

Abstract
AIM: Persistent cholestasis is a rare complication of severe trauma or infections. Little is known about the possible pathomechanisms and the clinical course.
METHODS: Secondary sclerosing cholangitis was diagnosed in five patients with persistent jaundice after severe trauma (one burn injury, three accidents, one power current injury). Medical charts were retrospectively reviewed with regard to possible trigger mechanisms for cholestasis, and the clinical course was recorded.
RESULTS: Diagnosis of secondary sclerosing cholangitis was based in all patients on the primary sclerosing cholangitis (PSC)-like destruction of the intrahepatic bile ducts at cholangiography after exclusion of PSC. In four patients, arterial hypotension with subsequent ischemia may have caused the bile duct damage, whereas in the case of power current injury direct thermal damage was assumed to be the trigger mechanism. The course of secondary liver fibrosis was rapidly progressive and proceeded to liver cirrhosis in all four patients with a follow-up >2 years. Therapeutic possibilities were limited.
CONCLUSION: Posttraumatic sclerosing cholangitis is a rare but rapidly progressive disease, probably caused by ischemia of the intrahepatic bile ducts via the peribiliary capillary plexus due to arterial hypotension. Gastroenterologists should be aware of this disease in patients with persistent cholestasis after severe trauma.
Keywords: Life-threatening trauma, Arterial hypotension, Cholestasis, Ischemia of intrahepatic bile ducts, Secondary sclerosing cholangitis, Posttraumatic sclerosing cholangitis
INTRODUCTION
For a long time it is known that after severe trauma marked cholestasis can occur[1-3]. Different reasons were held responsible for this including hepatic hypoxia, inflammation, and mass transfusions[1,4]. Unfortunately, cholangiographic studies to detect changes of the bile ducts to exclude obstructive jaundice were not done, primarily as most of these studies were performed in the pre-ERCP era.
On the other side, gastroenterologists are occasionally confronted with patients with a history of persistent cholestasis who have survived life-threatening events with long-term stays in intensive care units. In the diagnostic work-up ERC in some of these patients shows a PSC-like picture with strictures and dilatations of the intrahepatic bile ducts.
Except one recent report by Engler et al.[5], these patients are characterized badly in the literature. Therefore, we report on five patients with secondary sclerosing cholangitis after life-threatening injuries in order to demonstrate the clinical course, evaluate measures for diagnosis, and discuss possible trigger mechanisms for posttraumatic sclerosing cholangitis.
MATERIALS AND METHODS
All five patients (one woman and four men; age 18-71 years) presented with acute or persistent cholestasis. Three of them were seen as outpatients between January 2001 and June 2003, 4 to 22 mo after occurrence of cholestasis (Table 1). Patient 5 was seen as consultant during the initial stay of the patient on the intensive care unit of a burn clinic after occurrence of jaundice, patient 1 is a former case of one of the hospitals that has already been published and who was re-evaluated[6].
Table 1.
Summary of patients‘ characteristics
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | |
| Sex | Female | Male | Male | Male | Male |
| Age (yr) | 56 | 41 | 56 | 71 | 18 |
| Initial event | Room fire | Motorbike accident | Tractor accident | Fall from a raised hide | Power current accident |
| and fall | |||||
| Injuries | Burn injury: 34 % of | Polytrauma with avulsions | Polytrauma with serial | Polytrauma with burst | Burn injury (36 % of body |
| body surface (hands, | (left subclavian artery, | rib fracture, hemothorax, | fractures of lumbar spine, | surface), multiple rib | |
| head, neck, back) | forearm, anterior tibial | pleural effusion, fracture | paraplegia, rib fractures, | fractures, hemothorax, | |
| artery) and fractures | of right joint ankle | pneumothorax | fractures of the thoracic | ||
| (left humeral shaft, | vertebrae 5 to 9 | ||||
| pelvis, both lower legs) | |||||
| Intensive care (d) | 58 | 23 | 34 | 26 | 88 |
| Time from | 4 | 27 | 4 | 13 | 2 |
| occurrence | |||||
| of cholestasis until | |||||
| diagnosis (mo) | |||||
| Treatment start | mo 4 | mo 22 | mo 4 | mo 13 | mo 1 |
| with UDCA | |||||
| Follow-up(mo) | 55 (death) | 48 | 41 | 33 | 12 |
| Complications | Liver cirrhosis (Child- | Small intrahepatic | unbearable pruritus | liver cirrhosis | - |
| Pugh C), variceal | gallstones after 6 mo | during first year, | (Child-Pugh B), | ||
| bleeding, death due | (endoscopically extracted), | liver cirrhosis | recurrent stomal | ||
| to liver insufficiency | liver cirrhosis (Child-Pugh A) | (Child-Pugh A) | bleeding |
UDCA: urso deoxycholic acid.
