGGGTAGAGGAAGGCAGCTA; RegB_150_F: CGACGC GTCTAT- GGGCCTTTCTCCAAC and RegB_150_R: CCCAAGCTTGCCCTATTGGGAGGATCAAT; RegC_150_F: CGACGCGTCACCGCTACACATCT-GGCTA and RegC_150_R: CCCAAGCT- TTGTTTTAGCATGCCTCTGGA; RegA_125b_F: CGACGCGTTCATCT-TCCCATCTGCCT and RegA_125b_R: CCCAAGCTTCT-GCGGATTCTTTGAAGC; RegB_125b_F: CGACGCG- TGGGCAAAGTAGTGAGACCCT and RegB_125b_R: CCCAA-GCTTTTCCAATCCA GGT- CAGCACT; RegB_221_F: CG-ACGCGTTGCATAGTCATTTACCTTCTAGAATAA and RegB_221_R: CCCAAGCTTCCTTGCACTCACGGAAGT-TT
For control vectors in various transfections pEGFP-C1 (Cat No. 6084–1, Clontech), pDsRed-C1 (Cat No. 632466, Clontech), pRNAU61.Hygro (Cat No. SD1202, Genescript), pGL3 Basic vector (Cat No. E1751, Promega) and pcDNA3.1 (Cat No. V790–20, Invitrogen) were used.
RNA preparation
Total RNA was prepared from cultured cells using TriZol Reagent (Cat No. 15596–026, Invitrogen) according to manufacturer's protocol. RNA samples were quantitated using Biophotometer (Eppendorf, Germany).
Quantitative real time PCR for amplification of mature miRNAs and their analysis
For amplification of mature miRNA sequences, cDNA was prepared following standard procedure68 100 ng of total RNA using Taqman miRNA specific stem-loop primers (Applied Biosystems (AB), USA) or stem loop primers designed as per described protocol,69 Mulv-Reverse transcriptase (Cat No. EP0442, Fermentas) and dNTPs (Cat No. R1121, Fermentas). cDNA was then accordingly subjected to either Taqman Quantitative Real Time PCR which detects only mature miRNAs69 using Taqman PCR master mix (Cat No. 430447, Applied Biosystems) or quantitative real time PCR using SYBR Green master mix (Catalog Number 4309155, Applied Biosystems) with miRNA specific forward primer and universal reverse primer. The sequences of miRNA stem loop specific primers for cDNA preparation, miRNA specific forward primers and universal reverse primer for quantitative real time PCR using SYBR Green have been given in Supplementary Table S2. Real time PCR was carried on 7500 Real time PCR system (AB, USA). The small RNA U6 snRNA detected in both human and mouse were used as endogenous control to calculate fold change. The fold changes were calculated as per SDS software (AB, USA).
Semi-quantitative RT-PCR for amplication of pri-miRNAs and their analysis
For detection of primary miRNA expression levels 1 μg of total RNA was subjected to DNase treatment (Sigma) followed by cDNA preparation using random hexamer primer (Fermentas), dNTPs and MuLv- Reverse transcriptase (Fermentas). Semi-quantitative RT-PCR was next performed with the help of Bioline Taq (AB, USA) using primers designed to amplify region of the primary transcripts of the respective miRNAs. The primers were designed by Primer3 software and the sequences are given in Supplementary Table S3. Yield of the PCR products was estimated from the integrated optical density (IOD) using Image Master VDS software (Amersham Bioscience, UK) and mRNA expression levels were normalized relative to those of GAPDH.
