

Abstract
AIM: To conduct the proteomic analysis of human colorectal carcinoma cell line, SW480 by using two-dimensional electrophoresis (2-DE) and matrix-assisted laser desorption /ionization-time of flight mass spectrometry (MALDI-TOFMS).
METHODS: The total proteins of human colorectal carcinoma cell line, SW480 were separated with 2-DE by using immobilized pH gradient strips and visualized by staining with silver nitrate. The gel images were acquired by scanner and 2-DE analysis software, Image Master 2D Elite. Nineteen distinct protein spots were excised from gel randomly and digested in gel by TPCK-trypsin. Mass analysis of the tryptic digest peptides mixture was performed by using MALDI-TOF MS. Peptide mass fingerprints (PMFs) obtained by the MALDI-TOF analysis were used to search NCBI, SWISS-PROT and MSDB databases by using Mascot software.
RESULTS: PMF maps of all spots were obtained by MALDI-TOF MS and thirteen proteins were preliminarily identified.
CONCLUSION: The methods of analysis and identification of protein spots of tumor cells in 2-DE gel with silver staining by MALDI-TOF MS derived PMF have been established. Protein expression profile of SW480 has been obtained. It is demonstrated that a combination of proteomics and cell culture is a useful approach to comprehend the process of colon carcinogenesis.
Keywords: Colorectal carcinoma, SW480 cell line, Two-dimensional electrophoresis, MALDI-TOF MS, Peptide mass fingerprinting
INTRODUCTION
Colorectal cancer (CRC) is one of the three leading causes of morbidity and mortality worldwide[1]. It is believed that CRC develops through a multistep process involving the accumulation of genetic alterations. The genetic changes involved in colon carcinogenesis, including changes in proto-oncogenes, tumor suppressor genes, and DNA repair genes were well studied. But the exact mechanisms involved in this process are not well understood. Recently, changes in the transcriptome of colon tissues were studied by using DNA microarray analysis[2]. However, the value of mRNA changes may be limited in terms of understanding changes in cellular physiology. Instead, genetic alterations lead to altered expression patterns, modifications in protein structures and functions. Alterations in the proteome may reflect cellular changes more accurately since proteins are the actual mediators of intracellular processes as opposed to mRNAs. Proteomics is the study and analysis of the proteins of living organisms. In recent years, the development of research entailing the protein complement of the genome, the proteome, has evolved significantly as a result of improved technology for two-dimensional gel electrophoresis (2-DE) and mass spectrometry (MS) for protein identification. With these technologies, it is now possible to obtain a more holistic view of protein changes associated with colon carcinogenesis. Here, for the first time, a proteomic approach is used to display the protein profile of human colorectal carcinoma cell line, SW480 to understand the basis of colon carcinogenesis.
MATERIALS AND METHODS
Chemicals and materials
Immobilized pH gradient (IPG) strips (pH3-10, linear, 13 cm), IPG buffer (pH3-10, linear) were purchased from Amersham Biosciences (Uppsala, Sweden). Dithiothreitol (DTT), iodoacetamide (IAA), TPCK-trypsin, Trifluoroacetic acid (TFA), CHAPS, α-cyano-4-hydroxycinnamic acid (CHCA) were purchased from Sigma company (St. Louis, MO, USA). All the buffers were made by using high purity MilliQ water. IPGphor electrophoresis unit, Hoefer SE 600 vertical chambers, electrophoresis apparatus, Image Master 2D Elite 4.01 software and image scanner were purchased from Amersham Biosciences. Voyager-DE MALDI-TOF MS was product of Applied Biosystems (USA).
Cell line culture and sample preparation
The cell line, SW480 was purchased from Institute of Bioche-mistry and Cell Biology, Chinese Academy of Sciences (Shanghai, China). The cells were cultured in RPMI 1640 medium supplemented with 10 mL/L fetal bovine serum (FBS) and antibiotics. The cells were maintained in an incubator at 37°C in 50 mL/L CO2 humidified atmosphere. The cells grown at the exponential growth phase were harvested with trypsinization. After washing in Hanks’ solution and ice-cold PBS, the cells were counted, lysated in a cocktail of 9 mol/L urea, 40 g/L CHAPS, 40 mmol/L Tris and 40 mmol/L DTT and centrifuged at 12 000 g in for 1 h at 4°C. Protein concentrations were determined by the method of Bradford.
