

Abstract
Biological pharmaceuticals are growing rapidly and represent some of the most effective pharmaceuticals on the market, however, delivery of these agents remains a challenge. Melt processing is emerging as a viable protein encapsulation method because it is solvent free, is high throughput, and yields very high encapsulation efficiencies. Problematically though, proteins can denature and lose activity during melt processing due to high heat and shear forces. Covalent attachment of poly(ethylene glycol) (PEG), commonly referred to as PEGylation, has been widely used to increase the thermal stability and prevent aggregation of proteins in aqueous solutions. This study explored the effect of PEGylation on protein stability during melt processing using lysozyme and poly(lactic-co-glycolic acid) (PLGA). The results indicate that PEGylation can increase the retained activity of lysozyme post-processing, increase dispersion in the melt, and reduce the biphasic release profile exhibited by lysozyme in PLGA melt processed systems.
Keywords: poly(lactic-co-glycolic acid), PEGylation, melt processing, protein stability
1. Introduction
The use of protein therapeutics for disease treatment and tissue regeneration has increased greatly over the past ten years with advances in biotechnology and large-scale protein production methods.[1] The delivery of protein therapeutics remains a challenge as oral administration results in degradation and low bioavailability.[2][3] Intravenous injection is the most commonly used administration route, even though protein therapeutics are rapidly cleared from circulation via renal clearance and proteolytic degradation.[4] As a result, there has been an increase in research developing depot formulations, in which a protein therapeutic is encapsulated in a biodegradable polymer that can be injected or implanted.[5] Depot formulations are advantageous because proteins are protected from in vivo degradation and the release profile can be sustained and controlled over time based on the polymer composition.[6] Current protein encapsulation methods used to create microspheres,[7][8] electrospun fibers,[9][10] and hydrogels[11]–[13] have seen great progress both in vitro and in vivo. These techniques, though, often involve co-dissolution steps where the protein is dissolved or dispersed into organic media alongside polymer excipients and surfactants. The dissolution or dispersion of proteins into organic solvents often results in interfacial adsorption, unfolding, aggregation, and inactivation of the protein.[14][15] Solvent-based encapsulation processes have encapsulation efficiencies of 30 – 50%,[16][17] requiring the use of excess, expensive protein therapeutics.
Melt processing is a viable alternative to emulsification based encapsulation techniques and a handful of studies have explored protein stability after encapsulation via melt extrusion.[18][19] Melt processing is advantageous due to the lack of organic solvent, high protein loading, and the variety of geometries that can be produced.[20] Furthermore, melt processing is a high throughput industrial process and does not require solvent removal steps, allowing for rapid and economical encapsulation compared to other methods.[21][22] A limitation of melt processing is the exposure of proteins to high heat and shear stress which can result in denaturation and aggregation. Studies have shown that proteins can be successfully integrated into biodegradable polyester matrices and recovered intact and in active conformations despite the high stresses and heat during melt processing, depending on the processing temperature.[23] Additionally, stabilizing additives and multiblock copolymers have also been used to improve protein stability and lower temperature requirements during melt processing.[19][24] However, modifications to the protein to improve stability have not yet been explored.
Covalent attachment of poly(ethylene glycol) (PEG) to proteins, also known as PEGylation, has been widely used to improve the therapeutic value of many FDA approved protein drugs.[25] PEGylation of proteins reduces immunogenicity, increases circulation time,[26] improves thermal stability,[27] and can prevent denaturation during lyophilization.[28] In this study, poly(lactic-co-glycolic acid) (PLGA) was used as the excipient and lysozyme as a model protein in a melt-processing system. Lysozyme is a common model protein used in PEGylation studies due to the wide varieties of PEGylation chemistries that can be applied, well characterized secondary structure of lyszozyme, and the ability to quantify enzymatic activity via simple assays. The impact of PEGylation on the thermal stability of solubilized lysozyme[27] and adsorption onto solid substrates[29] has been explored, however no studies to date have investigated thermal stability in the solid/melt state. PLGA is a biodegradable polymer that has been widely researched and utilized for several FDA-approved biomedical applications.[30] Lysozyme has also been melt processed with PLGA and recovered with relative activities ranging from 60 to 90% depending on the processing temperature.[23]
The results presented herein detail the melt processing of PEGylated lysozyme. These studies reveal improved activity of recovered protein, increased dispersion within the melt, and a more linear release profile relative to unmodified protein. As a host of therapeutic proteins are PEGylated, we anticipate that the results of this study can be translated to therapeutic biopharmaceuticals.
