

Abstract
Solid state NMR is the primary tool for studying the quantitative, site-specific structure, orientation, and dynamics of biomineralization proteins under biologically relevant conditions. Two calcium phosphate proteins, statherin (43 amino acids) and leucine rich amelogenin protein (LRAP; 59 amino acids), have been studied in depth and have different dynamic properties and 2D- and 3D-structural features. These differences make it difficult to extract design principles used in nature for building materials with properties such as high strength, unusual morphologies, or uncommon phases. Consequently, design principles needed for developing synthetic materials controlled by proteins are not clear. Many biomineralization proteins are much larger than statherin and LRAP, necessitating the study of larger biomineralization proteins. More recent studies of the significantly larger full-length amelogenin (180 residues) represent a significant step forward to ultimately investigate the full diversity of biomineralization proteins. Interactions of amino acids, a silaffin derived peptide, and the model LK peptide with silica are also being studied, along with qualitative studies of the organic matrices interacting with calcium carbonate. Dipolar recoupling techniques have formed the core of the quantitative studies, yet the need for isolated spin pairs makes this approach costly and time intensive. The use of multidimensional techniques to study biomineralization proteins is becoming more common, methodology which, despite its challenges with these difficult-to-study proteins, will continue to drive future advancements in this area.
Keywords: Biomineralization, Immobilized proteins, Dipolar recoupling, Protein structure, Protein dynamics, Protein orientation, Multi-dimensional solid state NMR, Amelogenin, Statherin, Silaffin
1. Introduction
Biomineralization proteins direct the construction of hard tissues in the natural world, such as bones, teeth, pearl, nacre and egg shells [1]. The properties of the resulting tissues range significantly, even within a given mineral type, with the proteins present during construction allowing properties such as strength, hardness, morphology and/or phases that are not realized in their absence. This impressive control over inorganic minerals with the addition of what is effectively a polymer has incredible potential in aiding the development of new materials, if only the mechanisms underlying the formation of biominerals were understood.
Key to the study of any protein is an understanding of its structure, including primary, secondary, tertiary and quaternary structures. The primary structures of the identified proteins are relatively easily determined using biochemical methods, and quaternary structures (protein–protein interactions), if present, are studied with macroscopic techniques such as dynamic light scattering (DLS) and yeast-two hybrid systems as has been demonstrated for the biomineralization protein amelogenin [2,3], although these techniques don’t address questions about the molecular level description driving the interactions. Secondary and tertiary structures are critical components of a proteins function, and are typically studied with solution state Nuclear Magnetic Resonance (NMR) or X-ray crystallography. These techniques have been used to provide insight into potential protein function during biomineralization, however, the functional form of biomineralization proteins is immobilized on a surface. Therefore the best mechanistic insight into the biomineralization protein’s function will be gleaned from structural studies of the protein bound to its biologically relevant surface, a regime where solution state NMR and X-ray crystallography are not applicable. An additional challenge biomineralization proteins is that many of them are intrinsically disordered. IDP’s are thought to be structurally labile to accommodate multiple functions, and adopt a specific structure when performing that function [4]. Therefore, solution state NMR studies of biomineralization proteins which suggest a lack of structure are misleading or oversimplified since the protein is being evaluated outside of its functional environment; consequently, studying them bound to their biologically relevant surface becomes even more essential to extract physiologically relevant data.
1.1. Studying the interaction mechanism of biomineralization proteins with solid state NMR (SSNMR)
There are three aspects of a surface immobilized protein that define its interaction: structure, orientation relative to the surface, and dynamics. SSNMR is well suited to address each of these aspects of surface interaction. Secondary, tertiary, and quaternary structure can all be studied by SSNMR, though studies of secondary structure, and when achievable, tertiary structure have been the focus of biomineralization studies to date. The orientation refers to how a particular residue or region within a protein is positioned relative to the surface, i.e. the protein may lay with the backbone parallel to the surface, perpendicular to the surface, or some combination of parallel and perpendicular, achieved by the arrangement of secondary and tertiary structures. Both the orientation and the structure can be studied using either homonuclear or heteronuclear dipolar recoupling methods. The protein dynamics indirectly indicate the strength of the interaction, based on the hypothesis that proteins that bind strongly will be less mobile than those that are not tightly associated with the surface. Side chain and backbone dynamics can be studied using a combination of relaxation measurements (T1, T2, T1ρ, and cross polarization efficiency) as well as 2H dynamics.