Luciferase assay
For promoter luciferase assay of NFκB p65/RelA response element (p65-RE) and cloned upstream sequence constructs of miRNA genes, 1 × 105 HeLa cells or 1.5 × 105 STHdhQ7/HdhQ7 cells were plated a day before transfection per well in 6-well plates. The following day, 1 μg of p65-RE or upstream sequence constructs cloned into pGL3 vector were separately transfected either in presence or absence of pCMV-p53 or NFκB p65/RelA. Cells transfected with empty vector pGL3 were taken as control. Cells were collected 24 h post transfection and luciferase assays were performed (Sirius Luminometer, Berthold detection systems, USA) according to the manufacturer's protocol using Luciferase Reporter assay system (Cat No. E1500, Promega). Three μg of total protein was taken for each assay. Transfection efficiency was normalized by co-transfecting cells with GFP-C1 and measuring the Fluoresence at 510 nm (Fluoromax-3, Jobin Yvon Horiba, USA).22 The experiments were carried out in triplicate. For inhibiting p65-RE activity, cells were treated with 2 mM aspirin 24 hours prior to transfection and for knocking down endogenous p53, cells were transfected with 1 μg of p53 siRNA construct. Luciferase assay was performed in each case following the same procedure as discussed above. For measuring relative p53 and NFκB p65/RelA protein levels for luciferase assays 2.5 µg each of the plasmids was transfected either in isolation or in combination in 3 × 105 STHdhQ7/HdhQ7 cells in 60 mm plates. Immunoblot analysis with cell extracts revealed the protein levels.
Sub-cellular fractionation, immunoprecipitation and protein gel blot analysis
The methods used for sub-cellular fractionation, immunoprecipitation and western blot analysis were similar to those which have been published.70 Beta actin and lamin B were used as loading controls for proteins in cytoplasmic and nuclear extracts respectively. Calculations for nuclear protein contents were done normalized to nuclear Lamin B level.71 The various antibodies used for immunoprecipitation include anti P53 (Cat No. IMG-80061, Imgenex, USA) at 1:100 dilution, anti P65 (Cat No. MAB3026, Chemicon, USA) at 1:100 dilution and anti CBP (Cat No. sc-369, Santa Cruz Biotechnology, USA) at 1:100 dilution. For protein gel blot primary antibodies used were anti β actin (Cat No. A2228, Sigma Chemicals, USA) at 1:8000 dilution, anti Lamin B (Cat No. Ab8983, Abcam, USA) at 1:5000 dilution, Anti P53 (Cat No. IMG-80061, Imgenex, USA) at 1:4000 dilution, anti P65 (Cat No. MAB3026, Chemicon, USA) at 1:4000 dilution and anti CBP (Cat No. sc-369, Santa Cruz Biotechnology, USA) at 1:200 dilution. The various secondary antibodies used were Goat Anti-mouse IgG-HRP (Cat No. 105502, GENEI, India) and Goat anti-rabbit IgG-HRP (Cat No. 105499, GENEI, India) each at 1:8000 dilution. Integrated optical density (IOD) of each nuclear band compared to nuclear Lamin B was measured using Image Master VDS software (Amersham Biosciences, UK).
Chromatin immunoprecipitation and measure of promoter occupancy
Method used for ChIP experiments was described earlier.70 For each experiment, 200 μg of isolated chromatin was immunoprecipitated with 10 μg of anti p53 or anti p65 antibody. followed by PCR amplification to detect for the presence or absence of specific upstream sequence elements of miRNA genes bearing putative p53 or NFκB p65/RelA binding sites. PCR analysis with primers targeted to a region of GAPDH promoter32 and to a region on chromosome 1 (Chr1: 204,366,822–204,366,872)6 were taken as negative control in ChIP performed with anti p53 antibody and anti-p65 antibody respectively. The various antibodies used for immunoprecipitation include Anti P53 (Cat No. IMG-80061, Imgenex, USA) at 1:20 dilution and Anti P65 (Cat No. MAB3026, Chemicon, USA) at 1:20 dilution. The various primers used in ChIP with their sequences and purpose of use are given in Supplementary Table S4.
Statistical analysis
All the experiments were performed at least 3 times. Statistical analysis was done with the help of on-line software Graphpad QuickCalcs, (http://www.graphpad.com/quickcalcs/index.cfm). All data were reported into 2 groups, one for control and the other for experimental. The values of mean, standard deviation (SD) and exact number of independent experiments performed (n) for each group were provided. Student's unpaired 2-tailed -t-test was then performed by the software to compare between the means of control and experimental groups to determine their statistical significance. Error bars represent standard deviation, */† represents statistical significance (*P ≤ 0.05; ** P ≤ 0.01) or († P ≤ 0.05; †† P ≤ 0.01).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgements
We thank Dr. Marcy E. MacDonald of Massachusetts General Hospital, USA for providing STHdhQ7/HdhQ7 cells. We are thankful to Dr. Susanta Roy Choudhury, Indian Institute of Chemical Biology, Kolkata, India for providing us with p65 sub-unit of NFkB (NFκB p65/RelA) cloned into pLG3 vector and NFkB response element (p65-RE) construct cloned in basic pGL3 vector. We thank Mr. Utpal Basu and Mr. Saikat Mukhopadhyay, Crystallography & Molecular Biology Division, Saha Institute of Nuclear Physics for their technical support.
