

Abstract
AIM: To characterize cytochrome P4501A1 (CYP1A1), glutathione S-transferases (GSTs) and microsomal epoxide hydrolase (mEH) polymorphisms in Chinese esophageal cancer patients.
METHODS: Multiplex polymerase chain reaction (PCR) and PCR based restriction fragment length polymorphisms (PCR-RFLP) were used to detect polymorphism changes of CYP, GSTs and mEH on esophageal cancerous and precancerous lesions as well as in case control group. All the examination samples were obtained from Linzhou (formerly Linxian), Henan Province, the highest incidence area for esophageal cancer.
RESULTS: The frequency of CYP1A1 3’ polymorphism in case control group (26/38, 68%) was significantly higher than in esophageal squamous cell carcinoma group (ESCC) (29/62, 47%) (P < 0.05). A significant difference in the incidence of mEH slow allele variant was observed between case control group (15/38, 39%) and esophageal dysplasia group (22/32, 69%) or ESCC group (39/62, 63%) (P < 0.05). However, no significant difference was observed among different groups in the polymorphisms of CYP1A1 exon 7, GSTM1, GSTT1, GSTP1 and mEH fast allele.
CONCLUSION: The present results suggest that CYP1A1 3’ polymorphism may be one of the promising protective factors and its wild gene type may be an indicator for higher susceptibility to esophageal cancer. mEH slow allele variant, associated with the progression of esophageal precancerous lesions, may contribute to the high susceptibility to esophageal carcinoma.
INTRODUCTION
It has been revealed that carcinogenesis may be resulted from mutations or deletions in cancer-related genes. Meanwhile, a large proportion of human cancers is associated with diet, tobacco smoking and other environmental factors[1], suggesting that a combination of endogenous and exogenous factors is responsible for human carcinogenesis. In recent years, a relatively new field of cancer research has focused on the interaction between genes and enrironment to understand the aetiology of cancer[2]. Primary candidates for gene-environment interaction studies are those which encode enzymes related to the metabolism of established cancer risk factors. It has been known that most carcinogens require metabolic activation in the human body for the carcinogenic effects. There are two major enzyme systems that metabolize potential carcinogens, either synthetic or naturally occurring in the body, which have been classified as phase I and phase II. Generally, phase I enzymes can activate the carcinogen directly and produce more active metabolites. Phase II enzymes can detoxify and process the activated metabolites for final breakdown or excretion. Therefore, the genotypes with high phase I enzyme activity and low phase II enzyme level are considered to pose a high risk to cancer development[3].
Cytochrome P450 (CYP) isoenzymes are one major kind of phase I enzymes and play an important role in the oxidation of chemical compounds, such as polycyclic aromatic hydrocarbons (PAH), often resulting in the formation of highly reactive compounds that are the ultimate carcinogens[4]. Glutathione S-transferases (GSTs) are phase II enzymes and responsible for catalyzing the biotransformation of a variety of electrophiles, and have a central role in the detoxification of activated metabolites of procarcinogens produced by phase I reactions. GSTP1 is the main GST isoform expressed in esophageal mucosa[5,6]. Microsomal epoxide hydrolase (mEH) plays a dual role both in detoxication and activation of procarcinogens because it is not only involved in detoxication reaction but also generates some trans-dihydrodiols that could be metabolized to highly toxic, mutagenic and carcinogenic polycyclic hydrocarbon diol epoxides[7].
Esophageal cancer is one of the most common malignant diseases worldwide with a sharp variation in its geographic distribution[8]. The ratio in incidence between high- and low-risk areas could be as great as 500:1. The high incidence in special areas indicates the importance of environmental factors in esophageal carcinogenesis. However, only a small part of individuals in the high-risk area for esophageal cancer develop into esophageal cancer, although all the residents in that area share very similar environment-related risk factors and lifestyle, suggesting that host susceptibility factors, such as the polymorphisms of phase I and phase II enzymes, may play an important role in increased risk for esophageal cancer. Thus, the present study was undertaken to assess the genetic polymorphisms of CYP1A1, GSTM1, GSTT1, GSTP1 and mEH in esophageal precancerous and cancerous lesions as well as in case control group from the subjects in high-incidence area for esophageal cancer in Henan to correlate these genetic polymorphisms and susceptibility to esophageal cancer.