Advances in SSNMR in the past 20 years allow this technique to lend itself almost uniquely to the study of these remarkable proteins. By this time, Cross-polarization Magic Angle Spinning (CPMAS) had become well established, producing spectra which in many ways are “liquid-like”, providing high resolution spectra in minimal time [5,6]. Advances in spectrometer and probe reliability and stability also contributed, enabling the development of dipolar recoupling sequences which allow precise distance measurements to be made.
1.2. Dipolar recoupling methods
One of the most accurate ways to determine distances in SSNMR is through the use of dipolar recoupling methods [7]. Dipolar coupling between two spins is averaged during MAS, but can be recovered using dipolar recoupling. Techniques such as Dipolar Recoupling with a Windowless Sequence (DRAWS) [8], have been developed for recoupling the dipolar coupling between homonuclei, while techniques such as Rotational Echo DOuble Resonance (REDOR) are used to recouple the dipolar coupling between heteronuclei, and both have been commonly implemented for the study of biomineralization proteins [9]. Application of these techniques results in distance measurements between two unique spins. For 13C–13C, the maximum distance accurately measurable is 4–5 Å, with an error of ±0.5 Å at the longest distances. For heteronuclear coupling, the distance depends upon the nuclei being included. For 13C–15N, nuclei of interest for inter- and intramolecular protein distances, accurate distances can be measured to ~5–6 Å, while for 13C–31P, distances up to ~9–10 Å can be measured, with an accuracy of ~±0.5 to 1 Å at the longest distances. The accuracies of the resulting distances for both heteronuclear and homonuclear couplings depend on the spin system, the distance measured and the sample. Shorter distances can be as accurate as ±0.2 Å, for instance 13C–31P distances of 5 Å or less, as can samples with high signal to noise. Inherent in the technique is a higher accuracy for larger couplings, arising from the 1/r3 dependence in the Hamiltonian (r=distance), and observed by the enhanced distinction in dephasing curves for larger couplings (i.e. shorter distances; can be seen in Fig. 2).
Fig. 2.
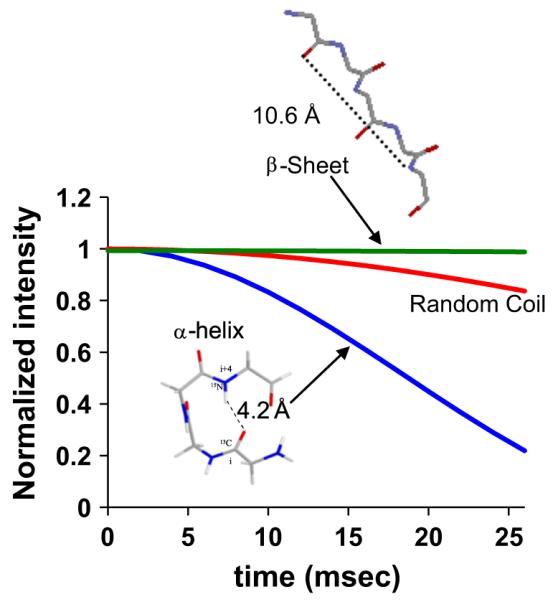
Labeling scheme and resulting REDOR dephasing curves for common structural motifs.
The most unambiguous distances resulting from these measurements are realized by measuring the distance between isolated isotopically labeled spin pairs. For the application of DRAWS to determine the structure of proteins, two 13C carbonyl carbons on adjacent amino acid residues are introduced (Fig. 1). The distance derived from this labeling scheme has a well-defined dependence on the torsion angle φ, which correlates well with structure (Fig. 1). Structural measurements with REDOR are facilitated by introducing a 13C backbone carbonyl and an 15N backbone amide on residues at the i and i+4 positions, respectively (Fig. 2). These two nuclei are the heavy atoms involved in a hydrogen bond if the structure is helical, and result in dephasing curves with distinct differences for α-helical and β-sheet structures, and also for a random coil, due to the resulting different distances (Fig. 2). REDOR is also used to study the distance to the surface, from backbone or side chain 13C or 15N to surface ions such as 31P in calcium phosphate or 29Si for silica. More complex spin systems, i.e. more than two spins, can also be studied, however, in all cases the proteins require precise control of residue labeling. The result of these studies is a site-specific, atomic level understanding of the residues of interest, including structure, orientation of the residue with respect to the surface, and dynamics.
Fig. 1.