Time between the occurrence of cholestasis and definitive diagnosis ranged between 2 and 27 mo.
All patients experienced life-threatening injuries and needed long-term treatment on intensive care units (ICU) (from 23 to 88 d, Table 1). Mechanical ventilation was necessary in all patients (from 13 to 62 d). Temporary acute renal failure occurred in patients 3 and 4 (maximum creatinine 3.6 mg/dL [-1.0] on d 5 and 1.6 mg/dL on d 4, respectively). In all patients several surgeries had to be performed (without abdominal surgery), in three of them pleural drainages were necessary.
The charts of these patients at their initial hospital stay were intensely reviewed, especially concerning periods of hypotension, therapy with vasopressors and RBC units, septic fever, antibiotics, mechanical ventilation, and liver function tests to find triggers for subsequent bile duct damage. Pre-injury liver function tests were documented if available. Diagnostic work-up included ERC, liver biopsy, imaging studies of the liver and vessels of the liver hilus (ultrasound, doppler ultrasound, CT, and/or MRI), and the exclusion of other liver diseases (e.g. viral hepatitis, hereditary, and autoimmune liver diseases). Liver biopsies (also from hospitals outside) were re-evaluated by one pathologist. The further course was observed and recorded.
For the purpose of the study an increase in liver function tests more than twice above the upper limit of normal was defined abnormal.
RESULTS
Before the trauma no patient had known liver disease. No comorbidities were present with the exception of patient 3 (arterial hypertension, cardiac arrhythmias). No patient reported about alcohol abuse. On hospital admission patient 5 (power current injury) had increased values of ALT (alanine aminotransferase) and AST (aspartate aminotransferase) due to direct thermal cell damage. In all other patients liver function tests were normal on admission. No other liver diseases or thrombosis of hepatic artery and veins were found.
Early course
In all patients as the first sign of bile duct damage a permanent increase of GGT (gamma glutamyl transferase) twice above the upper limit of normal was first documented between d 4 and 11 (Table 2). Alkaline phosphatase (AP) and bilirubin rose during the next 2 weeks in all but patient 1 in whom bilirubin increased after four weeks and patient 2 whose bilirubin levels according to a milder course of cholestasis reached twice the upper limit of normal only once. Aminotransferases always increased secondary to signs of cholestasis (Table 2). As an example the early course (first month) of AST, ALT, GGT, AP, and bilirubin in patient 3 is shown in Figure 1A.
Table 2.
