

Summary
Rationale
Previous investigations of cystic fibrosis (CF) incidence in Massachusetts, Colorado, and Minnesota (USA) yielded contradictory results, particularly regarding allele p.Phe508del; the racial compositions of the cohorts were not reported.
Objectives
To clarify discrepancies in reported incidence with the ultimate goal of improving screening and quality of care, we assessed CF incidence, stratified by race and mutations in cystic fibrosis transmembrane conductance regulator (CFTR), in Wisconsin (USA) from 1994 to 2011.
Methods
Data on patients diagnosed with CF (N=283), CFTR genotypes, CF carriers, and birth rate were collected. All data were categorized by racial background of the birth mother and the incidence of CF births was accordingly adjusted. Spearman’s nonparametric rank correlation and Fisher’s exact test were performed for continuous and categorical variables, respectively. Trends over time were fitted with a cubic spline.
Results
We detected a trending increase in CF cases (range within all data 1.67–2.98 per 10,000 births per year), homozygous p.Phe508del cases (0.57–1.79 per 10,000), heterozygous p.Phe508del cases (0.29–1.55 per 10,000), and cases lacking p. Phe508del (0–0.45 per 10,000). Both the number of cases lacking the p.Phe508del mutation per year and the number of cases lacking p.Phe508del per 10,000 births significantly increased (P=0.05) from 1994 to 2011; the increase in overall incidence was not significant. The number of carriers identified through newborn screening significantly increased within the non-Hispanic Black (P=0.0.021) and Hispanic (P=0.003) populations.
Conclusion
The racial composition of the CF cohort is changing in Wisconsin, possibly influencing disease detection, care, and outcome.
Keywords: race, epidemiology, incidence of CFTR, CFTR mutation
INTRODUCTION
Although cystic fibrosis (CF), caused by mutations in the gene encoding cystic fibrosis transmembrane conductance regulator (CFTR), is the most common life-limiting autosomal recessive disorder in the non-Hispanic White (NHW) population (1:3,500), it also seems to be prevalent in other racial groups, including non-Hispanic Blacks (NHBs) (1:10-15,000) and Hispanics (1:9,200–13,500).1–3 The Cystic Fibrosis Foundation estimates that there are 30,000 affected individuals in the USA, and that one in 29 people are carriers.4 Early diagnosis, enabled by newborn screening (NBS), and advances in medical care over the past 3 decades have helped improve both the quality and duration of life for CF patients, increasing the median life expectancy for the total CF population in the USA (41.1 years in 2012).5–9 However, this measure does not consider race.
Improvements in life expectancy for NHWs with CF have not necessarily benefited Hispanic or NHB patients.3,9 Hispanic populations do worse clinically with regard to forced expiratory volume levels and early acquisition of Pseudomonas aeruginosa infection compared to non-Hispanic populations.9 NHB patients display worse nutritional status and pulmonary function (forced expiratory volume and forced vital capacity) at diagnosis and later in life compared to NHWs.3 In addition to clinical heterogeneity, mutation frequencies vary considerably among racial groups; much remains to be learned about CFTR allele frequencies within minority populations.
To clarify discrepancies in reported incidence with the ultimate goal of improving screening and quality of care, here we describe CF incidence stratified by CFTR mutation status and race in Wisconsin over the last 17 years (1994–2011).10–12 NBS for CF using a combination of immunoreactive trypsinogen (IRT; sweat testing is performed if elevated IRT levels render the test necessary5) and DNA testing has been performed in Wisconsin since 1994, as recommended by the American College of Medical Genetics and the American College of Obstetricians and Gynecologists.13 Of the ~1,900 CFTR mutations identified to date, p.PheF508del is the most common, with an overall frequency of 75–87% and frequencies of 72% and 31–44% in the NHW and NHB populations in the USA, respectively.2,6,14–17 Detection rates through NBS differ between minorities and NHWs18,19; a previous retrospective study of ~3 million CF tests over an 80-year period reported a 77% detection rate in the pan-ethnic population of the USA, 90% among NHWs, 69% among Hispanics, and 63% among NHBs.20 In addition, the detection rate among Hispanic CFTR mutation carriers is 57% versus 80–97% in NHWs, whereas the Hispanic carrier frequency of 1:46 is only modestly lower than that in NHWs (1:29).21,22
Several studies have evaluated the impact of NBS on CF, leading to a general consensus that NBS has led to improved survival, better nutritional and thus cognitive outcomes, and has the potential to improve pulmonary outcomes if early common infections are prevented.23–26 The 27-year Wisconsin Randomized Control Trial of the CF Neonatal Screening Project determined that the introduction of NBS for CF had minimal risks as well as potential benefits on nutritional status and progression of lung disease.27,28 However, the heterogeneity in mutations among racial populations has presented challenges in optimizing sensitivity while encompassing minority populations in the NBS screening panels.