Left: Torsion angles of a protein fragment are shown. In most cases, ω is restricted to 180° due to the delocalized electron density of the O=C–N bond, leaving φ and ψ as the only variables in determining the secondary structure of the protein. Right: The Ramachandran plot maps φ and ψ combinations, illustrating the energetically favored dihedral angles found in common protein secondary structure. The dark gray areas are the most energetically favored, the light gray, less so and the white areas are energetically unallowed. The black markers represent experimentally determined φ and ψ angles, clustered in the dark gray regions. The special secondary structures (α-helix, β-sheet (parallel and anti-parallel), and the collagen triple helix) have also been indicated, according to their classically defined values. Bottom: The φ angles map out to distances between adjacent carbonyl carbons, as shown, allowing a direct correlation between the distance, or dipolar coupling, and the structure.
Solid phase peptide synthesis enables the incorporation of a specifically labeled residue in a specific location in the primary structure, allowing complete control of the labeling frequency and positioning. Most residues are commercially available with various labeled options (a single 13C, 15N, or 2H, various combinations of the three, or uniform 13C and 15N), and many are available already protected for solid phase synthesis, using either a BOC strategy or an FMOC strategy, the two common methodologies of solid phase synthesis [10]. The control achievable with solid phase synthesis is contrasted with recombinant protein expression in E. coli, where at a minimum, all the residues of a given type will be labeled. The limitation of solid phase synthesis is that high coupling efficiency can only be achieved for proteins up to ~60 residues, restricting the protein size available for study using this labeling technique.
There are reviews that cover early work in this area [11–14], which primarily focused on statherin, a 43 amino acid residue protein that blocks primary and secondary precipitation of calcium phosphate within the oral environment. This protein was the first biomineralization protein to have detailed, quantitative structural studies performed bound to its biologically relevant surface and remains one of the most well-studied biomineralization proteins to date. It was also thought that it served as a model for other biomineralization proteins, due to its high content of aspartic acid, glutamic acid and phosphoserine in the binding region. This review primarily focuses on studies published after the previously reported review articles on solid state NMR of biomineralization proteins [11–14], discussing other proteins which have now been well studied, describing new techniques which are being implemented, and providing a look to the future of both methodology and systems. Work investigating interactions of non-protein based molecules such as citrate [15], succinate [16], tethered chains [17], or liposomes [18] on or with inorganic materials provide interesting venues for comparison, have recently been reviewed [19], and are outside the scope of this review. Additionally, the inorganic mineral of the biominerals has been studied in some detail [16,20–25], but that will also not be covered here. The focus of this review is covering the proteins at protein–inorganic interfaces.
1.3. Sample preparation
A significant portion of the sample contains no protein (i.e. a large portion of the sample is the mineral surface to which it is bound), and therefore does not contribute to the signal of interest. By nature, this limits signal-to-noise, and optimizing signal-to-noise becomes essential to minimize experiment time. The ratio of protein to surface will vary depending on the surface area of the inorganic material, and the size and the coverage of the protein, but a good rule of thumb is to have a minimum of ~1 × 1018 spins in the rotor to get good dipolar recoupling data. Sample preparation can contribute significantly to this, by using high surface area inorganics. Studies of proteins bound to calcium phosphate and silica are facilitated by the high surface areas that can be prepared for these materials (~100 m2/g and ~1000 m2/g, respectively). Another way to increase signal to noise is to co-precipitate the protein with the crystal by growing the mineral in the presence of the protein, an approach to be used as dictated by the biological relevance. Signal-to-noise can also be increased by ensuring that the rotor is fully packed; however, packing can be challenging due to the desire to maintain hydrated samples, conditions which result in the highest biological relevance. One method for achieving high sample volumes in the rotor is described here: Transfer a portion of the protein–inorganic complex slurry or paste into the rotor and spin the rotor in the probe at the recoupling experiment spinning speed for ~2–5 min. Remove the endcap and remove excess water, compress the remaining solid, transfer more protein–inorganic complex, and continue this process until the rotor is completely full. However it is achieved, pains taken to ensure the highest signal-to-noise possible are well spent to minimize expensive instrument time and reduce errors bars in distance measurements.
2. Studies with hydroxyapatite
Initial studies in the area of surface immobilized proteins and SSNMR began with Jeff Reimers group. Unlabeled, polymeric models of the proteins polyglutamic acid and polylysine, proteins which also represent the amino acid content of many biomineralization proteins, were studied bound to hydroxyapatite (HAP) and silica [26]. The structural analysis in this study was based on a chemical shift analysis, and dynamics were evaluated by fitting 2H lineshapes, evaluating cross polarization dynamics, and measuring spin-lock relaxation time measurements (T1ρ). These results provided a strong foundation upon which the directed study of biomineralization proteins could be built, by demonstrating several key principles: (1) Standard NMR techniques could be used to provide insight into unlabeled immobilized proteins; (2) The protein structure can change when bound to the surface, in this case from helical to extended; and (3) Investigating dynamics has the potential to reveal significant insight about the strength of the interaction between the protein and the surface. What was also clear from this data, however, was that without isotopic labeling, or two- or three-dimensions to provide resolution, the data would only provide qualitative insight, based on an average of the entire sample.