Patients’ data and important findings during the early course at the intensive care unit
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | |
| Time of increase of liver | |||||
| function tests > 2× ULN (d) | |||||
| GGT | 101 | 6 | 5 | 4 | 11 |
| AP | 18 | 18 | 9 | 12 | 15 |
| Bilirubin | 36 | 62 | 10 | 8 | 13 |
| AST | 18 | 52 | 9 | 12 | 16 |
| ALT | 18 | 23 | 9 | 7 | 19 |
| Mechanical ventilation (d) | 36; | 13; | 25; | 16; | 62; |
| PEEPmax. 6 mmHg; | PEEPmax. 10 mmHg; | PEEPmax. 8 mmHg; | PEEPmax. 15 mmHg; | PEEPmax. 20 mmHg; | |
| FiO2 not documented | FiO2max. 0.6 | FiO2max. 0.6 | FiO2max. 0.7 | FiO2max. 0.6 | |
| Severe hypotension | d 1 (2 h) | d 1 (45 min) | d 2 (30 min) and | d 1 (30 min) | None |
| (systolic blood pressure | d 3 (60 min) | ||||
| < 70 mm Hg)3 | |||||
| Vasopressor therapy | Dobutamine d 1-8 | None | Norepinephrine d 3-5 | Norepinephrine d 2-4 | None |
| dopamine d 1-18 | and d 9-17 | ||||
| norepinephrine d 1-7 | |||||
| Minimum hemoglobin3 | 7.3 g/dL (d 3) | 2.4 g/dL (d 1) | 7.2 g/dL (d 3) | 8.6 g/dL (d 1) | 7.9 g/dL (d 4) |
| Transfusions (RBC units)3 | 30 | 34 | 10 | 10 | 24 |
| Fever (> 38.0 ) | d 7-16 | d 6-13 | d 2-25 | d 5-17 | d 2- >20 |
| Antibiotics3 | Cefotaxim, imipenem, | None | Piperacillin/sulbactam | None | Piperacillin/tazobactam, |
| sulbactam, mezlocillin | ciprofloxacin, meropenem, | ||||
| levofloxacin, fluconazol |
ALT: alanine aminotransferase; AP: alkaline phosphatase; AST: aspartate aminotransferase; FiO2max.: maximum fraction of inspired oxygen; GGT: gamma glutamyl transferase; PEEPmax.: maximum positive end-exspiratory pressure; ULN: upper limit of normal 1No control of liver function tests between d 4 and 10; 2A bilirubin value 2× ULN was noted only once at d 6;
Until increase of GGT ≥ 2× ULN.
Figure 1.
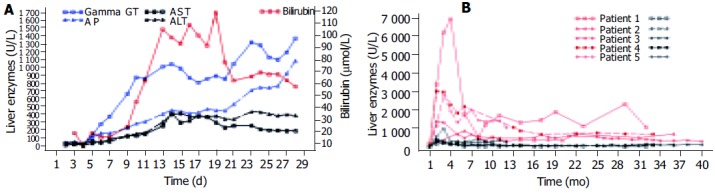
A: Liver function tests of patient 3 in the first month showing the strong increase of cholestasis followed by a milder rise of aminotransferases; B: Long-time course of AP (red) and ALT (black) of all patients showing the initial strong increase of AP with a subsequent slow decrease and only a mild initial rise of ALT with nearly normal values in the long term.
Possible reasons for bile duct damage
Details of different parameters during intensive care of the patients are shown in Table 2.
Severe arterial hypotension with a systolic blood pressure of ≤70 mmHg as a possible cause of bile duct ischemia occurred in four patients. Only patient 5 experienced no hemodynamic instability throughout the whole course. In contrast to all other patients aminotransferases were already increased at clinic admission and nearly normalized during the following days. According to the pattern of his burns current flow through the body was diagnosed. Therefore, a direct thermal damage of the intrahepatic bile ducts can be suspected.
Two patients (patients 2 and 3) suffered of sudden strong blood loss requiring mass transfusions in patient 2 (30 RBC units, 11 fresh frozen plasma units, and four platelet concentrates during the first four hours after hospital admission), whereas a moderate decrease of the hemoglobin occurred in the other patients. After ICU admission all patients received parenteral nutrition via central venous lines, and additional enteral feeding via a nasogastric tube was started in all cases between d 2 and 5.
Imaging findings
Imaging findings are shown in Table 3.
Table 3.