Funding
This work was supported by the Institutional Grant to Saha Institute of Nuclear Physics for the Project ‘Molecular and Structural Aspects of Cellular Regulatory Process (MSACR)' by the Department of Atomic Energy, Govt. of India. Besides, NPB acknowledges Department of Biotechnology (DBT), Govt. of India for partial financial assistance for the project.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Lee Y, Kim M, Han J, Yeom KH, Lee S, Baek SH, Kim VN. MicroRNA genes are transcribed by RNA polymerase II. EMBO J 2004; 23(20):4051-4060. Epub 2004 Sep 16; PMID:15372072; http://dx.doi.org/ 10.1038/sj.emboj.7600385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Borchert GM, Lanier W, Davidson BL. RNA polymerase III transcribes human microRNAs. Nat Struct Mol Biol 2006; 13:1097-1101; PMID:17099701; http://dx.doi.org/ 10.1038/nsmb1167 [DOI] [PubMed] [Google Scholar]
- 3.Gu TJ, Yi X, Zhao XW, Zhao Y, Yin JQ. Alu-directed transcriptional regulation of some novel miRNAs. BMC Genomics 2009; 10:563; PMID:19943974; http://dx.doi.org/ 10.1186/1471-2164-10-563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Monteys AM, Spengler RM, Wan J, Tecedor L, Lennox KA, Xing Y, Davidson BL. Structure and activity of putative intronic miRNA promoters. RNA 2010; 16:495-505; PMID:20075166; http://dx.doi.org/ 10.1261/rna.1731910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cai X, Hagedorn CH, Cullen BR. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. RNA 2004; 10:1957-1966; PMID:15525708; http://dx.doi.org/ 10.1261/rna.7135204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang TC, Yu D, Lee YS, Wentzel EA, Arking DE, West KM, Dang CV, Thomas-Tikhonenko A, Mendell JT. Widespread microRNA repression by Myc contributes to tumorigenesis. Nat Genet 2008; 40(1):43-50; PMID:18066065; http://dx.doi.org/ 10.1038/ng.2007.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laurenti E, Wilson A, Trumpp A. Myc's other life: stem cells and beyond. Curr Opin Cell Biol 2009; 21(6):844-854. Epub 2009 Oct 14; PMID:19836223; http://dx.doi.org/ 10.1016/j.ceb.2009.09.006 [DOI] [PubMed] [Google Scholar]
- 8.Yaguan X, Shalgi R, Fodstad O, Pilpel Y, Ju J. Differentially regulated micro-RNAs and actively translated messenger RNA transcripts by tumor suppressor p53 in colon cancer. Clin Cancer Res 2006; 12:2014-2024; PMID:16609010; http://dx.doi.org/ 10.1158/1078-0432.CCR-05-1853 [DOI] [PubMed] [Google Scholar]
- 9.Tarasov V, Jung P, Verdoodt B, Lodygin D, Epanchintsev A, Menssen A, Meister G, Hermeking H. Differential regulation of microRNAs by p53 revealed by massively parallel sequencing: miR-34a is a p53 target that induces apoptosis and G1-arrest. Cell Cycle 2007; 6:1586-1593; PMID:17554199; http://dx.doi.org/ 10.4161/cc.6.13.4436 [DOI] [PubMed] [Google Scholar]
- 10.Suzuki HI, Yamagata K, Sugimoto K, Iwamoto T, Kato S, Miyazono K. Modulation of microRNA processing by p53. Nature 2009; 460(7254):529-533; PMID:19626115; http://dx.doi.org/ 10.1038/nature08199 [DOI] [PubMed] [Google Scholar]