Longitudinal data from Massachusetts,10 Colorado,11 and Minnesota12 (USA) show conflicting trends in their states’ incidence of CF and p.Phe508del identified through their respective NBS programs. The number of NBS-identified cases of CF in Massachusetts decreased between 2003 and 2006, as did the number of infants homozygous for p.PheF508del.10 In contrast, there was no change in the number of infants diagnosed with CF from 1983 to 2006 in Colorado, and no trend in the frequency of p.PheF508del over time.11 In Minnesota, while the overall incidence of CF did not increase, the incidence of homozygous p.PheF508del increased from 2006–2009.12 None of these studies reported the racial composition within their cohort, which may underlie the disparity in their observations. These conflicting data raise many questions regarding the epidemiology of CF, specifically in terms of the effect of birth rates and racial diversity on CF incidence.
In order to address these questions, we evaluated the incidence of CF in Wisconsin from 1994 through 2011, as reported by the state’s NBS program, and compared these race-related data to previously reported incidence studies. We determined the number of infants carrying the homozygous p.PheF508del genotype and other CFTR mutations and investigated whether this incidence has changed over time, which could impact diagnosis and prognosis in minority populations.
METHODS
This retrospective study encompassed the start of Wisconsin’s CF NBS program (July 1994) through December 2011 (Fig. 1). This study was reviewed and granted exempt status by the Children’s Hospital of Wisconsin Institutional Review Board.
Fig. 1.
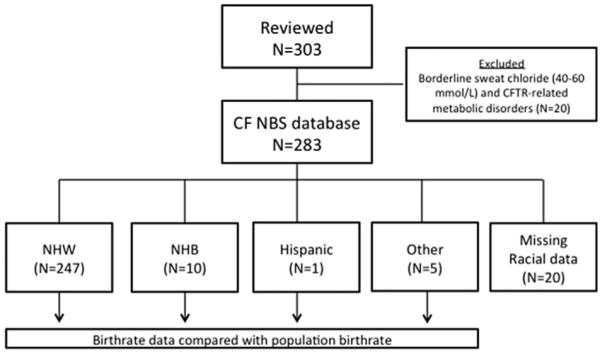
Description of cohort included in analyses. Data acquisition cutoff points were set as July 1994 and December 2011. Subjects with missing or incomplete data (N=20) were excluded from analysis.
Population Data
Annual Wisconsin birth, birth rate, and racial data (E-Table 1) were obtained from the Wisconsin Department of Health Services data query system.29 In 1994, there were 68,267 births, but this number was divided in half to calculate CF incidence for the year (NBS was started in July of that year). This calculation assumed that racial composition remained the same throughout the year.
CF Data
Data for this study were obtained through the NBS laboratory at the Wisconsin State Laboratory of Hygiene, the Wisconsin Department of Health Services, and the Milwaukee Cystic Fibrosis Center at the Children’s Hospital of Wisconsin in Milwaukee, Wisconsin, USA. Our current retrospective cohort study reviewed population-level CF NBS data, individual CF mutations, and state birth rates. The number of patients diagnosed with CF, their genotypes, and the number of CF carriers were collected by the Wisconsin NBS Program from 1994 onward. Individual demographics, including year of birth, sex, self-identified race, genotype, and IRT/sweat chloride levels, were available from the NBS Laboratory at the Wisconsin State Laboratory of Hygiene for 1994 through 2011.
CFTR Genotyping
CFTR genotyping was performed at the time of diagnosis or when available if the subject was diagnosed prior to the availability of genetic testing. Patients identified via NBS were evaluated by the Wisconsin NBS Laboratory for the recommended American College of Medical Genetics panel of 23 CFTR mutations.21 Additional genetic testing was carried out for patients with zero or one identified mutations, including expanded mutation panel testing (Genzyme Genetics, Cambridge, MA), modified temporal temperature gradient electrophoresis of CFTR (Ambry Genetics, Aliso Viejo, CA), and multiplex ligation-dependent probe amplification for deletions and duplications (Ambry Genetics).
NBS in Wisconsin
NBS for CF has been performed as a routine clinical service in Wisconsin since 1994, with DNA testing carried out for infants in the top 4% of daily IRT results.5 The DNA mutation panel consisted of p.Phe508del from 1994–2002; since 2002, Wisconsin has used the recommended American College of Medical Genetics panel of 23 CFTR mutations to detect the most common mutations associated with CF in the NHW population.30 A longitudinal analysis of the sensitivity of the IRT/DNA algorithm found that it continues to be 97% sensitive.30 Individuals with one or two CFTR mutations then underwent sweat chloride testing, with sweat chloride levels ≥60 mmol/L considered diagnostic of CF. Subjects with intermediate sweat chloride levels (30–60 mmol/L) had their sweat tests repeated. From 1994 through 2011, all infants were screened for CF, with 283 infants meeting diagnostic criteria (Fig. 1). DNA analysis of CFTR mutations was carried out on samples with elevated IRT values from these individuals. When two sweat chloride sample values were reported, the larger value was used in the current analysis. Individual data points were excluded when incomplete or missing. Individuals diagnosed with CF but with only one identified CFTR mutation were included in the analysis only when the Wisconsin NBS Laboratory verified the data and that diagnostic criteria had been met through elevated sweat chloride levels.20
Statistical Analyses
All data were categorized according to the self-reported racial background of the birth mother: NHW, NHB, Hispanic, or Other. The “Other” category captured participants who did not identify as NHW, NHB, or Hispanic. The incidence of CF births was then adjusted for racial background. Estimates of period prevalence were calculated as the number of events for a racial group divided by the size of the Wisconsin population self-reporting as that race or by the number of total births in Wisconsin (E-Table 1), then multiplied by 10,000. In general, missing or incomplete data points were excluded from analysis. However, we did examine prevalence trends for cases lacking self-reported race data.