2-D electrophoresis
2-DE was performed by using IPG strips. Briefly, first-dimensional isoeletric focusing (IEF) was performed on 13 cm strips (pH 3-10, linear) by using an Amersham IPGphor unit. IEF was carried out by using an IPGphor electrophoresis unit (Amersham Biosciences). Separation in the second dimension (SDS-PAGE) was carried out on a 1.0 mm-thick 125 g/L polyacrylamide gels at a constant current (20 mA/gel) and temperature (15°C) by using the Hoefer SE 600 vertical chambers. After 2-D separation, the gels were stained with silver nitrate. Image analysis was performed by using the Image Master 2D Elite software 4.01.
In-gel trypsin digestion of proteins
Proteins were in-gel digested as previously described by Wilm et al (Nature 1996; 379: 466-469) with some modification. Silver-stained spots were excised and washed with a 50 µL fresh bleaching liquid (100 mmol/L Na2S2O3: 30 mmol/L K3Fe (CN)6 = 1:1). Gel spots were dried in a vacuum centrifuge and reswelled in 50 µL of solution containing 10 mol/L DTT/100 mol/L NH4HCO3 and incubated at 57°C for 1 h. This solution was subsequently replaced with 50 µL of solution containing 55 mol/L IAA/100 mol/L NH4HCO3 and incubated at room temperature for 30 min. The gel spots were dried again and digested with TPCK-tryp sin at 37°C overnight. After the incubation, the liquid was removed from the gel piece and the liquid was transferred to a new-labeled tube. This solution contains the extracted tryptic peptides.
MALDI-TOF –MS analysis of tryptic peptide
Mass analysis was performed by using a Voyager-DE MALDI-TOF MS (Framingham, MA, USA), operated in the delayed extraction and linear mode. The tryptic digest mixture was mixed with CHCA matrix. The MALDI spectra averaged over 50 laser shots. All mass spectra were calibrated externally by using a standard peptide mixture (angiotensin II and ACTH 18-39). Internal calibration was performed by using auto digestion peaks of trypsin.
Database searching and identification of proteins
Peptide mass fingerprints obtained by the MALDI-TOF MS were used to search NCBInr, SWISS-PROT and MSDB databases by using Mascot software (http://www. matrixscience.com). The parameters used for the search were as follows: peptide mass ranged from 1 000 tp 3 000 U; modifications were allowed for carboxy-amidomethylation of cysteine and oxidation of methionine; one missed cleavage site was allowed; mass accuracy was±1 U; restriction was placed on the species of Homo sapiens. The criteria for positive identification of proteins were set as follows: (1) the MS match consisted of a minimum of four peptides; (2) the matched peptides covered at least 20% of the whole protein sequence; (3) 50 ppm or better mass accuracy.
RESULTS
Proteomic pattern of SW480 by 2-DE
Proteins of human colorectal cancer cell line SW480 was separated by 2-DE. Spots were visualized with silver staining. Three pairs of gels from SW480 were analyzed by using the Image Master 2D Elite software 4.01. Figure 1 shows a representative example of cell proteins separated on a 2-DE gel, where 100 µg of total protein was applied. Nearly 1 000 proteins spots were obtained in the range of Mr 14 400-9 4000 u, PI 3-10.
Figure 1.

2-DE map of human colorectal cancer cell line SW480 proteome (silver staining).
MALDI-TOF MS analysis of proteins
Nineteen distinct protein spots were excised from gels randomly and marked with Arabic numbers at the corresponding sites in Figure 1. All spots obtained the PMF maps by MALDI-TOF MS following in-gel digestion with TPCK-trypsin. Figure 2 shows the spectrum of trypsin digestion of protein spot 2.
Figure 2.
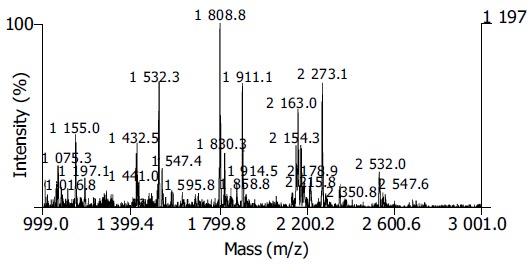
PMF of protein spot 2 in SW480 2-DE map.