2. Results and Discussion
This work investigated the effect of the covalent attachment of a single 20kDa PEG chain (i.e. monoPEGylation) on the thermal stability of lysozyme during melt processing within a PLGA matrix. Lysozyme has exhibited higher thermal stability after covalent attachment of at least one PEG chain, preventing aggregation[31] and limiting solvent accessibility to the secondary structure of the protein.[32] PEGylated therapeutic proteins are typically monoPEGylated,[33]–[35] hence monoPEGylation was used to represent the methods used in clinical formulations of therapeutic proteins. Attachment of a single, long PEG chain has been increasingly utilized due to enhanced circulation times and improved in vivo activity when compared to attachment of multiple, shorter PEG chains.[36] Furthermore, we expected that a longer PEG chain would more effectively shield the protein during melt processing compared to PEG chains of 5 or 10 kDa size. N-hydroxysuccinimide (NHS) functionalized PEG was used to PEGylate lysozyme because of the ease of the reaction and prior studies utilizing NHS chemistry to monoPEGylate lysozyme.[37][38] The NHS functionalized PEG was reacted with lysozyme in 0.1 M phosphate buffer (pH 8) at a substoichiometric ratio to prevent multiple PEGylations of the same protein. The monoPEGylated conjugates were purified from unreacted PEG and native lysozyme via FPLC purification, with an average yield of 10.15 mg (69%) per reaction mixture (Figure S1). SDS-PAGE results of the lyophilized and resuspended fractions indicated that the product was 95% monoPEGylated lysozyme and free of native lysozyme (Figure 1A). FPLC of the collected fractions further verified the removal of native lysozyme (Figure 1B). MALDI-TOF mass spectroscopy verified the molecular weight of the lysozyme-PEG conjugate (Figure S2).
Figure 1.

A. SDS-PAGE of protein ladder (L), native lysozyme (1) and the purified lysozyme-PEG conjugate (2). B. FPLC trace of the purified PEG-lysozyme conjugate. C. 2nd derivative of the Amide I band from FT-IR spectra of native (solid) and PEGylated (dotted) lysozyme. B. Circular dichroism spectra of native (solid) and PEGyated lysozyme (dotted).
The activity of lysozyme and lysozyme-PEG conjugates was measured colorimetrically as a function of the ability to degrade glycol chitosan,[39] and the activity of the PEG conjugate was 40.4% relative to native lysozyme (Figure S3). The loss in activity is expected for enzymes that act on macromolecular substrates,[27][38] as a large PEG chain will sterically hinder the active site from complexing with the macromolecule. Previous studies have determined the K33 residue to be the most reactive lysine and this is likely the predominate site of attachment.[40] However, diPEGylation and monoPEGylation at the K97 or K116 residue can result in a lower activity through steric effects.[27] The secondary structure of the lysozyme-PEG conjugate was analyzed via infrared spectroscopy (Figure S4) and circular dichroism spectroscopy (CD). Second derivative analysis of the Amide I band in the FT-IR spectra (Figure 1C) and the CD spectra (Figure 1D), both demonstrated that the secondary structure was maintained through the conjugation reaction and subsequent purification. The spectroscopy results therefore support that the decrease in relative activity of the lysozyme-PEG conjugate is due to steric hindrance of the active site by the PEG chain and not loss of active site conformation. Once protein activity and conformation were confirmed, implants were manufactured using a self-built syringe-die extruder driven by a syringe pump. The syringe extruder was built to allow for the melt processing of small sample masses. The syringe-die was heated using heating tape with a digital controller and the extrudate was air cooled to prevent die-swell and skin instabilities (Figure 2). All implants were loaded with 7.5 wt% of lysozyme or lysozyme-PEG conjugate and processed at 95°C with a volumetric flowrate rate of 2.35 mm3 s−1. The volumetric flowrate was selected as the upper limit that would still produce uniform samples and 95°C was selected as the lowest temperature that would homogeneously melt the sample in 10 minutes. The 10 minute processing time was chosen to represent the upper-limit of residence times encountered in industrial scale pharmaceutical extrusion.[41]
Figure 2.