Spearman’s nonparametric rank correlation coefficient (ρ) was calculated for correlations of continuous variables and Fisher’s exact test was performed to test categorical variables. Trends over time were fitted with a cubic spline. Statistical analyses were carried out with SPlus version 8.2 (TIBCO Software, Palo Alto, CA) and SPSS version 21 (IBM Software, Chicago, IL). Unadjusted P-values <0.05 were reported as significant; actual values are also reported.
RESULTS
As depicted in Figure 2a and Table 1, Wisconsin’s birthrate linearly and significantly declined from 1994 to 2011 (13.4–12.0 per 1,000 births per year; P=0.001), yet the state’s overall population continued to increase during this interval (P ≤ 0.001). Children born to NHW mothers in Wisconsin were associated with a trending decrease from 82.7% of total births in 1994 to 74.4% in 2010 (P ≤ 0.001). The percentage of children born to NHB mothers did not significantly change (9.9% of total births in 1994 and 10.0% in 2010; P=0.29). In contrast, children born to Hispanic mothers increased from 3.5% of total births in 1994 to 9.6% of total births in 2010 (P ≤ 0.001).
Fig. 2.
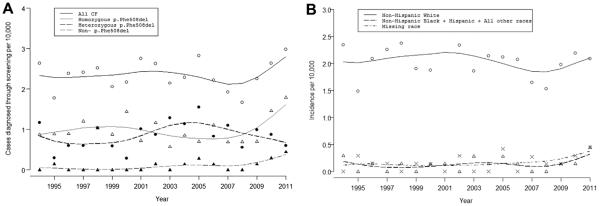
(A) Trends in Wisconsin CF incidence. Breakdown of incidence of CF cases in Wisconsin. Trend lines (black for all CF, gray for homozygous p.Phe508del, dashed black for heterozygous p. Phe508del, and gray dashed for mutations that did not include p.Phe508del) were fit with a cubic spline. (B) Incidence of CF cases stratified by self-reported race. Trend lines were fit with a cubic spline. Note that the incidence rates for cases lacking self-reported race data “missing data” (dash-dot line; Methods) did not substantially differ from the rate for NHB, Hispanic, and Other (dashed line). NHW, solid line.
TABLE 1.
CF Infants Born From 1994 to 2011 According to Self-Reported Maternal Race
| Year | Number of births |
Total CF N |
NHW N (%) |
NHB N (%) |
Hispanic N (%) |
Other N (%) |
Missing race N (%) |
Homozygous p.PheF508del N (%) |
Heterozygous p.PheF508del N (%) |
Other mutation N (%) |
Only one mutation known N (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1994 | 34,134 | 9 | 8 (89) | 1 (11) | – | – | – | 3 (33) | 4 (45) | – | 2 (22) |
| 1995 | 67,493 | 12 | 10 (83) | – | – | – | 2 (17) | 6 (50) | 2 (17) | 1 (8) | 3 (25) |
| 1996 | 67,150 | 16 | 14 (88) | – | – | 1 (6) | 1 (6) | 6 (38) | 4 (25) | – | 6 (38) |
| 1997 | 66,490 | 16 | 15 (94) | – | – | – | 1 (6) | 8 (50) | 4 (25) | – | 4 (25) |
| 1998 | 67,379 | 17 | 16 (94) | – | – | 1 (6) | – | 7 (41) | 7 (41) | – | 3 (18) |
| 1999 | 68,181 | 14 | 13 (93) | – | – | – | 1 (7) | 6 (42) | 4 (29) | – | 4 (29) |
| 2000 | 69,289 | 15 | 13 (86) | – | 1 (7) | – | 1 (7) | 10 (67) | 2 (13) | – | 3 (20) |
| 2001 | 69,012 | 19 | 18 (95) | 1 (5) | – | – | – | 5 (26) | 7 (37) | – | 7 (37) |
| 2002 | 68,510 | 18 | 16 (89) | – | – | – | 2 (11) | 8 (44) | 6 (33) | 1 (6) | 3 (17) |
| 2003 | 69,999 | 15 | 13 (86) | 1 (7) | – | 1 (7) | – | 4 (27) | 9 (60) | – | 2 (13) |
| 2004 | 70,130 | 16 | 15 (94) | 1 (6) | – | – | – | 6 (38) | 8 (50) | 1 (6) | 1 (6) |
| 2005 | 70,934 | 20 | 15 (75) | 1 (5) | – | 1 (5) | 3 (15) | 5 (25) | 11 (55) | 2 (10) | 2 (10) |
| 2006 | 72,302 | 16 | 15 (94) | – | – | 1 (6) | – | 8 (50) | 6 (38) | 1 (6) | 1 (6) |
| 2007 | 72,757 | 14 | 12 (86) | – | – | – | 2 (14) | 4 (29) | 8 (57) | – | 2 (14) |
| 2008 | 72,002 | 12 | 11 (92) | – | – | – | 1 (8) | 5 (42) | 4 (33) | – | 3 (25) |
| 2009 | 70,824 | 16 | 14 (88) | 1 (6) | – | – | 1 (6) | 5 (31) | 7 (44) | 1 (6) | 3 (19) |
| 2010 | 68,367 | 18 | 15 (83) | 1 (6) | – | – | 2 (11) | 10 (56) | 6 (33) | 2 (11) | – |