Diagnostic findings in posttraumatic sclerosing cholangitis during the course of the disease (ultrasound, MRT/MRCP, ERCP, and liver histology)
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | |
| 1 - 2 mo | |||||
| Ultrasound | L normal, | L normal, | L normal, | L normal, | L enlarged, |
| BD normal, | BD normal, | BD normal, | BD normal, | hyperechoic, | |
| GB sludge | GB sludge | GB sludge | GB sludge | BD normal, | |
| GB contracted, | |||||
| splenomegaly | |||||
| ERC | Stenoses and loss of IHBD, | - | IHBD irregular, | - | - |
| CBD normal, | CBD normal, | ||||
| GB normal | GB normal | ||||
| MRT/MRCP | - | - | - | L normal, BD | - |
| normal, GB normal | |||||
| 4 - 6 mo | |||||
| Ultrasound | L cirrhosis, | L inhomogeneous, | L inhomogeneous with | - | L enlarged, |
| hyperechoic areas | |||||
| alongside the IHBD, | BD normal, | ||||
| BD normal, | BD normal | BD normal, | BD normal, | ||
| GB stones | GB normal, | splenomegaly | |||
| ERC | - | IHBD: multifocal short | - | IHBD irregular, | |
| strictures and dilatations, | beaded | ||||
| CBD normal, | appearance, | ||||
| GB normal (Figure 2) | CBD normal, | ||||
| GB normal | |||||
| Liver histology | Substantial cholestasis; | Portal tract inflammation, | Canalicular bile thrombi; | - | - |
| inflammation of the portal | occasionally lymphocytes | edematously swollen | |||
| tracts, liver lobules, and | in bile duct epithelium; | portal tracts; | |||
| sporadically the bile ducts; | several necrotic foci with | inflammatory infiltrates, | |||
| feathery degeneration of | foamy macrophages in | esp. around bile ducts; | |||
| hepatocytes; | the liver acini; | occasionally feathery | |||
| fibrosis around bile ducts; | bridging fibrosis; | degeneration of hepatocytes; | |||
| regenerative bile duct | regenerative bile duct | minimal fibrosis | |||
| proliferations | proliferations | ||||
| 12 - 24 mo | |||||
| Ultrasound | L inhomogeneous cirrhosis | L enlarged, inhomogeneous, | L cirrhosis, | L cirrhosis, | - |
| hyperechoic areas segment | |||||
| 7/8, IHBD slightly dilated, | IHBD slightly dilated, | ||||
| BD normal, | CBD normal, | BD normal, | CBD normal, | ||
| GB stones and sludge, | GB stones, | GB normal, | GB normal, | ||
| splenomegaly, | splenomegaly | splenomegaly | splenomegaly | ||
| distinct ascites | |||||
| ERC | stenoses and loss of IHBD, | IHBD: loss and stenoses | - | IHBD: multifocal | - |
| of right-sided bile ducts, | high-grade strictures and | ||||
| a long, stretched running | dilatations on the left side, | ||||
| left bile duct (suitable to | bile ducts on the right | ||||
| liver hypertrophy), | side not presentable, | ||||
| CBD normal, | CBD normal, | CBD normal | |||
| GB normal | GB stones | ||||
| MRT/MRCP | - | L atrophy of right liver, | L macronodular cirrhosis, | - | - |
| hypertrophy of left liver; | |||||
| reduced signal intensity of | |||||
| segments 2 and 3 and | |||||
| partly 7 and 8; | |||||
| IHBD segmentally dilated, | |||||
| CBD pseudoobstruction, | CBD pseudoobstruction, | ||||
| GB stones, splenomegaly | splenomegaly | ||||
| Liver histology | No cholestasis; | - | Occasionally canalicular | Substantial cholestasis (bile | |
| complete liver cirrhosis | cholestasis; mild | thrombi in dilated canaliculi); | |||
| portal inflammation | occasionally lymphocytes in | ||||
| (predominantly | the epithelium of bile ducts; | ||||
| lymphocytes); some | numerous regenerative | ||||
| regenerative bile duct | bile duct proliferations in the | ||||
| proliferations; mild | periphery of portal tracts; | ||||
| fibrosis; (sampling error ?) | cirrhosis (Figure 4) | ||||
BD: bile ducts; CBD: common bile duct; GB: gallbladder; IHBD: intrahepatic bile ducts; L: liver.
Ultrasound Ultrasound was repeatedly performed in all patients. Depending on the time since occurrence of cholestasis different findings were recorded (Table 3).
In the early phase (first 2 mo) the liver was described as normal or enlarged with homogeneous or hyperechoic parenchyma, bile ducts were normal in all patients, gallbladder sludge was seen in four patients. Later on liver parenchyma got more and more inhomogeneous and signs of liver cirrhosis (nodularity/irregularity of liver surface, inhomogeneous parenchyma, splenomegaly) occurred. Intrahepatic bile ducts showed segmental dilatation in two patients after 12 to 24 mo, whereas the common bile duct remained normal in all patients.