A. Schematic of the syringe extruder set-up with heating tape. Inset shows samples of the extrudates; (1) PLGA/lysozyme, (2) PLGA/lysozyme/PEG6000, (3) PLGA/20kDaPEG-lysozyme. B. Top down view of syringe extruder. C. Front view of syringe extruder.
The thermal properties of lysozyme, NHS functionalized 20kDa PEG, and lysozyme-PEG conjugate were determined via differential scanning calorimetry (DSC). The melting temperatures of lysozyme, NHS functionalized 20kDa PEG, and lysozyme-PEG conjugate were 202°C, 60°C, and 222°C respectively, as determined from DSC thermographs (Figure S5). Therefore, the temperatures used during melt processing are not high enough to fully melt (or thermally denature) the protein or conjugate. DSC analysis of the PLGA indicated a glass transition temperature (Tg) of 37°C with enthalpic relaxation from 30°C to 50°. These results are consist with previous studies on PLGA[42] and indicate the melting temperature of polymer complexes instead of melting as a first-order transition.[43]
Previous studies have used low molecular weight PEG additives to enhance the release of proteins from a scaffold,[18][23] but did not examine if PEG additives have any stabilizing effect during processing. As an additional control PEG6000 was utilized as the PEG additive at 4 wt%. The size and percentage of PEG were chosen to be the highest that would yield uniform samples. Higher molecular weight PEG additives and higher loadings of PEG6000 decreased the viscosity of the blend and resulted in non-uniform samples. Therefore, samples processed with 4% by weight PEG6000 and 7.5 wt% lysozyme were used to distinguish the effect of covalent PEGylation versus the addition of free PEG to the blend and to account for the effect of small amounts of unreacted 20kDa PEG possibly present in the lyophilized conjugate. All samples produced had a uniform cross-section determined visually via optical microscopy and SEM. The dispersion and aggregation of protein in the implant was analyzed via EDX-SEM. The sulfur K series peak was mapped due to the unique atomic signature, derived from cysteines in the protein, and the spectral distance from the predominant carbon and oxygen peaks in the EDX spectrum. Under high heat and shear conditions, it was expected that native lysozyme would exhibit unfolding and aggregation behavior in response to increased temperatures, as has been previously observed.[23] In contrast, we expected a covalently attached PEG to act as a stabilizer when the protein and polymer matrix are in the melt state, mitigating protein unfolding and aggregation due to the amphiphilic nature of the PEG. The results indicate the formation of large aggregates when lysozyme is processed with no additives (Figure 3D). The simple blending of PEG6000 leads to decreased aggregation, but aggregation still remains significant in the EDX map (Figure 3E). Additionally, the melt viscosity is lowered by adding low molecular weight PEGs leading to non-uniform samples lacking in robust mechanical properties once processed. Finally, the lysozyme-PEG conjugates exhibited minimal aggregation when melt-extruded (Figure 3F). A simple covalent PEG modification imparted shielding behavior and significantly improved dispersion within the melt. As is detailed in activity and release studies, this improved dispersion had a substantial effect on protein stability and linearity of release.
Figure 3.