MATERIALS AND METHODS
Patients and controls
Sixty-two cases of esophageal squamous cell carcinoma (ESCC), including 32 males with a mean age of 55 (55 ± 9.8) and 30 females with a mean age of 60 (66 ± 10.5) were recruited from Yaocun Esophageal Cancer Hospital in Linzhou, who were histopathologically confirmed in 1999. All the cases were from Linzhou district and were interviewed to exclude other simultaneous malignancies. Thirty-eight subjects with matched age and sex frequencies were randomly selected as control group from the same region during the field surveys between 1998 and 1999. Thirty cases of esophageal basal cell hyperplasia (BCH), including 20 males with a mean age of 52 (52 ± 8) and 10 females with a mean age of 54 (54 ± 7) and thirty-two cases of esophageal dysplasia (DYS), including 18 males with a mean age of 54 (54 ± 8) and 14 females with a mean age of 55 (55 ± 7) were also randomly recruited from the same region during the field surveys between 1997 and 1999.
PCR analysis of CYP1A1 gene polymorphism
Genomic DNA was extracted from surgically resected ESCC specimen, BCH and DYS biopsies and buccal smear (for control group). The PCR was performed in a total volume of 25 μL with GeneAmp 9700 (Perkin-Elmer Corp., Norwalk. CT) in this study. The concentration of primers for GSTT1 was 0.3 μM, and others were 1.0 μM. The A to G transition polymorphism in exon 7 of the CYP1A1 gene was analyzed by primers 5’-GAAAGGCTGGGTCCACCCTCT and 5’-CCAGGAAGAAAGACCTCCCAGCGGGCCA. Briefly, 100 ng of the DNA sample was amplified in buffer (10 mM Tris-HCl, 50 mM KCl, 1.5 mM MgCl2 pH8.4) with 0.1 mM of each dNTP (Pharmacia, Piscatoway, NJ) and 1.25 U Taq polymerase (Perkin Elmer Corp., Norwalk. CT). Pre-heated at 80 °C for 3 sec, then initial denaturation was performed at 95 °C for 10 min, followed by 35 cycles of annealing for 1 min at 55 °C, extention for 1 min at 72 °C and denaturation for 1 min at 95 °C, finally extention for 5 min at 72 °C. The PCR products were digested with NcoI (New England Biolabs, Inc., Beverly, MA) at 37 °C overnight, subjected to electrophoresis in an ethidium-bromide-stained 3% agarose gel (Nusieve 3:1; American Bioanalytical, Natick, MA) in TBE buffer (89 mM Tris-HCl, 0.89 mM boric acid and 2 mM EDTA, pH8.0). PCR-RFLP analysis resulted in the following genotype classification: A predominant homozygote (Ile/Ile), a heterozygote (Ile/Val) and a rare homozygote (Val/Val).
For 3'-flank region polymorphism of CYP1A1, PCR was performed using the primers 5'-CAGTCAACAGGTGTAGC and 5'-GAGGCAGGTGGATCACTTGAGCTC. After preheated for 3 sec at 80 °C, initial denaturation was performed at 94 °C for 1 min, followed by 37 thermal cycles consisting of denaturation for 25 sec at 94 °C, annealing for 25 sec at 62 °C, extention for 25 sec at 72 °C and a final extention for 5 min at 72 °C. The PCR products were digested with MspI (New England Biolabs, Inc., Beverly, MA) at 37 °C overnight and subjected to electrophoresis on a 3% agarose gel. The genotypes of CYP1A1 3 were classified as follows: Wild-type, heterozygous variant and homozygous variant.
GSTM1 and GSTT1 genotyping
GSTM1 and GSTT1 genotyping for gene deletion was carried out by a multiplex PCR using primers 5'-GAACTCCCTGAAAAGCTAAAGC and 5'-GTTGGGCTCAAATATACGGTGG for GSTM1, which produced a 219 bp product, primers 5'-TTCCTTACTGGTCCTCACATCTC and 5'-TCACCGGATCATGGCCAGCA for GSTT1, which produced a 480 bp product. At the same time, amplification of the b-globin gene (5'-ACACAACTGTGTTCACTAGC and 5'-CTCAAAGAACCTCTGGGTCC) was used as an internal control and produced a 299 bp product. PCR was performed in a 25 μL mixture consisting of 100 ng sample DNA, 10 mM Tris-HCl, 50 mM KCl, 1.5 mM MgCl2 pH8.4, 0.1 mM of each dNTP and 1.25 U Taq polymerase. After initial denaturation for 3 min at 94 °C, 35 cycles were performed at 94 °C for 1 min (denaturation), at 62 °C for 2 min (annealing) and at 72 °C for 2 min (extention), followed by a final step for 5 min at 72 °C. The amplified products were visualized by electrophoresis in ethidium-bromide-stained 3% agarose gel in TBE buffer. For null deletions of GSTM1 and GSTT1, no amplified product could be observed.