Computed tomography CT scans performed in patient 1 18 mo and in patient 3 10 d and 4 mo after the onset of cholestasis confirmed sonographic findings.
MRT/MRCP MRT/MRCP in patient 3 in the early phase showed normal abdominal findings. In two patients MRT/MRCP was performed after 2 years. In patient 2 MRT/MRCP disclosed atrophy of the right liver, hypertrophy of the left liver, a reduced signal intensity in segments 2 and 3 and partly 7 and 8, and peripheral segmental dilatation of intrahepatic bile ducts rather unusual findings (Figure 2). In patient 3 signs of liver cirrhosis with large nodules and inhomogeneous liver parenchyma were present.
Figure 2.
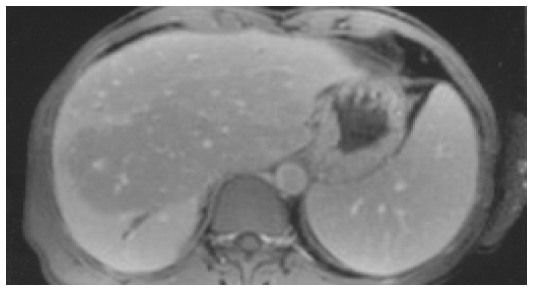
MRT of patient 2 (T1-weighted after contrast medium) showing an area centrally in the liver with reduced signal intensity, a dilatation of the bile duct in segment 6, and splenomegaly.
ERC Diagnostic ERC was performed in all patients. Together with the history it led to diagnosis in all patients. Already three weeks after first signs of cholestasis slight changes of the intrahepatic bile ducts were noticed (patient 3). After four to six months multifocal strictures and dilatations of the intrahepatic bile ducts (comparable to lesions in primary sclerosing cholangitis) occurred (Figure 3), that were also present after 12-24 mo. The common bile duct and the cystic duct were normal in all patients at all times.
Figure 3.
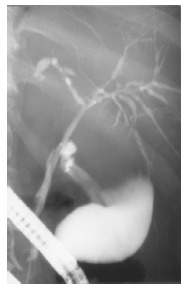
ERCP of patient 3 showing a normal cystic and common bile duct, but multifocal short strictures and dilatations of the intrahepatic bile ducts (4 mo after occurrence of cholestasis).
Liver histology
Liver biopsy was performed in four patients (in no one in the acute phase), at least once in three patients and three times in one patient (patient 3) (Table 3). After 4-6 mo portal inflammation (predominantly lymphocytes) with involvement of the bile ducts, necrosis of periportal hepatocytes, fibrosis of different degree, and depending on the biochemical extent of cholestasis bile thrombi were present. After 12-24 mo complete liver cirrhosis was diagnosed in patients 1 and 4, whereas in patient 3 only mild fibrosis was diagnosed, possibly due to a sampling error (clinically cirrhosis). Inflammation of the portal tracts with involvement of bile ducts was still present. Regenerative bile duct proliferations were a prominent feature. As an example liver histology of patient 4 is shown in Figure 4.
Figure 4.
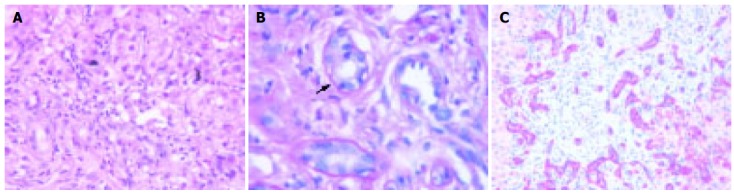
Liver histology of patient 4 (13 mo after occurrence of cholestasis). A: Portal inflammation, severe cholestasis with bile thrombi, HE (20x); B: Lymphocytic infiltration of a small bile duct (arrow). Periodic acid Schiff’s reaction after treatment with diastase (100x); C: Immunostaining for cytokeratin 7. Regenerative bile duct proliferations show a strong expression of cytokeratin 7 (25x).
Follow-up, treatment, and complications
The patients were followed between 12 and 55 mo after trauma (Table 1).