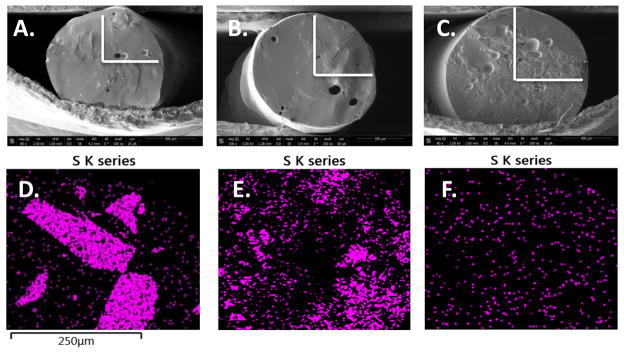
A–C: SEM micrographs. A. PLGA with native lysozyme, B. PLGA with native lysozyme and PEG6000 additive, and C. PLGA with PEGylated lysozyme with the region examined via EDX marked in white. D–F: EDX mapping of the sulfur peak for D. PLGA with native lysozyme, E. PLGA with native lysozyme and PEG6000 additive, and F. PLGA with PEGylated lysozyme.
The protein content of the implants was extracted using ethyl acetate, a method that has been used to recover lysozyme from similar systems with full activity.[44] The activity of the extracted and resuspended protein was measured via the glycol chitosan assay (Figure S7). Samples of released protein from day 1, 10, and 20 were analyzed for relative activity to unprocessed lysozyme using the glycol chitosan assay (Figure S8). The extracted and released sample activities were averaged and the results demonstrated that lysozyme processed with and without the PEG6000 additive lost 48% of initial activity after melt-extrusion and release, while covalently PEGylated lysozyme retained 93% of initial activity (Figure 4A). The enhanced retention of enzymatic activity is consistent with the EDX-SEM results, as the lysozyme processed with and without the PEG6000 additive exhibited aggregation that can result in loss of activity.[16] These results demonstrate that protein PEGylation, with a single large PEG chain, allows for almost full retention of pre-processed activity. As more protein:polymer conjugates are coming to market, these findings have significant implications in the high-throughput encapsulation of protein depot formulations.
Figure 4.

A. Initial and post-processing (denoted ‘Initial’ and ‘Post’ respectively) relative activities of protein samples measured via glycol chitosan assay. B. Release profiles of melt processed samples for native lysozyme (bottom), native lysozyme with PEG6000 additive (middle), and PEGylated lysozyme (top) over 32 days.
The effect of covalent PEGylation on release from the implants was investigated by monitoring the release at 37°C with agitation at 80 rpm over a 32 day period. Lysozyme exhibits the highest stability in sodium acetate buffer at pH 5 and was used to eliminate any potential reduction in activity due to buffer or pH effects.[27] The release profiles indicate that covalent PEGylation resulted in a more continuous release throughout the study period in comparison to lysozyme processed with and without PEG additive, with a reduction in the typical biphasic kinetic profile (Figure 4B). Covalently PEGylated lysozyme samples and samples processed with PEG6000 additive both exhibited accelerated release rates when compared to samples with solely native lysozyme. The release of covalently PEGylated lysozyme was similar to lysozyme processed with 4 wt% PEG6000 after 10 days, with the major differences occurring during the initial hydration stage of release over the first 24 hours. Samples with PEG additive released 38% of total protein mass during initial hydration over the first 24 hours. This burst release was attributed to expansion of the matrix and pore formation by diffusion of PEG out of the matrix, as PEG is a known porogen in PLGA implants.[45] The release after this point was controlled by the degradation of the matrix and diffusion of lysozyme through the matrix. Covalently attached PEG reduces the initial release to 16% and continued to release up to 51% after 5 days. This resulted in a near exponential release profile as opposed to the distinct two phase release observed in the other samples. The release study was stopped after 32 days when the implants were completely dissolved.