- 11.Taganov KD, Boldin MP, Chang KJ, Baltimore D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci 2006; 103:12481-12486; PMID:16885212; http://dx.doi.org/ 10.1073/pnas.0605298103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou R, Hu G, Liu J, Gong AY, Drescher KM, Chen XM. NF-kappaB p65-Dependent Transactivation of miRNA Genes following Cryptosporidium parvum Infection Stimulates Epithelial Cell Immune Responses. PLoS Pathog 2009; 5(12):e1000681; PMID:19997496; http://dx.doi.org/ 10.1371/journal.ppat.1000681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou R, Hu G, Gong AY, Chen XM. Binding of NF-kappaB p65 subunit to the promoter elements is involved in LPS-induced transactivation of miRNA genes in human biliary epithelial cells. Nucleic Acids Research 2010; 38(10):3222-3232; PMID:20144951; http://dx.doi.org/ 10.1093/nar/gkq056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fukao T, Fukuda Y, Kiga K, Sharif J, Hino K, Enomoto Y, Kawamura A, Nakamura K, Takeuchi T, Tanabe M. An evolutionarily conserved mechanism for microRNA-223 expression revealed by microRNA gene profiling. Cell 2007; 129:617-631; PMID:17482553; http://dx.doi.org/ 10.1016/j.cell.2007.02.048 [DOI] [PubMed] [Google Scholar]
- 15.Conaco C, Otto S, Han J, Mandel G. Reciprocal actions of REST and a microRNA promote neuronal identity. Proc Natl Acad Sci 2006; 103(7):2422-2427; PMID:16461918; http://dx.doi.org/ 10.1073/pnas.0511041103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Davis-Dusenbery BN, Hata A. MicroRNA in Cancer: The Involvement of Aberrant MicroRNA Biogenesis Regulatory Pathways. Genes Cancer 2010; 1(11):1100-1114; PMID:21533017; http://dx.doi.org/ 10.1177/1947601910396213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arora S, Rana R, Chhabra A, Jaiswal A, Rani V. miRNA-transcription factor interactions: a combinatorial regulation of gene expression. Mol Genet Genomics 2013; 288(3-4):77-87; PMID:23334784; http://dx.doi.org/ 10.1007/s00438-013-0734-z [DOI] [PubMed] [Google Scholar]
- 18.Wang D, Chang PS, Wang Z, Sutherland L, Richardson JA, Small E, Krieg PA, Olson EN. Activation of cardiac gene expression by myocardin, a transcriptional cofactor for serum response factor. Cell 2001; 105:851-862; PMID:11439182; http://dx.doi.org/ 10.1016/S0092-8674(01)00404-4 [DOI] [PubMed] [Google Scholar]
- 19.Choy MK, Movassagh M, Siggens L, Vujic A, Goddard M, Sánchez A, Perkins N, Figg N, Bennett M, Carroll J, Foo R. High-throughput sequencing identifies STAT3 as the DNA-associated factor for p53-NF-kappaB-complex-dependent gene expression in human heart failure. Genome Med 2010; 2(6):37-50; PMID:20546595; http://dx.doi.org/ 10.1186/gm158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liang M, Yao G, Yin M, Lü M, Tian H, Liu L, Lian J, Huang X, Sun F. Transcriptional cooperation between p53 and NF-jB p65 regulates microRNA-224 transcription in mouse ovarian granulosa cells. Molecular and Cellular Endocrinology 2013; 370:119-129; PMID:23474441; http://dx.doi.org/ 10.1016/j.mce.2013.02.014 [DOI] [PubMed] [Google Scholar]
- 21.Yan B, Li H, Yang X, Shao J, Jang M, Guan D, Zou S, Van Waes C, Chen Z, Zhan M. Unraveling Regulatory Programs for NF-kappaB, p53 and MicroRNAs in Head and Neck Squamous Cell Carcinoma. PLoS One 2013; 8(9):e73656; PMID:24069219; http://dx.doi.org/ 10.1371/journal.pone.0073656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sinha M, Ghose J, Das E, Bhattarcharyya NP. Altered microRNAs in STHdh(Q111)/Hdh(Q111) cells: miR-146a targets TBP. Biochem Biophys Res Commun 2010; 396(3):742-747; PMID:20451497; http://dx.doi.org/ 10.1016/j.bbrc.2010.05.007 [DOI] [PubMed] [Google Scholar]