| 2011 | 67,057 | 20 | 14 (70) | 3 (15) | – | – | 3 (15) | 12 (60) | 4 (20) | 3 (15) | 1 (5) |
| Total | 1,212,010 | 283 | 247 (87) | 10 (4) | 1 (<1) | 5 (2) | 20 (7) | 118 (42) | 103 (36) | 12 (4) | 50 (18) |
A total of 1,212,010 newborns were screened for CF through Wisconsin NBS from July 1994 to December 2011 (E-Table 1), resulting in 283 meeting the diagnostic criteria for CF (Table 1). The annual Wisconsin CF incidence for all CF cases ranged from 1.67 to 2.98 cases per 10,000 births per year, with a median of 2.23 cases per 10,000 births per year. The incidence of total CF cases was relatively stable until 2005, dipped until 2008, then increased (Fig. 2a).
The incidence of homozygous p.Phe508del, heterozygous p.Phe508del, and cases whose mutations did not include p.Phe508del (“non-p.Phe508del”) followed a similar pattern to the overall cases, with an upswing in the last 4–5 years (Fig. 2a, Table 1). Homozygous p.Phe508del incidence ranged from 0.57 to 1.79 cases per 10,000 births per year, with a median of 0.88. Heterozygous p.Phe508del incidence ranged from 0.29 to 1.55 cases per 10,000 births per year, with a median of 0.88. The incidence of mutations not including p.Phe508del ranged from 0 to 0.45 cases per 10,000 births per year with a median of 0. p.Phe508del was the most common mutation (78%) within this dataset, with 42% of all characterized CFTR mutations being homozygous p.Phe508del, a frequency that varied by year (from 25% to 67%).
Eighty-seven percent of the identified CF patients were self-reported NHW, 4% NHB, <1% Hispanic, and 2% Other (Fig. 2b, Table 2). NHWs made up the highest percentage of affected individuals each year, with incidences of 1.48–2.61 cases per 10,000 births per year. The incidence of CF in Wisconsin’s NHW and NHB populations trended upward from 1994–2011; there were too few Other (N=5) and Hispanic (N=1) CF cases to establish trends for these racial groups. However, the incidence trend for cases lacking self-reported race data did not substantially differ from the trend for NHB, Hispanic, and Other (Fig. 2b).
TABLE 2.
Demographics of Wisconsin CF Patients (N = 283) Stratified by Self-Reported Race
| Mutation |
Gender |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Race | Total N | Total p.Phe508del |
Homozygous p.Phe508del |
Heterozygous p.Phe508del |
Other/other | Only one mutation | Male | Female | Missing |
| NHW, N (%) | 247 (87) | 313 (64) | 106 (43) | 89 (36) | 8 (3) | 44 (18) | 118 (48) | 125 (51) | 4 (1) |
| NHB, N (%) | 10 (4) | 12 (63) | 3 (30) | 5 (50) | 1 (10) | 1 (10) | 3 (30) | 7 (70) | – |
| Hispanic, N (%) | 1 (<1) | 2 (100) | 1 (100) | – | – | – | 1 (100) | – | – |
| Other, N (%) | 5 (2) | 5 (56) | 1 (20) | 2 (40) | 1 (20) | 1 (20) | 2 (40) | 3 (60) | – |
Mutations differed among racial groups in our cohort (Table 3; mutations with no protein name available are instead listed by their variant legacy name31). The most common identified mutations were p.Phe508del (51%), p.Gly542X (5%), 3849+10KbC>T (4%), p.Arg117His (3%), 2789+5G>A (2%), 3120+1G>A (2%), and p.Arg553X (2%). From 1994 to 2011, p.Phe508del was the most common mutation overall and within each race, accounting for 62.7% of alleles. Forty-two percent of the CF patients were homozygous p.Phe508del; three patients (3%) were NHB, one Hispanic (<1%), one other (<1%), and the remaining NHW (96%). Seven patients were homozygous p.Phe508del. three mutations (1679+1634A>G, p.Ala559Thr, and p.Ser466X) were unique to the NHB CF population and three mutations to the Other population (p.Glu831X, 1812-1G>A, and c.4259_4263delTGGAT) (Table 3). Fifty patients had only one CFTR mutation detected (Table 1).