The results of this study shed light on the ability to formulate proteins in a solvent-free and high throughput process. Simple PEGylation, as is often seen in the pharmaceutical industry, leads to enhanced dispersion of proteins in the melt, allows for stabilization of protein activity at high temperatures, and mitigates some of the burst release associated with depot formulations. In our system, we developed a syringe extruder to process extremely small masses of implants, but industrial scale extruders can process in excess of 1000 kg h−1,[22] perhaps leading to a more robust manufacturing process as the biopharmaceutical field advances. The activity of lysozyme was diminished upon PEGylation, however, there are a host of therapeutic proteins that can be PEGylated where this isn’t the case, for instance N-terminal PEGylation of bone morphogenetic protein 2.[46] Proteins such as this could benefit from the improved retention of activity post-processing, as the high temperatures of melt processing had little effect on lysozyme activity. Lastly, an additional advantage of melt-processing is the 100% encapsulation efficiency. This can be compared to solvent-based encapsulation methods where encapsulation can be below 50%,[16][17] both wasting a costly active pharmaceutical and introducing organic solvents which could potentially have residual biological effects.
3. Conclusions
The results of this study indicate that covalent attachment of PEG prevented loss of activity of lysozyme and minimal aggregation during the melt-extrusion process. PEGylation also reduced the biphasic characteristic of the release profile when compared to implants processed with native lysozyme and with a PEG additive. These initial results demonstrate that the stability of proteins during melt processing can be enhanced using PEGylation and the release profile is dependent on polymer attachment. Further studies are planned to investigate how the release profile can be tuned depending on the conjugated PEG length and the protein behavior in response to temperature and time spent in the melt state. PEGylating therapeutically relevant proteins to increase stability in the melt state will also be explored.
Supplementary Material
Acknowledgments
J.K.P acknowledges funding from an NIH Pathway to Independence Award (R00EB011530) and thanks the National Science Foundation Center for Layered Polymeric Systems (DMR 0423914), for start-up funds. P.W.L. acknowledges a GAANN fellowship for financial support. This material is based upon work supported by the National Science Foundation under Grant No. MRI-0821515 (for the purchase of the MALDI-TOF-TOF). The authors would like to thank the Swagelok Center for Surface Analysis of Materials for the EDX-SEM facilities.
Footnotes
Supporting Information is available online from the Wiley Online Library or from the author.
Contributor Information
Parker Lee, Department of Macromolecular Science and Engineering, Case Western Reserve University, Cleveland, OH 44106, United States.
Jenna Towslee, Department of Macromolecular Science and Engineering, Case Western Reserve University, Cleveland, OH 44106, United States.