- 23.Ruepp A, Kowarsch A, Schmidl D, Buggenthin F, Brauner B, Dunger I, Fobo G, Frishman G, Montrone C, Theis FJ. PhenomiR: a knowledgebase for microRNA expression in diseases and biological processes. Genome Biol 2010; 11(1):R6; PMID:20089154; http://dx.doi.org/ 10.1186/gb-2010-11-1-r6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sinha M, Mukhopadhyay S, Bhattacharyya NP. Mechanism(s) of Alteration of Micro RNA Expressions in Huntington's Disease and Their Possible Contributions to the Observed Cellular and Molecular Dysfunctions in the Disease. Neuromolecular Med 2012; 14:221-243; PMID:22581158; http://dx.doi.org/ 10.1007/s12017-012-8183-0 [DOI] [PubMed] [Google Scholar]
- 25.Kim H, Kokkotou E, Na X, Rhee SH, Moyer MP, Pothoulakis C, Lamont JT. Clostridium difficile Toxin A–Induced Colonocyte Apoptosis Involves p53-Dependent p21(WAF1/ CIP1) Induction via p38 Mitogen-Activated Protein Kinase. Gastroenterology 2005; 129:1875-1888; PMID:16344056; http://dx.doi.org/ 10.1053/j.gastro.2005.09.011 [DOI] [PubMed] [Google Scholar]
- 26.Kopp E, Ghosh S. Inhibition of NF-kappa B by sodium salicylate and aspirin. Science 1994; 265:956-959; PMID:8052854; http://dx.doi.org/ 10.1126/science.8052854 [DOI] [PubMed] [Google Scholar]
- 27.Duan Q, Wang X, Gong W, Ni L, Chen C, He X, Chen F, Yang L, Wang P, Wang DW. ER Stress Negatively Modulates the Expression of the miR-199a/214 Cluster to Regulates Tumor Survival and Progression in Human Hepatocellular Cancer. PLoS One 2012; 7(2):e31518; PMID:22359598; http://dx.doi.org/ 10.1371/journal.pone.0031518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li J, Wang K, Chen X, Meng H, Song M, Wang Y, Xu X, Bai Y. Transcriptional activation of microRNA-34a by NF-kappa B in human esophageal cancer cells. BMC Mol Biol 2012; 13:4-9; PMID:22292433; http://dx.doi.org/ 10.1186/1471-2199-13-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin YZ, Yao SY, Veach RA, Torgerson TR, Hawiger J. Inhibition of nuclear translocation of transcription factor NF-κB by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. J Biol Chem 1995; 270:14255-14258; PMID:7782278; http://dx.doi.org/ 10.1074/jbc.270.24.14255 [DOI] [PubMed] [Google Scholar]
- 30.Hermeking H. MicroRNAs in the p53 network: micromanagement of tumour suppression. Nat Rev Cancer 2012; 12(9):613-626. Epub 2012 Aug 17; PMID:22898542; http://dx.doi.org/ 10.1038/nrc3318 [DOI] [PubMed] [Google Scholar]
- 31.Datta M, Choudhury A, Lahiri A, Bhattacharyya NP. 2012 Genome wide gene expression regulation by HIP1 Protein Interactor, HIPPI: Prediction and validation. BMC Genomics 2012; 12:463-479; http://dx.doi.org/ 10.1186/1471-2164-12-463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yan H, Xue G, Mei Q, Wang Y, Ding F, Liu MF, Lu MH, Tang Y, Yu H, Sun S. Repression of the miR-17-92 cluster by p53 has an important function in hypoxia-induced apoptosis. EMBO J 2009; 28:2719-2732; PMID:19696742; http://dx.doi.org/ 10.1038/emboj.2009.214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Galardi S, Mercatelli N, Farace MG, Ciafrè SA. NF-kB and c-Jun induce the expression of the oncogenic miR-221 and miR-222 in prostate carcinoma and glioblastoma cells. Nucleic Acids Res 2011; 39(9):3892-3902; PMID:21245048; http://dx.doi.org/ 10.1093/nar/gkr006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Melchjorsen J, Paludan SR. Induction of RANTES/CCL5 by herpes simplex virus is regulated by nuclear factor kappa B and interferon regulatory factor 3. J Gen Virol 2003; 84(Pt 9):2491-2495; PMID:12917470; http://dx.doi.org/ 10.1099/vir.0.19159-0 [DOI] [PubMed] [Google Scholar]