PCR-RFLP analysis of GSTP1 gene polymorphism
The primers 5'-GTATTTTGCCCAAGGTCAAG and 5'-AGCCACCTGAGGGGTAAG were used to amplify exon 5 of GSTP1 gene that includes the BsmAI enzyme recognition site. The same reaction mixture as above was used, after digestion with BsmAI at 55°Covernight, the following genotypes could be shown: Wild type (one restriction site yielded two fragments of 329 bp and 113 bp), variant with 2 restriction sites, heterozygous variant yielded 3 fragments (329, 216, 113 bp), homozygous one yielded 2 fragments (216, 113 bp).
Analysis of mEH gene polymorphism
Primers 5'-GATCGATAAGTTCCGTTTCACC and 5'-ATCCTTAGTCTTGAAGTGAGGAT were used to amplify mEH slow allele (113 code). Primers 5'-ACATCCACTTCATCCACGTT and 5'-ATGCCTCTGAGAAGCCAT were used to amplify mEH fast allele (139 code). After two separate PCR reactions, the varant, correlated with decreased mEH activity (His113) was identified through the presence of EcoR V restriction site, and the allele correlated with increased activity (Arg139) was identified through the presence of RsaI site.
Statistical analysis
The χ2 test was used to examine the differences in genotype distribution between patients and controls. The difference was considered significant in case of a two-tailed P value less than 0.05.
RESULTS
CYP1A1 genetic polymorphism (Table 1)
Table 1.
Distribution of CYP1A1 genetic polymorphism in controls and subjects with cancer and different severity of lesions n(%)
| Control (n = 38) | BCH (n = 30) | DYS (n = 32) | ESCC (n = 62) | |
| CYP1A1 3' | ||||
| Wild type | 12 (32) | 8 (27) | 9 (28) | 33 (53) |
| Heterozygous | 22 (58) | 16 (53) | 17 (53) | 25 (40) |
| Homozygous variant | 4 (10) | 3 (10) | 3 (9) | 4 (6) |
| Heterozygous+Homozygous | 26 (68) | 19 (63) | 20 (62) | 29 (47)a |
| CYP1A1 exon 7 | ||||
| Ile/Ile | 20 (53) | 14 (47) | 16 (50) | 30 (48) |
| Val/Ile | 16 (42) | 14 (47) | 15 (47) | 28 (45) |
| Val/Val | 2 (5) | 2 (7) | 1 (3) | 4 (6) |
| Val/Ile + Val/Val | 18 (47) | 16 (53) | 16 (50) | 32 (52) |
P < 0.05, vs case control group.
DNA samples subjected to PCR and enzymatic digestion with MspI revealed the expected fragment lengths and resulted in three genotypes of CYP1A1 3’ noncoding area (Figure 1). The frequency of combined heterozygous and homozygous variant genotype detected in the groups of control, BCH, DYS and ESCC was 68%, 63%, 62%, 47%, respectively (Table 1), the difference was significant between control group and ESCC group (P < 0.05). However, no significant difference was observed for heterozygous and homozygous variant incidence among the different groups (P > 0.05). CYP1A1 exon 7 polymorphisms in the groups of control, BCH, DYS and ESCC were observed with an incidence of 47%, 53%, 50% and 52%, respectively, but there was no significant difference among these groups (P > 0.05). The corresponding heterozygous and homozygous variant frequency did not show a significant difference among these groups.
Figure 1.
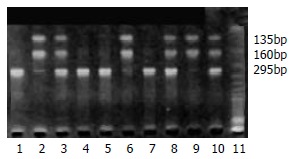
Examples of CYP1A1 3’ polymorphism. The RFLPs of PCR-amplified fragments obtained using MspI and subjected to agarose gel electrophoresis. Wild type without MspI restriction site shows a 295 bp band (lanes 1, 4, 5, 7), variant with MspI restriction site results in two bands of 135 and 160 bp (homozygous variant, lanes 2, 6, 9) or all three bands (heterozygous variant, lanes 3, 8, 10).