Prof. João Maia, Department of Macromolecular Science and Engineering, Case Western Reserve University, Cleveland, OH 44106, United States
Prof. Jonathan Pokorski, Email: jon.pokorski@case.edu, Department of Macromolecular Science and Engineering, Case Western Reserve University, Cleveland, OH 44106, United States
References
- 1.Walsh G. Nat Biotechnol. 2014;32:992. doi: 10.1038/nbt.3040. [DOI] [PubMed] [Google Scholar]
- 2.Woodley JF. Crit Rev Ther Drug Carrier Syst. 1994;11:61. [PubMed] [Google Scholar]
- 3.Cleland Jeffrey L, Langer Robert. Formul Deliv Proteins Pept. Vol. 1 American Chemical Society; 1994. [Google Scholar]
- 4.Park K, Kwon IC, Park K. React Funct Polym. 2011;71:280. [Google Scholar]
- 5.Jain RA. Biomaterials. 2000;21:2475. doi: 10.1016/s0142-9612(00)00115-0. [DOI] [PubMed] [Google Scholar]
- 6.Giteau A, Venier-Julienne MC, Aubert-Pouëssel A, Benoit JP. Int J Pharm. 2008;350:14. doi: 10.1016/j.ijpharm.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 7.Okada H. Adv Drug Deliv Rev. 1997;28:43. doi: 10.1016/s0169-409x(97)00050-1. [DOI] [PubMed] [Google Scholar]
- 8.Gao G, Yan Y, Pispas S, Yao P. Macromol Biosci. 2010;10:139. doi: 10.1002/mabi.200900186. [DOI] [PubMed] [Google Scholar]
- 9.Chew SY, Wen J, Yim EKF, Leong KW. Biomacromolecules. 2005;6:2017. doi: 10.1021/bm0501149. [DOI] [PubMed] [Google Scholar]
- 10.Nepal D, Minus ML, Kumar S. Macromol Biosci. 2011;11:875. doi: 10.1002/mabi.201000490. [DOI] [PubMed] [Google Scholar]
- 11.Woo BH, Jiang G, Jo YW, DeLuca PP. Pharm Res. 2001;18:1600. doi: 10.1023/a:1013090700443. [DOI] [PubMed] [Google Scholar]
- 12.Hammer N, Brandl FP, Kirchhof S, Messmann V, Goepferich AM. Macromol Biosci. 2015;15:405. doi: 10.1002/mabi.201400379. [DOI] [PubMed] [Google Scholar]
- 13.Manokruang K, Lee DS. Macromol Biosci. 2013;13:1195. doi: 10.1002/mabi.201300236. [DOI] [PubMed] [Google Scholar]
- 14.Manning MC, Chou DK, Murphy BM, Payne RW, Katayama DS. Pharm Res. 2010;27:544. doi: 10.1007/s11095-009-0045-6. [DOI] [PubMed] [Google Scholar]
- 15.Manning MC, Patel K, Borchardt RT. Pharm Res. 1989;6:903. doi: 10.1023/a:1015929109894. [DOI] [PubMed] [Google Scholar]
- 16.Wang W, Nema S, Teagarden D. Int J Pharm. 2010;390:89. doi: 10.1016/j.ijpharm.2010.02.025. [DOI] [PubMed] [Google Scholar]
- 17.Fan Y, Li X, Zhou Y, Fan C, Wang X, Huang Y, Liu Y. Int J Pharm. 2011;416:323. doi: 10.1016/j.ijpharm.2011.06.029. [DOI] [PubMed] [Google Scholar]
- 18.Ghalanbor Z, Körber M, Bodmeier R. Int J Pharm. 2012;438:302. doi: 10.1016/j.ijpharm.2012.09.015. [DOI] [PubMed] [Google Scholar]
- 19.Stankovi M, de Waard H, Steendam R, Hiemstra C, Zuidema J, Frijlink HW, Hinrichs WLJ. Eur J Pharm Sci. 2013;49:578. doi: 10.1016/j.ejps.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 20.Qian F, Szymanski A, Gao J. J Biomed Mater Res. 2001;55:512. doi: 10.1002/1097-4636(20010615)55:4<512::aid-jbm1044>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 21.Maniruzzaman M, Boateng JS, Snowden MJ, Douroumis D. ISRN Pharm. 2012;2012:1. doi: 10.5402/2012/436763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Repka MA, Shah S, Lu J, Maddineni S, Morott J, Patwardhan K, Mohammed NN. Expert Opin Drug Deliv. 2012;9:105. doi: 10.1517/17425247.2012.642365. [DOI] [PubMed] [Google Scholar]