2.1. Salivary statherin
Salivary statherin (Table 1) is a 43-residue salivary protein which blocks the precipitation and growth of calcium phosphates [27,28]. The size of statherin made it an ideal protein to begin studying biomineralization proteins with dipolar recoupling techniques, and the features of the N-terminal binding region, high in positively charged and phosphorylated residues, allow it to serve as a model for other biomineralization proteins. There are several reviews available on the earlier studies of statherin [11–14,29–32], so only the highlights of this important body of work will be mentioned to establish ground work and provide context.
Table 1.
The primary structures of the biomineralization peptides/proteins studied by SSNMR. The charged residues are shown in bold.
| Peptide/ protein |
Primary structure |
|---|---|
| Statherin | DpSpSEEKFLRRIGRFGYGYGPYQPVPEQPLYPQPYQPQYQQYTF |
| LRAP | MPLPPHPGSPGYINLpSYEVLTPLKWYQSMIRQPPLSPILPELPLE AWPATDKTKREEVD |
| Amelogenin | MPLPPHPGSPGYINLpSYEVLTPLKWYQSMIRQPYPSYGYEPMGG WLHHQIIPVLSQQHPPSHTLQPHHHLPVVPAQQPVAPQQPMMPV PGHHSMTPTQHHQPNIPPSAQQPFQQPFQPQAIPPQSHQPMQPQSPLH PMQPLAPQPPLPPLFSMQ PLSPILPELPLEAWPATDKTKREEVD |
| Silaffin peptide (R5) |
SSKKSGSYSGSKGSKRRIL |
| LK-peptide | LKKLLKLLKKLLKL |
Initial studies of statherin utilized either the full-length protein, or a peptide consisting of the first 15 residues (SN15) which also contained the binding region, the first five residues [33–35]. These results identified that the entire region was helical when bound to the surface in the presence of water, including the binding region, and that increased motion was observed as distance increased from the binding region. One of the disadvantages of this approach is that it requires a separate, costly sample for each individual measurement. To address this problem, a more complex spin system was introduced to obtain multiple constraints from a single sample [36]. This study focused on the C-terminal region and revealed that the protein folded itself to form a 3-dimensional structure (Fig. 3), as well as revealing a helical structure that is induced upon binding to calcium phosphate, a structure that may be important for bacterial binding recognition or lubrication.
Fig. 3.
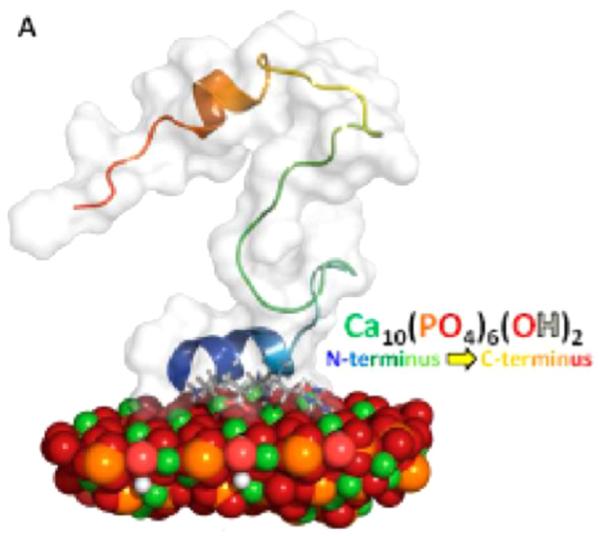
A representative lowest energy structure of statherin bound to the 100 face of HAP, obtained with RosettaSurface and consistent with SSNMR constraints. The N-terminus is helical and associated with the surface, while the C-terminus folds back on itself and is thought to serve a role in lubrication of bacterial binding. Reproduced with permission from Ref. [44]. Copyright 2014 American Chemical Society.