Database searching and identification of proteins
PMFs obtained by the MALDI-TOF MS were used to search NCBInr, SWISS-PROT and MSDB databases by using Mascot software (http://www.matrixscience.com). Figure 3 shows the searching result of spot 2.
Figure 3.
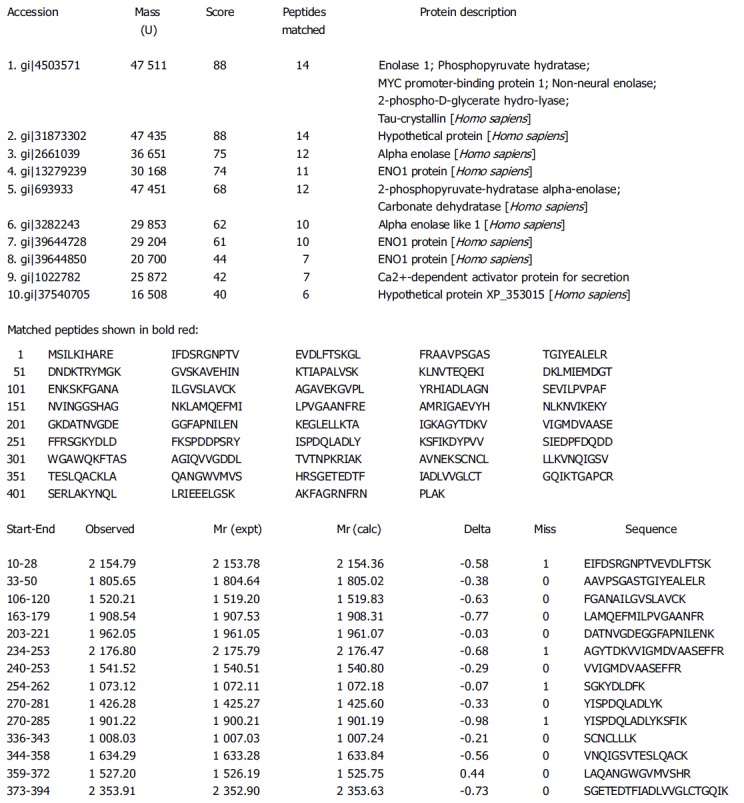
Searching results of spot 2.
Thirteen proteins were preliminarily identified on the basis of peptide mass matching by database searching. Some PMFs such as 1, 8, 9, 10, 11, 16 were not searched by the satisfactory proteins. Table 1 lists the identified proteins. Among these proteins, some were correlated with signal transduction (e.g. zinc finger protein 79 and PR-domain zinc finger protein 7 isoform B), some with cell metabolism (e.g. enolase 1, phosphopyruvate hydratase, carboxypeptidase A5 precursor and peptidylprolyl isomerase), some with cell growth and adhesion (e.g. paired box transcription factor and intermediate filament-binding fragments of Desmoplakin), and some with the immunological function of tumor cells (e.g. MHC class I promoter binding protein, immunoglobulin heavy chain VHDJ region and human B7-1 CTLA-4 co-stimulatory complex).
Table 1.
Database searching results of proteins of SW480 cell line
| Spot ID | NCBI ID | Peptides matched | Sequence covered (%) | Theoretic Mr/pI | Protein name |
| 1 | Unidentified | ||||
| 2 | gi|4503571 | 14/40 | 44 | 47 511/7.01 | Enolase 1; Phosphopyruvate hydratase; |
| MYC promoter-binding protein 1; Non-neural enolase; | |||||
| 2-phospho-D-glyceratehydro-lyase; Tau-crystallin | |||||
| 3 | gi|24307937 | 19/35 | 26 | 56 918/8.48 | Zinc finger protein 79 (pT7) |
| 4 | gi|37723146 | 4/11 | 24 | 33 130/7.04 | Paired box transcription factor |
| 5 | SwissProt ID: | 3/12 | 15 | 394 787.81 | Carboxypeptidase A5 precursor (EC 3.4.17.1) |
| Q8WXQ8-01-00-00 | |||||
| 6 | gi|22219251 | 4/13 | 21 | 23 385/6.23 | Chain A, structures of two intermediate |
| filament-binding fragments of Desmoplakin | |||||
| 7 | gi|2135648 | 6/38 | 31 | 25 794/6.42 | MHC class I promoter binding protein (fragment) |
| 8 | Unidentified | ||||
| 9 | Unidentified | ||||
| 10 | Unidentified | ||||
| 11 | Unidentified | ||||
| 12 | gi|13786754 | 4/24 | 34 | 24093/5.38 | Chain A, human B7-1 CTLA-4 co-stimulatory complex |
| 13 | MSDB ID: 1AK4B | 6/36 | 33 | 18010/7.85 | Peptidylprolyl isomerase (EC 5.2.1.8) A, chain B |
| 14 | gi|21670535 | 3/19 | 50 | 13285/8.64 | Immunoglobulin heavy chain VHDJ region [Homo sapiens] |
| 15 | gi|8575802 | 5/26 | 35 | 18715/8.29 | PR-domain zinc finger protein 7 isoform B; |
| PR-domain family protein 4 isoform B; PRDM7B; PFM4B | |||||
| 16 | Unidentified | ||||
| 17 | gi|2222804 | 4/31 | 55 | 10467/4.85 | HLA-DPB1 |
| 18 | gi|24475861 | 4/25 | 42 | 14004/5.65 | Phosphohistidine phosphatase; |
| Sex-regulated protein janus-a | |||||
| 19 | gi|2134526 | 3/43 | 52 | 9785/5.02 | Gene MHC DQ-beta 1 protein (fragment) |
DISCUSSION
To further understand cellular functioning, the next logical level of analysis is proteomics. Proteomics is the study of global protein expression patterns in a cell, tissue, or organism. In general, it deals with the large-scale determination of gene and cellular function directly at the protein level[3]. The most commonly used method is a combination of two-dimensional electrophoresis (2-DE) and mass spectrometry (MS)[4]. Protein mixtures from cells are separated by 2-DE, stained, and each observed protein spot is quantified by its staining intensity. Selected spots are excised, digested and analyzed by MS[5]. 2-DE was the first technique capable of supporting the concurrent quantitative analysis of large numbers of gene products. Proteins are separated by charge in the first dimension and then by size in the second dimension, yielding spots on a polyacrylamide gel. Then, each protein spot was individually recovered and cleaved into short peptide fragments. 