GSTs genetic polymorphism (Table 2)
Table 2.
Genotypes of GSTM1, GSTT1 and GSTP1 in controls and subjects with different severity of lesions and cancer n(%)
| Control (n = 38) | BCH (n = 30) | DYS (n = 32) | ESCC (n = 62) | |
| GSTM1 | ||||
| + | 19 (50) | 16 (53) | 18 (56) | 35 (56) |
| - | 19 (50) | 14 (47) | 14 (44) | 27 (44) |
| GSTT1 | ||||
| + | 18 (47) | 17 (57) | 15 (47) | 28 (45) |
| - | 20 (53) | 13 (43) | 17 (53) | 34 (55) |
| GSTP1 | ||||
| Ile/Ile | 24 (63) | 15 (50) | 15 (47) | 29 (47) |
| Val/Ile | 13 (34) | 15 (50) | 16 (50) | 30 (48) |
| Val/Val | 1 (3) | 0 (0) | 1 (3) | 3 (5) |
| Val/Ile + Val/Val | 14 (37) | 15 (50) | 17 (53) | 33 (53) |
Table 2 shows the homozygous deletion of GSTM1 and GSTT1. A similar percentage (around 50%) for GSTM1 and T1 homozygous deletion in the groups of control, BCH, DYS and ESCC was observed. GSTP1 polymorphism incidence in control group (37%) was a little lower than that in other groups (about 50%), but the difference was not significant (P > 0.05). There was also no apparent difference for their corresponding heterozygous and homozygous variant distribution in all groups.
mEH genetic polymorphism (Table 3)
Table 3.
Slow and fast allele polymorphism of mEH in controls and subjects with different severity of lesions and cancer n(%)
| Control (n = 38) | BCH (n = 30) | DYS (n = 32) | ESCC (n = 62) | |
| mEH slow allele | ||||
| Tyr/Tyr | 23 (61) | 14 (47) | 10 (31) | 23 (37) |
| His/Tyr | 10 (26) | 13 (43) | 15 (47) | 22 (35) |
| His/His | 5 (13) | 2 (7) | 7 (22) | 17 (27) |
| His/Tyr+His/His | 15 (39) | 16 (53) | 22 (69)a | 39 (63)a |
| mEH fast allele | ||||
| His/His | 32 (84) | 22 (73) | 23 (72) | 50 (81) |
| Arg/His | 5 (13) | 6 (20) | 7 (22) | 11 (18) |
| Arg/Arg | 1 (3) | 2 (7) | 2 (6) | 1 (2) |
| Arg/His + Arg/Arg | 6 (16) | 8 (27) | 9 (28) | 12 (19) |
P < 0.05, vs case control group.
mEH polymorphism was observed occurring frequently in exons 3 and 4, which resulted in substitution of amino acid histidine to tyrosine at residue 113 (slow allele) and arginine to histidine at residue 139 (fast allele), respectively. The variant of mEH fast allele was observed with a relatively low frequency in all groups. The homozygous and heterozygous variant for mEH was also detected with a low incidence and no significant difference was observed among different groups (Table 3, P > 0.05). However, the different distribution of mEH slow allele variant was observed in the present study. The frequency for mEH slow allele variant (39%) was the lowest in the control group and increased in BCH (53%), DYS (69%) and ECSS (63%), the difference was significant between control group and BCH, or DYS and ESCC (P < 0.05). Figure 2 shows slow allele polymorphism for mEH.
Figure 2.

Example of mEH slow allele polymorphism. The RFLPs of PCR-amplified fragments obtained using EcoR V and subjected to agarose gel electrophoresis. Lane 5 is homozygote, lanes 1, 2, 3, 4, 6, 7 wild type.
Another interesting result was that none of polymorphic variants of all detected genes was found to be associated with ESCC differentiation in this study.
DISCUSSION
In this study, a similar frequency of CYP1A1 exon 7 polymorphism was observed in the groups of control, ESCC and precancerous lesions, but the CYP1A1 3 flank polymorphic variants most frequently occurred in control group, 1.4 times that in ESCC group (68% vs 47%), indicating that CYP1A1 3 variants could be a protective factor for ESCC in this population. Two major relevant genetic polymorphisms have been demonstrated in the CYP1A1 gene: One is a T to C substitution in the 3 flanking region altering protein folding, whereas an Ile to Val substitution may occur in exon 7. Both substitutions were considered to result in the enhancement of enzyme activity[9], but polymorphism in the noncoding region of CYP1A1 was unlikely to have direct functional consequences on CYP1A1 activity[10], even the variant of CYP1A1 exon 7 was not sure to induce an increased enzyme activity[11]. These controversial reports suggest that the effect of CYP1A1 polymorphism on cancer development remains to be characterized.