TABLE 3.
Mutations by Year for Minority Patients
| Race | Year | Mutation 1 | Mutation 2 |
|---|---|---|---|
| NHB | 1994 | p.Phe508del | p.Tyr1092X |
| 2001 | p.Phe508del | – | |
| 2003 | p.Phe508del | p.Phe508del | |
| 2004 | p.Phe508del | 3849+10KbC>T | |
| 2005 | p.Phe508del | p.Phe508del | |
| 2009 | p.Phe508del | p.Ser466X | |
| 2010 | p.Phe508del | p.Ala559Thr | |
| 2011 | p.Phe508del | 3120+1G>A | |
| 2011 | p.Phe508del | p.Phe508del | |
| 2011 | p.Gly551Asp | 1679+1634 A>G | |
| Hispanic | 2000 | p.Phe508del | p.Phe508del |
| Other | 1996 | p.Phe508del | – |
| 1998 | p.Phe508del | p.Phe508del | |
| 2003 | p.Phe508del | p.Glu831X | |
| 2005 | p.Gly542X | 1812-1G>A | |
| 2006 | p.Phe508del | c.4259_4263delTGGAT |
The number of infant carriers identified by NBS decreased from 2003 to 2010 among Wisconsin’s total population, although this decrease was not significant (data not shown). From 1999 to 2011, the incidence of CF cases increased, as did the incidences within all mutation subsets, in Wisconsin. In the last 4 years (2007–2011) there was a marked increase in all CF cases and mutation subsets, except heterozygous p.Phe508del (Fig. 2a). The number of CF carriers identified through NBS declined significantly within the NHB (P=0.021) and Hispanic (P=0.003) populations.
DISCUSSION
This study provides the first detailed incidence data for CF in Wisconsin for 17 years; this large sample enabled stratification by race and CFTR mutation status. Three recent estimates of longitudinal incidence were reported for Massachusetts,10 Colorado,11 and Minnesota12 (USA), with non-congruent results. None of these studies reported the racial composition within their cohort, which may underlie the disparity in their observations.
The population-level data analyzed in the current investigation suggest that the CF incidence increased in Wisconsin in 1994–2011 (Fig. 2a). Our findings agree with those of the Minnesota12 NBS study and conflict with the reports from Massachusetts10 and Colorado.11 Minnesota only reported an increase in homozygous p.Phe508del,12 while we found an increasing trend in all CF cases and within each mutation subset from 1994–2011 in Wisconsin, with a significant increase in the non-p.Phe508del cases (Table 1). Our results oppose both the absence of significant longitudinal changes in CF incidence in Colorado11 and the decrease in CF incidence in Massachusetts.10 These trends highlight the need to not only better understand CF within minority populations, but also to improve the identification and care of CF patients within the growing minority populations.32 A study from California, which is home to a large Hispanic population, predicted that 33% of all new CF cases will be Hispanic and 2% will be NHB.33 Further investigation into racial and mutation profiles on a state or regional basis would provide insight into such trends.
The causative factors underlying the observed change in CF incidence in Wisconsin are not obvious. By controlling for changes in birth rate and the racial makeup of Wisconsin’s population, we determined that these factors did not significantly affect CF incidence (Table 1). Likewise, net migration in Wisconsin is not likely to have a large impact on these trends, as the 2005–2010 mover rate was the lowest in United States Census history.34
Prenatal screening and subsequent education may influence the parental mindset regarding having a child with CF, leading to pregnancy termination or choosing to have no further children after the birth of the initial CF child. A longitudinal study assessing NBS in Italy reported a significant correlation in the number of screened carriers and the observed decrease in CF birth incidence.35 This significant decrease was observed only when CF carrier testing was offered to the general public versus a subset of the population, such as relatives of patients using in vitro fertilization.35 In addition, increasing awareness of CF in the general public coupled with increased prenatal counseling may influence CF incidence.