- 35.Gupta S, Radha V, Furukawa Y, Swarup G. Direct transcriptional activation of human Caspase-1 by tumor suppressor p53. J Biol Chem 2001; 276(14):10585-10588. Epub 2001 Feb 13; PMID:11278253; http://dx.doi.org/ 10.1074/jbc.C100025200 [DOI] [PubMed] [Google Scholar]
- 36.Ravi R, Mookerjee B, van Hensbergen Y, Bedi GC, Giordano A, El-Deiry WS, Fuchs EJ, Bedi A. p53-mediated repression of nuclear factor-kappa B RelA via the transcriptional integrator p300. Cancer Res 1998; 58:4531-4536; PMID:9788595 [PubMed] [Google Scholar]
- 37.Webster GA, Perkins ND. Transcriptional cross talk between NF-kB and p53. Mol Cell Biol 1999; 19:3485-3495; PMID:10207072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wadgaonkar R, Phelps KM, Haque Z, Williams AJ, Silverman ES, Collins T. CREB-binding protein is a nuclear integrator of nuclear factor-κB and p53 signaling. J Biol Chem 1999; 274:1879-1882; PMID:9890939; http://dx.doi.org/ 10.1074/jbc.274.4.1879 [DOI] [PubMed] [Google Scholar]
- 39.Schneider G, Krämer OH. NFκB/p53 crosstalk-a promising new therapeutic target. Biochim Biophys Acta 2011; 1815(1):90-103; PMID:20951769 [DOI] [PubMed] [Google Scholar]
- 40.He L, He X, Lim LP, Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, et al.. A microRNA component of the p53 tumour suppressor network 34b. Nature 2007; 447:1130-1135; PMID:17554337; http://dx.doi.org/ 10.1038/nature05939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, Feldmann G, Yamakuchi M, Ferlito M, Lowenstein CJ, et al.. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell 2007; 26(5):745-752; PMID:17540599; http://dx.doi.org/ 10.1016/j.molcel.2007.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sachdeva M, Zhu S, Wu F, Wu H, Walia V, Kumar S, Elble R, Watabe K, Mo Y. p53 represses c-Myc through induction of the tumor suppressor miR-145. Proc Natl Acad Sci USA 2008; 106(9):3207-3212; http://dx.doi.org/ 10.1073/pnas.0808042106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ghose J, Sinha M, Das E, Bhattacharyya NP. Regulation of miR-146a by NFkB and p53 in STHdhQ111/HdhQ111 cells, a cell model of Huntington's disease. PLos ONE 2011; 6:e23837; PMID:21887328; http://dx.doi.org/ 10.1371/journal.pone.0023837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Corney DC, Flesken-Nikitin A, Godwin AK, Wang W, Nikitin AY. MicroRNA-34b and MicroRNA-34c are targets of p53 and cooperate in control of cell proliferation and adhesion-independent growth. Cancer Res 2007; 67(18):8433-8438. Epub 2007 Sep 6; PMID:17823410; http://dx.doi.org/ 10.1158/0008-5472.CAN-07-1585 [DOI] [PubMed] [Google Scholar]
- 45.Braun CJ, Zhang X, Savelyeva I, Wolff S, Moll UM, Schepeler T, Ørntoft TF, Andersen CL, Dobbelstein M. p53-Responsive MicroRNAs 192 and 215 Are Capable of Inducing Cell Cycle Arrest. Cancer Res 2008; 68:10094-10104; PMID:19074875; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamakuchia M, Lottermanb CD, Baob C, Hrubane RH, Karime B, Mendell JT, Husof D, Lowensteina CJ. P53-induced microRNA-107 inhibits HIF-1 and tumor angiogenesis. Proc Natl Acad Sci 2010; 107(14):6334-6339; PMID:20308559; http://dx.doi.org/ 10.1073/pnas.0911082107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kim T, Veronese A, Pichiorri F, Lee