The second interesting observation in the present study was that a high rate of GSTM1 and GSTT1 null genotype and GSTP1 polymorphic variant occurred not only in the control group but also in ESCC patients and the subjects with different precancerous lesions (BCH and DYS), suggesting that GSTs polymorphism may be responsible for the higher-risk for esophageal cancer in this population. GSTM1 and GSTT1 null genotypes have been reported to enhance the risk of developing gastric, colorectal, and lung cancers[12,13], although other studies did not show such a genetic predisposition[14]. Again, in normal esophageal epithelium, GSTP1 was the mean isoform for GST[5]. An Ile to Val substitution in the GSTP1 gene has been found more often in patients with bladder and testicular cancer[5].
The third interesting observation was that mEH sequence alteration from tyrosine to histidine at residue 113 (slow allele), but not polymorphism at residue 139, was associated with progression of esophageal lesions from normal to BCH to DYS and ESCC, and might contribute to a high susceptibility to esophageal carcinoma. mEH is involved in the metabolism of carcinogens found in cigarette smoke and cooked meat, such as PAH. Expression studies of cDNA in vitro indicated that mEH enzymatic activities were decreased by exon 3 polymorphism and increased by exon 4 polymorphism. Reactive and toxic epoxides are frequently generated during PAH oxidative metabolism. Epoxides can be detoxicated partly by mEH, which catalyzes their hydrolysis, thereby yielding the corresponding trans-dihydrolysis. Although hydrolysis is generally considered to represent a detoxication reaction, some trans-dihydrodiols are substrates for additional metabolic changes to highly toxic polycyclic hydrocarbon diol epoxides[7]. It has been suggested that mEH gene His113 variant allele increases the risk of hepatocarcinoma[15] and lung cancer[16] but decreases the risk of ovarian cancer[17]. Further characterization of mEH polymorphism in a larger scale population will be needed to clarify the significance of mEH in high-risk area for esophageal cancer.
Finally, it is noteworthy that the genetic variants of all detected genes in this study were not associated with ESCC differentiation, suggesting that genetic polymorphism may represent susceptibility to ESCC.
ACKNOWLEDGMENTS
We acknowledge the help given by graduate students in Laboratory for Cancer Research, College of Medicine, Zhengzhou University (Drs. Ya-Nan Jiang, Ran Wang) in preparation of the manuscript.
Footnotes
Supported by National Outstanding Young Scientist Award of China 30025016 (China), State Key Project for Basic Research G1998051206 (China), Foundation of Henan Education Committee 1999125 and the U.S. NIH Grant CA65871
Edited by Zhu L and Wang XL
References
- 1.Doll R, Peto R. The causes of cancer: quantitative estimates of avoidable risks of cancer in the United States today. J Natl Cancer Inst. 1981;66:1191–1308. [PubMed] [Google Scholar]
- 2.Mucci LA, Wedren S, Tamimi RM, Trichopoulos D, Adami HO. The role of gene-environment interaction in the aetiology of human cancer: examples from cancers of the large bowel, lung and breast. J Intern Med. 2001;249:477–493. doi: 10.1046/j.1365-2796.2001.00839.x. [DOI] [PubMed] [Google Scholar]
- 3.Kihara M, Kihara M, Noda K. Risk of smoking for squamous and small cell carcinomas of the lung modulated by combinations of CYP1A1 and GSTM1 gene polymorphisms in a Japanese population. Carcinogenesis. 1995;16:2331–2336. doi: 10.1093/carcin/16.10.2331. [DOI] [PubMed] [Google Scholar]
- 4.Guengerich FP. Roles of cytochrome P-450 enzymes in chemical carcinogenesis and cancer chemotherapy. Cancer Res. 1988;48:2946–2954. [PubMed] [Google Scholar]