We suspect that factors such as access to prenatal screening and reproductive technologies, which varies among socio–economic classes and geographic regions, impact CF incidence. Both variation in CF incidence by geographical region and use of prenatal screening have been noted previously. The Wisconsin Cystic Fibrosis Neonatal Screening Project found that prenatal diagnosis was employed by 26% of families with a CF child for subsequent pregnancies.36 In contrast, a prenatal study from New England reported that 77% of families at risk for having another CF child had had or were considering prenatal counseling.37 In keeping with recommendations from the American College of Obstetricians and Gynecologists, Wisconsin obstetricians have routinely offered prenatal CFTR screening to pregnant women since 2005. In a national survey of practice patterns among obstetrician-gynecologists regarding prenatal screening for CF, 13% offered screening to all prenatal patients and 68% offered screening only if the patient requested it, if a positive family history for CF was present, or if a patient/significant other was in a high-risk racial group.38
While there is an increasing awareness that disease varies between racial groups, not only in its genetic bases but also in clinical course, prognosis, and outcomes, access to care, language barriers, delayed diagnosis, inconsistent follow-up, historical provider mindset of CF as a “Caucasian disease,” and other unknown factors also impact disease in a race-dependent fashion. Our findings suggest that the racial composition of the CF cohort is changing in Wisconsin, possibly influencing CF detection, care, and outcome.
In summary, despite a decrease in the NHW birthrate, we detected an increase in NHW CF incidence in Wisconsin from 1994 to 2011. Although there was no change in NHB birthrates, we detected an increase in NHB CF incidence. In contrast, there was an increasing trend in Hispanic birthrates and only one Hispanic CF patient. These findings suggest that Wisconsin population birth rates are not causative factors in the increase in overall CF incidence. Understanding CF complexity by state is complicated by the changes in race demographics over time in each state and in the USA as a whole; additionally, the number of CF cases identified in minority populations via NBS varies according to each state’s screening protocols, IRT methodology, and cut-off values. These trends highlight the need to not only better understand CF within minority populations nationally and worldwide, but also to improve the identification and clinical care of CF patients within growing minority populations.32
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the assistance of the Cystic Fibrosis Center staff at Children’s Hospital of Wisconsin in Milwaukee and American Family Children’s Hospital in Madison, Wisconsin, USA. We thank all of our CF patients and their families; we appreciate their involvement and support. We also appreciate Gregory Kopish and Karen Kennedy-Parker in the Wisconsin State Laboratory of Hygiene dedication to CF NBS. We are grateful to Anita Laxova for assisting with coordination and patient data management. We also thank Danuta Stachiw-Hietpas for her assistance with data collection. H.L. is supported by NIH/NHLBI grant DP2OD007031, the Children’s Hospital of Wisconsin Research Institute and the Medical College of Wisconsin Scholarship to K. Parker-McGill. Grants to M.R. and P.M.F. that supported the development and launching of the Wisconsin IRT/DNA method as well as the establishment of the surveillance method for C.F. in Wisconsin include Cystic Fibrosis Foundation grant A001-5-01 and National Institutes of Health grant D.K. 34108.
Footnotes
Conflict of interest: None.
AUTHOR CONTRIBUTIONS
K.P.M. assisted with data collection, analysis, and assisted in the writing of the manuscript. M.N. analyzed the data, and K.P.M., M.B., D.H., and R.B. assisted with data collection. R.B. helped collect samples and assisted with data collection. K.P.M., M.N., P.S., and P.M.F. assisted with data analysis and edited the manuscript. H.L. delineated the hypothesis, conceived and designed the study, performed and oversaw the data analyses, and assisted in the writing of the manuscript.
SUPPORTING INFORMATION
Additional supporting information may be found in the online version of this article at the publisher’s web-site.
REFERENCES
- 1.Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005;352:1992–2001. doi: 10.1056/NEJMra043184. [DOI] [PubMed] [Google Scholar]