- 23.Ghalanbor Z, Körber M, Bodmeier R. Pharm Res. 2010;27:371. doi: 10.1007/s11095-009-0033-x. [DOI] [PubMed] [Google Scholar]
- 24.Stankovi M, Tomar J, Hiemstra C, Steendam R, Frijlink HW, Hinrichs WLJ. Eur J Pharm Biopharm. 2014;87:329. doi: 10.1016/j.ejpb.2014.02.012. [DOI] [PubMed] [Google Scholar]
- 25.Alconcel SNS, Baas AS, Maynard HD. Polym Chem. 2011;2:1442. [Google Scholar]
- 26.Hershfield MS, Buckley RH, Greenberg ML, Melton AL, Schiff R, Hatem C, Kurtzberg J, Markert ML, Kobayashi RH, Kobayashi AL. N Engl J Med. 1987;316:589. doi: 10.1056/NEJM198703053161005. [DOI] [PubMed] [Google Scholar]
- 27.da Freitas DS, Abrahão-Neto J. Int J Pharm. 2010;392:111. doi: 10.1016/j.ijpharm.2010.03.036. [DOI] [PubMed] [Google Scholar]
- 28.Yamasaki N, Matsuo A, Hatakeyama T, Funatsu G. Agric Biol Chem. 1990;54:2635. [Google Scholar]
- 29.Daly Susan, Przybycien Todd, Tilton Robert. Langmuir. 2005;21:1328. doi: 10.1021/la048316y. [DOI] [PubMed] [Google Scholar]
- 30.Lü J-M, Wang X, Marin-Muller C, Wang H, Lin PH, Yao Q, Chen C. Expert Rev Mol Diagn. 2009;9:325. doi: 10.1586/erm.09.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malzert A, Boury F, Renard D, Robert P, Lavenant L, Benoît JP, Proust JE. Int J Pharm. 2003;260:175. doi: 10.1016/s0378-5173(03)00258-8. [DOI] [PubMed] [Google Scholar]
- 32.Pai SS, Hammouda B, Hong K, Pozzo DC, Przybycien TM, Tilton RD. Bioconjug Chem. 2011;22:2317. doi: 10.1021/bc2003583. [DOI] [PubMed] [Google Scholar]
- 33.Chapman AP, Antoniw P, Spitali M, West S, Stephens S, King DJ. Nat Biotechnol. 1999;17:780. doi: 10.1038/11717. [DOI] [PubMed] [Google Scholar]
- 34.Bailon P, Palleroni A, Schaffer CA, Spence CL, Fung WJ, Porter JE, Ehrlich GK, Pan W, Xu ZX, Modi MW, Farid A, Berthold W, Graves M. Bioconjug Chem. 2001;12:195. doi: 10.1021/bc000082g. [DOI] [PubMed] [Google Scholar]
- 35.Shin BS, Jung JH, Lee KC, Yoo SD. Chem Pharm Bull (Tokyo) 2004;52:957. doi: 10.1248/cpb.52.957. [DOI] [PubMed] [Google Scholar]
- 36.Harris JM, Chess RB. Nat Rev Drug Discov. 2003;2:214. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 37.Zalipsky S. Bioconjug Chem. 1995;6:150. doi: 10.1021/bc00032a002. [DOI] [PubMed] [Google Scholar]
- 38.Nodake Y, Yamasaki N. Biosci Biotechnol Biochem. 2000;64:767. doi: 10.1271/bbb.64.767. [DOI] [PubMed] [Google Scholar]
- 39.Imoto KYT. Agric Biol Chem. 1971;35 [Google Scholar]
- 40.Lee H, Park TG. J Pharm Sci. 2003;92:97. doi: 10.1002/jps.10270. [DOI] [PubMed] [Google Scholar]
- 41.Nikitine C, Rodier E, Sauceau M, Fages J. Chem Eng Res Des. 2009;87:809. [Google Scholar]
- 42.Park Peter In Pyo, Jonnalagadda Sriramakamal. J Appl Polym Sci. 2006;100:1983. [Google Scholar]
- 43.Köhler Karen, Shchukin Dmitry G, Möhwald Helmuth, Sukhorukov Gleb B. J Phys Chem B. 2005;109:18250. doi: 10.1021/jp052208i. [DOI] [PubMed] [Google Scholar]
- 44.Körber M, Bodmeier R. Eur J Pharm Sci. 2008;35:283. doi: 10.1016/j.ejps.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 45.Rahman CV, Ben-David D, Dhillon A, Kuhn G, Gould TWA, Müller R, Rose FRAJ, Shakesheff KM, Livne E. J Tissue Eng Regen Med. 2014;8:59. doi: 10.1002/term.1497. [DOI] [PubMed] [Google Scholar]
- 46.Hu J, Sebald W. Int J Pharm. 2011;413:140. doi: 10.1016/j.ijpharm.2011.04.043. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.