2-DE has been a mature technique for more than 25 years. Compared to classical 2-DE with carrier ampholytes (O’ Farrell 1975), the employment of immobilized pH gradient (IPG) gel strips as the first dimension, isoelectric focusing (IEF), has produced significant improvements in 2-D electrophoretic separation, permitting higher resolution and greater reproducibility. It become an effective method for separation of complex proteomes such as human cancer cells and tissues, facilitating spot identification by peptide mass fingerprinting (PMF), MALDI or tandem mass spectrometry, amino acid composition analysis, N-terminal and/or internal peptide microsequencing. We have developed a comprehensive strategy for reproducible, robust, gel-based separation of >1 000 protein components for quantitative protein profile. This strategy uses improved sample preparation methods and an optimized in-gel proteolysis protocol.
Protein identification is the most important step in expression proteomics. MS has increasingly become the method of choice for analysis of complex protein samples. MS-based proteomics is a discipline made possible by the availability of gene and genome sequence databases and technical and conceptual advances in many areas, most notably the discovery and development of protein ionization methods, as recognized by the 2002 Nobel prize in chemistry. The two most widely used ion-producing methods are called Electro Spray Ionization (ESI) and Matrix-Assisted Laser Desorption Ionization (MALDI)[6]. The method of choice for rapid, high-volume sequence analysis is MALDI, developed in 1987 by Hillenkamp and Karas[7]. In this process, peptides are first made soluble in a solvent containing an organic acid, such as nicotinic acid, and are then deposited onto a metal stage, as the solvent is evaporated. The organic acid plays a dual role - it is capable of absorbing light energy and it serves as a matrix that holds the protein molecules in place. Energy from a short laser pulse is absorbed by the matrix, which vaporizes, releasing stable precharged peptide ions into the ion chamber. Ionized molecules released in this manner may remain intact or break down into smaller pieces en route to the detector. MALDI’ s tendency to deliver intact protein masses to the detector is very useful for determining the full molecular weight of a protein, but provides little information about the amino acid sequence that composes it. This limitation can be overcome by enzymatically or chemically cleaving the protein to obtain a mixture of peptides that is then analyzed by mass spectrometry. The peptide mass pattern obtained in this manner is characteristic of the original protein and constitutes a sort of “fingerprint” that can aid in its identification. Each peptide in the fingerprint can be further analyzed by fragmentation to yield complex patterns of ion mass that may be used to identify the molecule. Once a suitable mass spectrum is obtained, it is compared against a protein mass spectral database to see if the sample pattern matches the fingerprint of any known peptides. Commonly used protein sequence databases include the SWISSPROT, OWL and NCBInr databases, which are publicly available. Several software programs for protein identification are available online such as Mascot (http://www.matrixscience.com), MOWSE (http://srs.hgmp.mrc.ac.uk/cgi-bin/mowse), PeptIdent2, and ProFound (http://prowl.rockefeller.edu/cgi-bin/ ProFound or http://www.proteometrics.com/prowl-cgi/ ProFound.exe), Prowl (http://prowl.rockefeller.edu/ contents/resource.htm), and Protein Prospector (http:// prospector.ucsf.edu/). PMF by MALDI-TOF has become highly efficient in the identification of gel-separated proteins[8].