The long-time courses of AP and ALT of all patients are shown in Figure 1B.
All patients were treated with ursodeoxycholic acid after different periods of time (Table 1).
Liver cirrhosis was diagnosed clinically and/or histologically in all four patients with a follow-up of >2 years. It occurred already at mo 5 in patient 4. In this case it was diagnosed macroscopically during colostomy which was performed due to fecal incontinence following paraplegia. Decompensation of liver cirrhosis with large amounts of ascites occurred in patient 1 already after 18 mo. This patient died after 55 mo due to hepatic insufficiency.
Liver transplantation was planned for patient 3 after 4 mo, but due to improvement of liver function tests and clinical symptoms transplantation was cancelled. Patient 5 is designated for liver transplantation.
DISCUSSION
Cholestasis is common in long-term ICU patients. However, in most cases it is mild and resolves within weeks after clinical improvement of the patient.
Secondary sclerosing cholangitis after trauma (“posttraumatic SC”) is a rare and probably underdiagnosed complication of severe trauma with to our knowledge only two publications in the literature[5,6]. Our patients shared the following findings, thus leading to the diagnosis: (1) a severe, life-threatening trauma; (2) slowly increasing signs of cholestasis with a secondary moderate rise of aminotransferases; (3) the typical PSC-like appearance of the intrahepatic bile ducts with multifocal strictures and dilatations; and (4) the exclusion of other liver diseases. All patients except patient 5 had normal liver function tests at hospital admission. After a few days a slow, but constant increase of signs of cholestasis was noted with a subsequent rise of aminotransferases that never exceeded 15 times of ULN. This differs from “shock liver” where an acute elevation of the aminotransferases to at least 20 times ULN is required for diagnosis[7]. According to our results ERC is of essential importance for the diagnosis. Probably earlier than MRC, ERC best shows the PSC-like bile duct changes (multifocal strictures and dilatations, beaded appearance) that occur only intrahepatically in posttraumatic SC. The possibility to aspirate bile for microbiological examination is another advantage of ERC compared to all non-invasive imaging procedures as to our experience and some case reports in the literature in some intensive care patients with severe, long-lasting infections requiring mechanical ventilation jaundice may be caused by secondary bacterial or fungal cholangitis without preexisting biliary obstruction[8,9]. The value of ultrasound, CT, and MRT in the early course is to exclude obvious biliary obstruction, later they may help to diagnose secondary development of liver cirrhosis.
Posttraumatic SC seems to be rapidly progressive. Liver cirrhosis was diagnosed clinically or by liver biopsy in all patients with a follow-up of more than 2 years, in one patient already after 5 mo, in the others after 18-24 mo. After decompensation of liver cirrhosis at mo 18 and esophageal variceal bleeding at mo 22, one patient died after 55 mo due to hepatic insufficiency. This rapidly progressive course compares well with the experience of Engler et al.[5]. In their series all four patients with a follow-up of more than 2 years developed liver cirrhosis, three of them during the second year.
All patients were treated with UDCA after 1-22 mo. As this was an uncontrolled retrospective study clear conclusions concerning the therapeutic value of UDCA are not possible, but the efficacy seems to be rather limited. In patients with posttraumatic SC and end-stage liver disease liver transplantation may be the only therapeutic option. In contrast to Engler et al. we are not convinced that endoscopic interventions for treatment of strictures (dilatation or stenting) are beneficial as bile duct strictures were multifocal and localized intrahepatically in all of our patients. This is comparable to PSC where only patients with a dominant extrahepatic stricture appear to have some potential benefit from endoscopic treatment[10]. Even in the patients of Engler et al.[5], endoscopic therapy (with the additional risk of secondary bacterial cholangitis) did not stop disease progression. However, for diagnostic reasons early ERC appears necessary.
Interestingly, bilirubin levels of all patients decreased without intervention after 2-6 mo. Proliferation of bile ducts in the portal tracts with metaplasia of hepatocytes may have improved bile flow. This phenomenon was also described in drug-induced prolonged cholestasis with ductopenia[11]. However, posttraumatic SC progresses in spite of decreasing bilirubin levels.