TJ, Jeon YJ, Volinia S, Pineau P, Marchio A, Palatini J, Suh SS, et al.. p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J Exp Med 2011; 208(5):875-883. Epub 2011 Apr 25; PMID:21518799; http://dx.doi.org/ 10.1084/jem.20110235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jin L, Hu WL, Jiang CC, Wang JX, Han CC, Chu P, Zhang LJ, Thorne RF, Wilmott J, Scolyer RA, Hersey P, Zhang XD, Wu M. MicroRNA-149*, a p53-responsive microRNA, functions as an oncogenic regulator in human melanoma. Proc Natl Acad Sci USA 2011; 108(38):15840-15845. Epub 2011 Sep 6; PMID:21896753; http://dx.doi.org/ 10.1073/pnas.1019312108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xiao J, Lin H, Luo X, Luo X, Wang Z. miR-605 joins p53 network to form a p53:miR-605:Mdm2 positive feedback loop in response to stress. EMBO J 2011; 30(3):524-532. Epub 2011 Jan 7; PMID:21217645; http://dx.doi.org/ 10.1038/emboj.2010.347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang Y, Liao JM, Zeng SX, Lu H. 2011 p53 downregulates Down syndrome-associated DYRK1A through miR-1246. EMBO Rep 2011; 12(8):811-817; PMID:21637297; http://dx.doi.org/ 10.1038/embor.2011.98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Barsotti AM, Beckerman R, Laptenko O, Huppi K, Caplen NJ, Prives C. p53-Dependent induction of PVT1 and miR-1204. J Biol Chem 2012; 287(4):2509-2519. Epub 2011 Nov 22; PMID:22110125; http://dx.doi.org/ 10.1074/jbc.M111.322875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bazzoni F, Rossato M, Fabbri M, Gaudiosi D, Mirolo M, Mori L, Tamassia N, Mantovani A, Cassatella MA, Locati M. Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc Natl Acad Sci USA 2009; 106(13):5282-5287. doi: 10.1073/pnas.0810909106; PMID:19289835; http://dx.doi.org/ 10.1073/pnas.0810909106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lu Z, Li Y, Takwi A, Li B, Zhang J, Conklin DJ, Young KH, Martin R, Li Y. miR-301a as an NF-κB activator in pancreatic cancer cells. EMBO J 2011; 30:57-67; PMID:21113131; http://dx.doi.org/ 10.1038/emboj.2010.296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Campbell KJ, Rocha S, Perkins ND. Active repression of antiapoptotic gene expression by RelA(p65) NF-κB. Mol Cell 2004; 13:853-865; PMID:15053878; http://dx.doi.org/ 10.1016/S1097-2765(04)00131-5 [DOI] [PubMed] [Google Scholar]
- 55.Shao J, Fujiwara T, Kadowaki Y, Fukazawa T, Waku T, Itoshima T, Yamatsuji T, Nishizaki M, Roth JA, Tanaka N. Overexpression of the wild-type p53 gene inhibits NF-kB activity and synergizes with aspirin to induce apoptosis in human colon cancer cells. Oncogene 2000; 19:726-736; PMID:10698490; http://dx.doi.org/ 10.1038/sj.onc.1203383 [DOI] [PubMed] [Google Scholar]
- 56.Kawauchi K, Araki K, Tobiume K, Tanaka N. Activated p53 induces P65 DNA binding but suppresses its transcriptional activation. Biochem Biophys Res Commun 2008; 372:137-141; PMID:18477470; http://dx.doi.org/ 10.1016/j.bbrc.2008.05.021 [DOI] [PubMed] [Google Scholar]
- 57.Ryan KM, Ernst MK, Rice NR, Vousden KH. Role of NF-kB in p53-mediated programmed cell death. Nature 2000; 404(6780):892-897; PMID:10786798; http://dx.doi.org/ 10.1038/35009130 [DOI] [PubMed] [Google Scholar]
- 58.Bohuslav J, Chen LF, Kwon H, Mu Y, Greene WC. p53 induces NF- kappaB activation by an IkappaB kinase-independent mechanism involving phosphorylation of p65 by ribosomal S6 kinase 1. J Biol Chem 2004; 279:26115-26125; PMID:15073170; http://dx.doi.org/ 10.1074/jbc.M313509200 [DOI] [PubMed] [Google Scholar]