The identified proteins were abundantly expressed in SW480 cells including enzymes (enolase), cytoskeletal components (intermediate filaments, IF), transcription factors (PR-domain family), and immunological molecules (MHC class and human B7-1 CTLA-4 co-stimulatory complex). These proteins maybe involved in colorectal carcinogenesis. Enolase is a key enzyme of glycolysis related with the energy metabolism[9]. This gene encodes one of three enolase isoenzymes found in mammals. It encodes alpha-enolase, a homodimeric soluble enzyme, and also encodes a shorter monomeric structural lens protein, tau-crystallin. The two proteins are made from the same message. The full-length protein, the isoenzyme, is found in the cytoplasm. The shorter protein is produced from an alternative translation start, is localized to the nucleus, and has been found to bind to an element in the c-myc promoter. A pseudogene has been identified, that is located on the other arm of the same chromosome. Intermediate filaments are the most abundant cytoskeletal proteins in cells and regulate the migration of normal and transformed epithelial cells and can be used to define a specific cancer tissue[10]. The PR domain (PRDI-BF1-RIZ homology region), a distant relative of the SET domain functioning in chromatin-mediated gene expression, defines a small family of zinc-finger type DNA-binding transcription factors. Three human members are presently known, and they are RIZ, MDS1-EVI1, and PRDI-BF1 or BLIMP1. A characteristic feature of these genes is the unusual yin-yang involvement in human cancers. Two products are normally produced from PR-domain family members who differ by the presence or absence of the PR domain; the PR-plus product is disrupted or under expressed whereas the PR-minus product is present or over expressed in cancer cells. This imbalance in the amount of the two products, a result of either genetic or epigenetic events, appears to be an important cause of malignancy[11].
Cancer cells can be detected and destroyed by cytotoxic T lymphocytes in many tumors. In humans however, most diagnosed tumors are not eliminated by T cells but grow steadily, invading and metastasizing until the host is destroyed. Evidence is accumulating that progressive tumor growth occurs not because the immune system is defective or deteriorated, but because the cancer cell is capable of developing a variety of strategies to escape immune recognition. Major histocompatibility complex (MHC) molecules are of central importance in regulating the immune response against tumors. Proper HLA class I antigen processing and presentation is a prerequisite for the recognition of tumor cells by cytotoxic T lymphocytes[12]. MHC II molecules are designated DP, DQ or DR and consist of two transmembrane polypeptides: an alpha (α) chain and a beta (β) chain. HLA-DPB1 is located on chromosome 6p12.3 and codes for Human Leukocyte Antigen, a Major Histocompatibility Class II (MHC II) molecule. MHC II molecules are heterodimeric, having α and β chains, DPB1 codes for the β chain. These molecules are responsible for initiation of the immune response through antigen presentation to T cells[13,14]. B7 family members play a central co-stimulatory role in T cell activation[15]. These two signals are necessary for T cell activation. It is generally accepted that human and experimental tumor cells can lose Major Histocompatibility Complex (MHC) class I molecules and this represents the major mechanism of tumor escape from T-cell immune responses. This is now mandatory, considering the high frequency of total or partial MHC class I losses observed in different tumor types as a result of escape mechanism from infiltrating-infiltrating lymphocytes: 90% in cervical carcinomas[16], 73% in colorectal carcinomas[17], 88% in breast carcinomas[18,19], 51% in melanomas[20,21], and 66% in laryngeal carcinomas[22] and renal cell carcinoma[23]. The expression of MHC I, MHC II and B7-1 molecules by SW480 cells might imply that such a kind of tumor cells had all the components necessary for evoking T lymphocyte activation and generating a positive anti-tumor immune response. Why the hosts cannot eliminate the malignancies? Recent findings discovered that the lymphocyte population of a patient with CRC contained at least two types of lymphocytes with tumor specificity, i.e., CTLs and regulatory T cells. They might be induced by a tumor-associated self antigen(s)[24,25]. However, the exact immunological pathomechanism(s) of these immu-nological molecules remains to be established. The regulatory T cells may suppress autologous CTL functions, resulting in escape of the tumor from immune surveillance by CTLs. This may explain why tumors often grow despite the presence of CTLs in the same individual.