- 2.Palomaki GE, FitzSimmons SC, Haddow JE. Clinical sensitivity of prenatal screening for cystic fibrosis via CFTR carrier testing in a United States panethnic population. Genet Med. 2004;6:405–414. doi: 10.1097/01.gim.0000139505.06194.39. [DOI] [PubMed] [Google Scholar]
- 3.Hamosh A, FitzSimmons SC, Macek M, Jr, Knowles MR, Rosenstein BJ, Cutting GR. Comparison of the clinical manifestations of cystic fibrosis in black and white patients. J Pediatr. 1998;132:255–259. doi: 10.1016/s0022-3476(98)70441-x. [DOI] [PubMed] [Google Scholar]
- 4.Cystic Fibrosis Foundation [Internet] [accessed 2013 June 17]. Available from: http://www.cff.org/AboutCF/
- 5.Cystic Fibrosis Foundation Newborn Screening [Internet] [accessed 2013 June 17]. Available from: http://www.cff.org/AboutCF/Testing/NewbornScreening/ScreeningforCF/
- 6.Cystic Fibrosis Foundation Patient Registry Report [Internet] [accessed 2013 June 19]. Available from: http://www.cff.org/livingwithcf/qualityimprovement/patientregistryreport/
- 7.Strausbaugh SD, Davis PB. Cystic fibrosis: a review of epidemiology and pathobiology. Clin Chest Med. 2007;28:279–288. doi: 10.1016/j.ccm.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 8.Watts KD, Layne B, Harris A, McColley SA. Hispanic infants with cystic fibrosis show low CFTR mutation detection rates in the Illinois newborn screening program. J Genet Couns. 2012;21:671–675. doi: 10.1007/s10897-012-9481-2. [DOI] [PubMed] [Google Scholar]
- 9.Watts KD, Seshadri R, Sullivan C, McColley SA. Increased prevalence of risk factors for morbidity and mortality in the US Hispanic CF population. Pediatr Pulmonol. 2009;44:594–601. doi: 10.1002/ppul.21037. [DOI] [PubMed] [Google Scholar]
- 10.Hale JE, Parad RB, Comeau AM. Newborn screening showing decreasing incidence of cystic fibrosis. N Engl J Med. 2008;358:973–974. doi: 10.1056/NEJMc0707530. [DOI] [PubMed] [Google Scholar]
- 11.Sontag ML, Wagener JS, Accurso F, Sagel SD. Consistent incidence of cystic fibrosis in a long-term newborn screen population. North American Cystic Fibrosis Conference Meeting Abstract.2008. [Google Scholar]
- 12.Temme R, McNamara J, Johnson M, Read L, Lui M. Incidence of cystic fibrosis and CFTR mutation distribution in Minnesota newborns. 2008 Meeting Abstract. [Google Scholar]
- 13.American College of Obstetricians and Gynecologists Committee on Genetics ACOG committee opinion no. 486: update on carrier screening for cystic fibrosis. Obstet Gynecol. 2011;117:1028–1031. doi: 10.1097/AOG.0b013e31821922c2. [DOI] [PubMed] [Google Scholar]
- 14.Palomaki GE, Haddow JE, Bradley LA, FitzSimmons SC. Updated assessment of cystic fibrosis mutation frequencies in non-Hispanic Caucasians. Genet Med. 2002;4:90–94. doi: 10.1097/00125817-200203000-00007. [DOI] [PubMed] [Google Scholar]
- 15.Sugarman EA, Rohlfs EM, Silverman LM, Allitto BA. CFTR mutation distribution among U.S. Hispanic and African American individuals: evaluation in cystic fibrosis patient and carrier screening populations. Genet Med. 2004;6:392–399. doi: 10.1097/01.gim.0000139503.22088.66. [DOI] [PubMed] [Google Scholar]
- 16.Heim RA, Sugarman EA, Allitto BA. Improved detection of cystic fibrosis mutations in the heterogeneous U.S. population using an expanded, pan-ethnic mutation panel. Genet Med. 2001;3:168–176. doi: 10.1097/00125817-200105000-00004. [DOI] [PubMed] [Google Scholar]
- 17.Alibakhshi R, Kianishirazi R, Cassiman JJ, Zamani M, Cuppens H. Analysis of the CFTR gene in Iranian cystic fibrosis patients: identification of eight novel mutations. J Cyst Fibros. 2008;7:102–109. doi: 10.1016/j.jcf.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 18.Ross LF. Newborn screening for cystic fibrosis: a lesson in public health disparities. J Pediatr. 2008;153:308–313. doi: 10.1016/j.jpeds.2008.04.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carles S, Desgeorges M, Goldman A, Thiart R, Guittard C, Kitazos CA, de Ravel TJ, Westwood AT, Claustres M, Ramsay M. First report of CFTR mutations in black cystic fibrosis patients of southern African origin. J Med Genet. 1996;33:802–804. doi: 10.1136/jmg.33.9.802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Strom CM, Crossley B, Buller-Buerkle A, Jarvis M, Quan F, Peng M, Muralidharan K, Pratt V, Redman JB, Sun W. Cystic fibrosis testing 8 years on: lessons learned from carrier screening and sequencing analysis. Genet Med. 2011;13:166–172. doi: 10.1097/GIM.0b013e3181fa24c4. [DOI] [PubMed] [Google Scholar]
- 21.Watson MS, Cutting GR, Desnick RJ, Driscoll DA, Klinger K, Mennuti M, Palomaki GE, Popovich BW, Pratt VM, Rohlfs EM, Strom CM, Richards CS, Witt DR, Grody WW. Cystic fibrosis population carrier screening: 2004 revision of American College of Medical Genetics mutation panel. Genet Med. 2004;6:387–391. doi: 10.1097/01.GIM.0000139506.11694.7C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schrijver I. Mutation distribution in expanded screening for cystic fibrosis: making up the balance in a context of ethnic diversity. Clin Chem. 2011;57:799–801. doi: 10.1373/clinchem.2011.164673. [DOI] [PubMed] [Google Scholar]