- 59.Wu H, Lozano SG. NF-kB Activation of p53. J Biol Chem 1994; 269(31):20067-20074; PMID:8051093 [PubMed] [Google Scholar]
- 60.Qin ZH, Chen RW, Wang Y, Nakai M, Chuang D, Chase TN. Nuclear Factor κB Nuclear Translocation Upregulates c-Myc and p53 Expression during NMDA Receptor-Mediated ApoptosiS in Rat Striatum. J Neurosci 1999; 19(10):4023-4033; PMID:10234031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ikeda A, Sun X, Li Y, Zhang Y, Eckner R, Doi TS, Takahashi T, Obata Y, Yoshioka K, Yamamoto K. p300/CBP-dependent and -independent transcriptional interference between NF-kappaB RelA and p53. Biochem Biophys Res Commun 2000; 272(2):375-379; PMID:10833421; http://dx.doi.org/ 10.1006/bbrc.2000.2786 [DOI] [PubMed] [Google Scholar]
- 62.Jeong SJ, Radonovich M, Brady JN, Pise-Masison CA. HTLV-I Tax induces a novel interaction between p65/RelA and p53 that results in inhibition of p53 transcriptional activity. Blood 2004; 104:1490-1497; PMID:15155458; http://dx.doi.org/ 10.1182/blood-2003-12-4174 [DOI] [PubMed] [Google Scholar]
- 63.Boldin MP, Baltimore D. MicroRNAs, new effectors and regulators of NF-κB. Immunol Rev 2012; 246(1):2005-2020; PMID:22435557; http://dx.doi.org/ 10.1111/j.1600-065X.2011.01089.x [DOI] [PubMed] [Google Scholar]
- 64.Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, MacDonald ME. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Hum Mol Genet 2000; 9:2799-2809; PMID:11092756; http://dx.doi.org/ 10.1093/hmg/9.19.2799 [DOI] [PubMed] [Google Scholar]
- 65.Griffiths-Jones S. The microRNA Registry. Nucleic Acids Res 2004; 32(Database issue):D109-111; PMID:14681370; http://dx.doi.org/ 10.1093/nar/gkh023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res 2002; 12(6):996-1006; PMID:12045153; http://dx.doi.org/ 10.1101/gr.229102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rosenbloom KR, Sloan CA, Malladi VS, Dreszer TR, Learned K, Kirkup VM, Wong MC, Maddren M, Fang R, Heitner SG, et al.. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res 2013; 41(Database issue):D56-63; PMID:23193274; http://dx.doi.org/ 10.1093/nar/gks1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen X, Ba Y, Ma L, Cai X, Yin Y, Wang K, Guo J, Zhang Y, Chen J, Guo X, et al.. Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases. Cell Research 2008; 18:997-1006; PMID:18766170; http://dx.doi.org/ 10.1038/cr.2008.282 [DOI] [PubMed] [Google Scholar]
- 69.Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, et al.. Real-time quantification of microRNAs by stem–loop RT–PCR. Nucleic Acids Res 2005; 33(20):e179; PMID:16314309; http://dx.doi.org/ 10.1093/nar/gni178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Banerjee M, Datta M, Majumder P, Mukhopadhyay D, Bhattacharyya NP. Transcription regulation of caspase-1 by R393 of HIPPI and its molecular partner HIP-1. Nucleic Acids Res 2010; 38(3):878-892. Epub 2009 Nov 24; PMID:19934260; http://dx.doi.org/ 10.1093/nar/gkp1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mendoza-Vilanueva D, Deng W, Lopez-Camacho C, Shore P. The Runx transcriptional co-activator CBFbeta, is essential for invasion of breast cancer cells. Mol Cancer 2010; 9:171-181; PMID:20591170; http://dx.doi.org/ 10.1186/1476-4598-9-171 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.