Peptide mass fingerprinting (PMF) is a powerful tool for identification of proteins separated by 2-DE. However, there are several limitations to peptide mass fingerprinting, including a lack of complete and accurately annotated genome- and protein-sequence databases for a great number of highly homologous human proteins. Beyond identification, confirmation and validation of the protein of interest are also important steps in proteomic approaches.
Footnotes
Science Editor Pan BR and Guo SY Language Editor Elsevier HK
Supported by the Natural Science Foundation, Y100-573006 and Doctoral Foundation of Xi’an Jiaotong University, DFXJTU2002-11
References
- 1.Crawford NP, Colliver DW, Galandiuk S. Tumor markers and colorectal cancer: utility in management. J Surg Oncol. 2003;84:239–248. doi: 10.1002/jso.10325. [DOI] [PubMed] [Google Scholar]
- 2.Bertucci F, Salas S, Eysteries S, Nasser V, Finetti P, Ginestier C, Charafe-Jauffret E, Loriod B, Bachelart L, Montfort J, et al. Gene expression profiling of colon cancer by DNA microarrays and correlation with histoclinical parameters. Oncogene. 2004;23:1377–1391. doi: 10.1038/sj.onc.1207262. [DOI] [PubMed] [Google Scholar]
- 3.Mann M, Hendrickson RC, Pandey A. Analysis of proteins and proteomes by mass spectrometry. Annu Rev Biochem. 2001;70:437–473. doi: 10.1146/annurev.biochem.70.1.437. [DOI] [PubMed] [Google Scholar]
- 4.Long YZ, Fan XG, Li N. Progress in tumor proteomics. Shijie Huaren Xiaohua Zazhi. 2002;10:1436–1440. [Google Scholar]
- 5.Katayama H, Nagasu T, Oda Y. Improvement of in-gel digestion protocol for peptide mass fingerprinting by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun Mass Spectrom. 2001;15:1416–1421. doi: 10.1002/rcm.379. [DOI] [PubMed] [Google Scholar]
- 6.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 7.Karas M, Hillenkamp F. Laser desorption ionization of proteins with molecular masses exceeding 10,000 daltons. Anal Chem. 1988;60:2299–2301. doi: 10.1021/ac00171a028. [DOI] [PubMed] [Google Scholar]
- 8.Berndt P, Hobohm U, Langen H. Reliable automatic protein identification from matrix-assisted laser desorption/ionization mass spectrometric peptide fingerprints. Electrophoresis. 1999;20:3521–3526. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3521::AID-ELPS3521>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 9.Thoden JB, Taylor Ringia EA, Garrett JB, Gerlt JA, Holden HM, Rayment I. Evolution of enzymatic activity in the enolase superfamily: structural studies of the promiscuous o-succinylbenzoate synthase from Amycolatopsis. Biochemistry. 2004;43:5716–5727. doi: 10.1021/bi0497897. [DOI] [PubMed] [Google Scholar]
- 10.Herzig KH, Altmannsberger M, Fölsch UR. Intermediate filaments in rat pancreatic acinar tumors, human ductal carcinomas, and other gastrointestinal malignancies. Gastroenterology. 1994;106:1326–1332. doi: 10.1016/0016-5085(94)90026-4. [DOI] [PubMed] [Google Scholar]
- 11.Jiang GL, Huang S. The yin-yang of PR-domain family genes in tumorigenesis. Histol Histopathol. 2000;15:109–117. doi: 10.14670/HH-15.109. [DOI] [PubMed] [Google Scholar]