What are the possible mechanisms for posttraumatic sclerosing cholangitis? As shown in Table 2 we examined several factors that are discussed in the literature, which could have contributed to bile duct damage[5,6,12]. In our opinion the dominant cause is arterial hypotension with subsequent ischemia of the intrahepatic bile ducts which occurred in four of the five patients in the early course after the accident (in patient 5 according to the pattern of burns current flow through the body was diagnosed suggesting a direct thermal damage of the intrahepatic bile ducts). It can be assumed that through temporary hypoperfusion, possibly aggravated by vasopressor therapy, an ischemic damage of the intrahepatic bile duct epithelium occurred which led to secondary scarring of the bile ducts and subsequent cholestasis. In contrast to shock liver the initial damage is not characterized by rapidly increasing aminotransferases as the liver via the portal vein - seems to be perfused sufficiently. After the acute ischemic phase, parallel to bile duct changes, increasing signs of cholestasis occur. Later, aminotransferases raise secondary to bile duct obstruction as in other obstructive bile duct diseases. Possibly due to the non-reversible scarring of the bile ducts with persistence of cholestasis and comparable to experimental bile duct ligation liver fibrosis and cirrhosis develop rather quickly[13].
In all of our patients only the intrahepatic bile ducts were affected. The reason may be the blood supply of the bile ducts[14-16]. Whereas the extrahepatic biliary system seems to be well supplied, mainly by two parallel to the common bile duct running arteries which are feeded by branches of eight arteries on the average (retroduodenal artery, gastroduodenal artery, left and right hepatic artery, etc.), the intrahepatic ducts are supplied by a nonaxial network of small arteries deriving only from the right and left hepatic artery. They form a plexus of arterioles, venules, and capillaries within the peribiliary adventitia with a second plexus within the biliary wall primarily composed of capillaries (“peribiliary capillary plexus”). This structure, possibly especially the blood supply via the left and right hepatic artery as functional end arteries, may cause the vulnerability of the intrahepatic bile ducts to ischemic events. Except in the periphery of the liver portal-venous blood plays no role in the supply of the bile ducts.
Other factors like systemic inflammation or transfusions may contribute to progressing cholestasis in some patients, but may be of minor importance.
In conclusion, after severe, life-threatening injuries a small number of patients develop jaundice that is caused by multiple intrahepatic bile duct strictures. The main trigger may be ischemia and subsequent damage of the bile ducts due to transient arterial hypotension. Slowly progressive cholestasis after severe injury in patients without prior liver disease is the hallmark of posttraumatic sclerosing cholangitis. The diagnosis is confirmed by ERC. The major characteristics of posttraumatic SC are summarized in Table 4. The course of this form of secondary sclerosing cholangitis appears to be rather progressive without promising therapeutic options (except probably liver transplantation). Gastroenterologists should be aware of this disease in patients with cholestasis after severe trauma
Table 4.
Characteristics of posttraumatic sclerosing cholangitis
| No former liver disease |
| Severe life-threatening injury with temporary severe arterial hypotension |
| Slowly increasing signs of cholestasis |
| Secondary moderate rise of aminotransferases |
| PSC-like appearance of intrahepatic bile ducts (multifocal strictures and dilatations) |
| Exclusion of other liver diseases (esp. hepatic artery thrombosis) |
ACKNOWLEDGMENTS
The authors thank the surgical hospitals, the Berufsgenos-senschaftliche Unfallklinik Ludwigshafen and the Clinic Nuremberg, both Germany, for the permission to analyze the medical files and therefore enabling this paper.