- 5.Peters WH, Roelofs HM, Hectors MP, Nagengast FM, Jansen JB. Glutathione and glutathione S-transferases in Barrett's epithelium. Br J Cancer. 1993;67:1413–1417. doi: 10.1038/bjc.1993.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakajima T, Wang RS, Nimura Y, Pin YM, He M, Vainio H, Murayama N, Aoyama T, Iida F. Expression of cytochrome P450s and glutathione S-transferases in human esophagus with squamous-cell carcinomas. Carcinogenesis. 1996;17:1477–1481. doi: 10.1093/carcin/17.7.1477. [DOI] [PubMed] [Google Scholar]
- 7.Sims P, Grover PL, Swaisland A, Pal K, Hewer A. Metabolic activation of benzo(a)pyrene proceeds by a diol-epoxide. Nature. 1974;252:326–328. doi: 10.1038/252326a0. [DOI] [PubMed] [Google Scholar]
- 8.Wang LD, Zhou Q, Feng CW, Liu B, Qi YJ, Zhang YR, Gao SS, Fan ZM, Zhou Y, Yang CS, et al. Intervention and follow-up on human esophageal precancerous lesions in Henan, northern China, a high-incidence area for esophageal cancer. Gan To Kagaku Ryoho. 2002;29 Suppl 1:159–172. [PubMed] [Google Scholar]
- 9.Landi MT, Bertazzi PA, Shields PG, Clark G, Lucier GW, Garte SJ, Cosma G, Caporaso NE. Association between CYP1A1 genotype, mRNA expression and enzymatic activity in humans. Pharmacogenetics. 1994;4:242–246. doi: 10.1097/00008571-199410000-00002. [DOI] [PubMed] [Google Scholar]
- 10.Bailey LR, Roodi N, Verrier CS, Yee CJ, Dupont WD, Parl FF. Breast cancer and CYPIA1, GSTM1, and GSTT1 polymorphisms: evidence of a lack of association in Caucasians and African Americans. Cancer Res. 1998;58:65–70. [PubMed] [Google Scholar]
- 11.Zhang ZY, Fasco MJ, Huang L, Guengerich FP, Kaminsky LS. Characterization of purified human recombinant cytochrome P4501A1-Ile462 and -Val462: assessment of a role for the rare allele in carcinogenesis. Cancer Res. 1996;56:3926–3933. [PubMed] [Google Scholar]
- 12.Chenevix-Trench G, Young J, Coggan M, Board P. Glutathione S-transferase M1 and T1 polymorphisms: susceptibility to colon cancer and age of onset. Carcinogenesis. 1995;16:1655–1657. doi: 10.1093/carcin/16.7.1655. [DOI] [PubMed] [Google Scholar]
- 13.Deakin M, Elder J, Hendrickse C, Peckham D, Baldwin D, Pantin C, Wild N, Leopard P, Bell DA, Jones P, et al. Glutathione S-transferase GSTT1 genotypes and susceptibility to cancer: studies of interactions with GSTM1 in lung, oral, gastric and colorectal cancers. Carcinogenesis. 1996;17:881–884. doi: 10.1093/carcin/17.4.881. [DOI] [PubMed] [Google Scholar]
- 14.Katoh T, Nagata N, Kuroda Y, Itoh H, Kawahara A, Kuroki N, Ookuma R, Bell DA. Glutathione S-transferase M1 (GSTM1) and T1 (GSTT1) genetic polymorphism and susceptibility to gastric and colorectal adenocarcinoma. Carcinogenesis. 1996;17:1855–1859. doi: 10.1093/carcin/17.9.1855. [DOI] [PubMed] [Google Scholar]
- 15.McGlynn KA, Rosvold EA, Lustbader ED, Hu Y, Clapper ML, Zhou T, Wild CP, Xia XL, Baffoe-Bonnie A, Ofori-Adjei D. Susceptibility to hepatocellular carcinoma is associated with genetic variation in the enzymatic detoxification of aflatoxin B1. Proc Natl Acad Sci U S A. 1995;92:2384–2387. doi: 10.1073/pnas.92.6.2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benhamou S, Reinikainen M, Bouchardy C, Dayer P, Hirvonen A. Association between lung cancer and microsomal epoxide hydrolase genotypes. Cancer Res. 1998;58:5291–5293. [PubMed] [Google Scholar]
- 17.Lancaster JM, Brownlee HA, Bell DA, Futreal PA, Marks JR, Berchuck A, Wiseman RW, Taylor JA. Microsomal epoxide hydrolase polymorphism as a risk factor for ovarian cancer. Mol Carcinog. 1996;17:160–162. doi: 10.1002/(SICI)1098-2744(199611)17:3<160::AID-MC8>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]