- 12.Cabrera T, López-Nevot MA, Gaforio JJ, Ruiz-Cabello F, Garrido F. Analysis of HLA expression in human tumor tissues. Cancer Immunol Immunother. 2003;52:1–9. doi: 10.1007/s00262-002-0332-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edelshtein D, Sidebottom D, Chen DF, Baxter-Lowe LA. A novel HLA-DPB1 allele identified by sequence-based typing and confirmed by SSP. Hum Immunol. 2003;64(10 Suppl):S162. [Google Scholar]
- 14.Zino E, Frumento G, Marktel S, Sormani MP, Ficara F, Di Terlizzi S, Parodi AM, Sergeant R, Martinetti M, Bontadini A, et al. A T-cell epitope encoded by a subset of HLA-DPB1 alleles determines nonpermissive mismatches for hematologic stem cell transplantation. Blood. 2004;103:1417–1424. doi: 10.1182/blood-2003-04-1279. [DOI] [PubMed] [Google Scholar]
- 15.Chen L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat Rev Immunol. 2004;4:336–347. doi: 10.1038/nri1349. [DOI] [PubMed] [Google Scholar]
- 16.Ryu KS, Lee YS, Kim BK, Park YG, Kim YW, Hur SY, Kim TE, Kim IK, Kim JW. Alterations of HLA class I and II antigen expression in preinvasive, invasive and metastatic cervical cancers. Exp Mol Med. 2001;33:136–144. doi: 10.1038/emm.2001.24. [DOI] [PubMed] [Google Scholar]
- 17.Cabrera T, Collado A, Fernandez MA, Ferron A, Sancho J, Ruiz-Cabello F, Garrido F. High frequency of altered HLA class I phenotypes in invasive colorectal carcinomas. Tissue Antigens. 1998;52:114–123. doi: 10.1111/j.1399-0039.1998.tb02274.x. [DOI] [PubMed] [Google Scholar]
- 18.Palmisano GL, Pistillo MP, Capanni P, Pera C, Nicolò G, Salvi S, Perdelli L, Pasciucco G, Ferrara GB. Investigation of HLA class I downregulation in breast cancer by RT-PCR. Hum Immunol. 2001;62:133–139. doi: 10.1016/s0198-8859(00)00241-x. [DOI] [PubMed] [Google Scholar]
- 19.Redondo M, García J, Villar E, Rodrigo I, Perea-Milla E, Serrano A, Morell M. Major histocompatibility complex status in breast carcinogenesis and relationship to apoptosis. Hum Pathol. 2003;34:1283–1289. doi: 10.1016/j.humpath.2003.06.001. [DOI] [PubMed] [Google Scholar]
- 20.Ferrone S, Marincola FM. Loss of HLA class I antigens by melanoma cells: molecular mechanisms, functional significance and clinical relevance. Immunol Today. 1995;16:487–494. doi: 10.1016/0167-5699(95)80033-6. [DOI] [PubMed] [Google Scholar]
- 21.Marincola FM, Ferrone S. Immunotherapy of melanoma: the good news, the bad ones and what to do next. Semin Cancer Biol. 2003;13:387–389. doi: 10.1016/j.semcancer.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 22.Cabrera T, Salinero J, Fernandez MA, Garrido A, Esquivias J, Garrido F. High frequency of altered HLA class I phenotypes in laryngeal carcinomas. Hum Immunol. 2000;61:499–506. doi: 10.1016/s0198-8859(00)00097-5. [DOI] [PubMed] [Google Scholar]
- 23.Atkins D, Ferrone S, Schmahl GE, Störkel S, Seliger B. Down-regulation of HLA class I antigen processing molecules: an immune escape mechanism of renal cell carcinoma? J Urol. 2004;171:885–889. doi: 10.1097/01.ju.0000094807.95420.fe. [DOI] [PubMed] [Google Scholar]
- 24.Jordan MS, Boesteanu A, Reed AJ, Petrone AL, Holenbeck AE, Lerman MA, Naji A, Caton AJ. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat Immunol. 2001;2:301–306. doi: 10.1038/86302. [DOI] [PubMed] [Google Scholar]
- 25.Sakaguchi S. Regulatory T cells: key controllers of immunologic self-tolerance. Cell. 2000;101:455–458. doi: 10.1016/s0092-8674(00)80856-9. [DOI] [PubMed] [Google Scholar]