- 23.Drumm ML, Konstan MW, Schluchter MD, Handler A, Pace R, Zou F, Zariwala M, Fargo D, Xu A, Dunn JM, Darrah RJ, Dorfman R, Sandford AJ, Corey M, Zielenski J, Durie P, Goddard K, Yankaskas JR, Wright FA, Knowles MR, Gene Modifier Study Group Genetic modifiers of lung disease in cystic fibrosis. N Engl J Med. 2005;353:1443–1453. doi: 10.1056/NEJMoa051469. [DOI] [PubMed] [Google Scholar]
- 24.Southern KW, Merelle MM, Dankert-Roelse JE, Nagelkerke AD. Newborn screening for cystic fibrosis. Cochrane Database Syst Rev. 2009;21:CD001402. doi: 10.1002/14651858.CD001402.pub2. doi(1):CD001402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farrell PM, Lai HJ, Li Z, Kosorok MR, Laxova A, Green CG, Collins J, Hoffman G, Laessig R, Rock MJ, Splaingard ML. Evidence on improved outcomes with early diagnosis of cystic fibrosis through neonatal screening: enough is enough! J Pediatr. 2005;147:S30–S36. doi: 10.1016/j.jpeds.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 26.Farrell PM, Kosorok MR, Rock MJ, Laxova A, Zeng L, Lai HC, Hoffman G, Laessig RH, Splaingard ML. Early diagnosis of cystic fibrosis through neonatal screening prevents severe malnutrition and improves long-term growth. Wisconsin cystic fibrosis neonatal screening study group. Pediatrics. 2001;107:1–13. doi: 10.1542/peds.107.1.1. [DOI] [PubMed] [Google Scholar]
- 27.Farrell PM, Kosorok MR, Laxova A, Shen G, Koscik RE, Bruns WT, Splaingard M, Mischler EH. Nutritional benefits of neonatal screening for cystic fibrosis. Wisconsin cystic fibrosis neonatal screening study group. N Engl J Med. 1997;337:963–969. doi: 10.1056/NEJM199710023371403. [DOI] [PubMed] [Google Scholar]
- 28.Kosorok MR, Zeng L, West SE, Rock MJ, Splaingard ML, Laxova A, Green CG, Collins J, Farrell PM. Acceleration of lung disease in children with cystic fibrosis after Pseudomonas aeruginosa acquisition. Pediatr Pulmonol. 2001;32:277–287. doi: 10.1002/ppul.2009.abs. [DOI] [PubMed] [Google Scholar]
- 29.Wisconsin Department of Health Services [Internet] [accessed 2013 June 17]. Available from: http://www.dhs.wisconsin.gov/wish/
- 30.Baker MW, Groose M, Hoffman G, Rock M, Levy H, Farrell PM. Optimal DNA tier for the IRT/DNA algorithm determined by CFTR mutation results over 14 years of newborn screening. J Cyst Fibros. 2011;10:278–281. doi: 10.1016/j.jcf.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.The Clinical and Functional Translation of CFTR (CFTR2) [Internet] Johns Hopkins University, The Hospital for Sick Children: US CF Foundation; c2014 [accessed 2014 April 30]. Available from: http://cftr2.org.
- 32.State of Wisconsin-Department of Administration [Internet] [accessed 2013 June 17]. Available from: http://www.doa.state.wi.us/subcategory.asp?linksubcatid=354&linkcatid=11&linkid=64&locid=9.
- 33.Alper OM, Wong LJ, Young S, Pearl M, Graham S, Sherwin J, Nussbaum E, Nielson D, Platzker A, Davies Z, Lieberthal A, Chin T, Shay G, Hardy K, Kharrazi M. Identification of novel and rare mutations in California Hispanic and African American cystic fibrosis patients. Hum Mutat. 2004;24:353. doi: 10.1002/humu.9281. [DOI] [PubMed] [Google Scholar]
- 34.Ihrke DK, Faber CS. Geographical mobility: 2005 to 2010 population characteristics. 2012 U.S. Department of Commerce, Economics and Statistics Administration, U.S. Census Bureau. Report nr P20–567. [Google Scholar]
- 35.Castellani C, Picci L, Tamanini A, Girardi P, Rizzotti P, Assael BM. Association between carrier screening and incidence of cystic fibrosis. JAMA. 2009;302:2573–2579. doi: 10.1001/jama.2009.1758. [DOI] [PubMed] [Google Scholar]
- 36.Mischler EH, Wilfond BS, Fost N, Laxova A, Reiser C, Sauer CM, Makholm LM, Shen G, Feenan L, McCarthy C, Farrell PM. Cystic fibrosis newborn screening: impact on reproductive behavior and implications for genetic counseling. Pediatrics. 1998;102:44–52. doi: 10.1542/peds.102.1.44. [DOI] [PubMed] [Google Scholar]
- 37.Wertz DC, Janes SR, Rosenfield JM, Erbe RW. Attitudes toward the prenatal diagnosis of cystic fibrosis: factors in decision making among affected families. Am J Hum Genet. 1992;50:1077–1085. [PMC free article] [PubMed] [Google Scholar]
- 38.Morgan MA, Driscoll DA, Mennuti MT, Schulkin J. Practice patterns of obstetrician-gynecologists regarding preconception and prenatal screening for cystic fibrosis. Genet Med. 2004;6:450–455. doi: 10.1097/01.gim.0000139509.04177.4b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.