Footnotes
Science Editor Li WZ Language Editor Elsevier HK
References
- 1.Nunes G, Blaisdell FW, Margaretten W. Mechanism of hepatic dysfunction following shock and trauma. Arch Surg. 1970;100:546–556. doi: 10.1001/archsurg.1970.01340230012003. [DOI] [PubMed] [Google Scholar]
- 2.Champion HR, Jones RT, Trump BF, Decker R, Wilson S, Stega M, Nolan J, Crowley RA, Gill W. Post-traumatic hepatic dysfunction as a major etiology in post-traumatic jaundice. J Trauma. 1976;16:650–657. doi: 10.1097/00005373-197608000-00010. [DOI] [PubMed] [Google Scholar]
- 3.Hartley S, Scott AJ, Spence M. Benign postoperative jaundice complicating severe trauma. N Z Med J. 1977;86:174–178. [PubMed] [Google Scholar]
- 4.Te Boekhorst T, Urlus M, Doesburg W, Yap SH, Goris RJ. Etiologic factors of jaundice in severely ill patients. A retrospective study in patients admitted to an intensive care unit with severe trauma or with septic intra-abdominal complications following surgery and without evidence of bile duct obstruction. J Hepatol. 1988;7:111–117. doi: 10.1016/s0168-8278(88)80514-2. [DOI] [PubMed] [Google Scholar]
- 5.Engler S, Elsing C, Flechtenmacher C, Theilmann L, Stremmel W, Stiehl A. Progressive sclerosing cholangitis after septic shock: a new variant of vanishing bile duct disorders. Gut. 2003;52:688–693. doi: 10.1136/gut.52.5.688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmitt M, Kölbel CB, Müller MK, Verbeke CS, Singer MV. Sclerosing cholangitis after burn injury. Z Gastroenterol. 1997;35:929–934. [PubMed] [Google Scholar]
- 7.Seeto RK, Fenn B, Rockey DC. Ischemic hepatitis: clinical presentation and pathogenesis. Am J Med. 2000;109:109–113. doi: 10.1016/s0002-9343(00)00461-7. [DOI] [PubMed] [Google Scholar]
- 8.Scheppach W, Druge G, Wittenberg G, Mueller JG, Gassel AM, Gassel HJ, Richter F. Sclerosing cholangitis and liver cirrhosis after extrabiliary infections: report on three cases. Crit Care Med. 2001;29:438–441. doi: 10.1097/00003246-200102000-00042. [DOI] [PubMed] [Google Scholar]
- 9.Domagk D, Bisping G, Poremba C, Fegeler W, Domschke W, Menzel J. Common bile duct obstruction due to candidiasis. Scand J Gastroenterol. 2001;36:444–446. doi: 10.1080/003655201300051397. [DOI] [PubMed] [Google Scholar]
- 10.Lee YM, Kaplan MM. Management of primary sclerosing cholangitis. Am J Gastroenterol. 2002;97:528–534. doi: 10.1111/j.1572-0241.2002.05585.x. [DOI] [PubMed] [Google Scholar]
- 11.Degott C, Feldmann G, Larrey D, Durand-Schneider AM, Grange D, Machayekhi JP, Moreau A, Potet F, Benhamou JP. Drug-induced prolonged cholestasis in adults: a histological semiquantitative study demonstrating progressive ductopenia. Hepatology. 1992;15:244–251. doi: 10.1002/hep.1840150212. [DOI] [PubMed] [Google Scholar]
- 12.Labori KJ, Bjørnbeth BA, Raeder MG. Aetiology and prognostic implication of severe jaundice in surgical trauma patients. Scand J Gastroenterol. 2003;38:102–108. doi: 10.1080/00365520310000519. [DOI] [PubMed] [Google Scholar]
- 13.Johnstone JM, Lee EG. A quantitative assessment of the structural changes the rat's liver following obstruction of the common bile duct. Br J Exp Pathol. 1976;57:85–94. [PMC free article] [PubMed] [Google Scholar]
- 14.Batts KP. Ischemic cholangitis. Mayo Clin Proc. 1998;73:380–385. doi: 10.1016/S0025-6196(11)63706-3. [DOI] [PubMed] [Google Scholar]
- 15.Northover JM, Terblanche J. A new look at the arterial supply of the bile duct in man and its surgical implications. Br J Surg. 1979;66:379–384. doi: 10.1002/bjs.1800660603. [DOI] [PubMed] [Google Scholar]
- 16.Cho KJ, Lunderquist A. The peribiliary vascular plexus: the microvascular architecture of the bile duct in the rabbit and in clinical cases. Radiology. 1983;147:357–364. doi: 10.1148/radiology.147.2.6836115. [DOI] [PubMed] [Google Scholar]