More recent studies on statherin have focused on surface orientation and dynamics. The distance of a number of the side chain residues from HAP were studied for statherin, including phenyalanines [37,38], lysines [39], arginines [40], and glutamic acids [41]. The results from these studies begin to provide a three-dimensional view of the protein–surface complex by revealing not only distances of the side chains of these residues from the surface, but also modifications in dynamics. In many cases, the results were not intuitive. For example, only one of the three glutamic acid residues (two in the N-terminus, one in the middle of the protein; Table 1) interacts strongly with the surface based on REDOR measurements (Fig. 4), although the side chain 13C′ resonances for both E4 and E5 shift 5 ppm upon binding, suggesting some inconsistency between the less rigorous chemical shift measurements and the more quantitative REDOR measurements. That only one Glu interacts is a somewhat surprising result, but it is consistent with the helical structure of the N-terminus which would only position one of the N-terminal residues next to the surface, and indirectly provides support of that structure. It is unclear why the C-terminal glutamic acid does not bind. Two hypotheses on the interaction of biomineralization proteins with surfaces include either an electrostatic interaction for negatively charged residues with a positively charged surface that may encourage unfolding, or a lattice-matching mechanism, whereby proteins adopted a specific structure resulting in side chains aligned with the corresponding oppositely charged atoms in the surface [1,42,43]. For statherin, maintaining the helical structure in favor of maximizing electrostatic interactions is consistent with a lattice matching mechanism. There are also three arginines present and studies of the distance of the side chain Cζ to the 31P in HAP showed the R9 and R10 were next to the surface (~4.5 Å; Fig. 5), while R13 had a much weaker interaction [40]. For a helical structure, these residues, along with K6 and E5 are all positioned to interact with the surface, consistent with the proposed structure and interaction mechanism. The reason for the weakly interacting R13 with the surface is less clear from the perspective of electrostatic protein–surface interactions, but was suggested to be oriented away from R9 and R10 to optimize peptide electrostatics. This new data and previously reported data were incorporated into RosettaSurface to help predict the complete structure of the protein bound to the surface [44]. Using the existing constraints, RosettaSurface suggested new experimental constraints to obtain, and the combined data resulted in very nearly complete agreement between the computationally derived and NMR based structure (Fig. 3) [32].
Fig. 4.
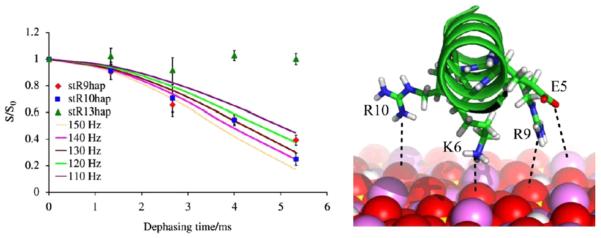
Left: REDOR dephasing curves for the 13Cζ labeled arginine in statherin to 31P in HAP collected under identical conditions. There are data points for each of the three different arginine positions: 9, 10 and 13. The lack of dephasing for R13 suggests that it is further from the surface than R9 and R10, which are ~4.5 Å. Right: Summary of the N-terminal charge groups surface interaction. Reproduced with permission from Ref. [40]. Copyright 2010 Elsevier.
Fig. 5.
SSNMR, NEXAFS, and SFG data suggest the following orientations of the two phenylalanines in the SN15 peptide of statherin. Reproduced with permission from Refs. [37] Copyright 2006, and 38 Copyright 2012 American Chemical Society.
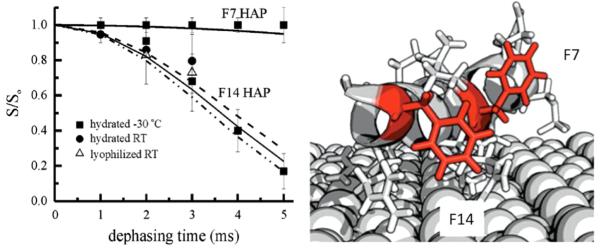
The dynamics of the phenylalanine rings in positions 7 and 14 provided critical insight further supporting the idea of a more complex interaction mechanism than one dominated merely by protein–surface electrostatics [37]. Both rings were found to have somewhat restricted motion, however the motion at F14 had a significantly longer correlation time, suggesting that this residue, further from the binding domain, was more restricted. This side chain of F14 was also found to be only ~4 Å from the surface, consistent with an interaction between the ring and the surface. In nicely complementary studies, Near Edge X-ray Absorption Fine Structure (NEXAFS) spectroscopy and sum frequency generation (SFG) provide data consistent with this interpretation [38]. The results from the study of F14 suggest that while the interaction in this region of the protein is weaker, there is still a preference, possibly entropic, for the protein to be associated with the surface. While much is left to be understood about the details of the statherin:HAP interface, SSNMR is critical for providing the molecular level details.
2.2. Amelogenin
Amelogenins are a family of proteins which are the predominant protein (>95%) present during enamel formation. The family consists of the full-length protein, a series of proteolytically cleaved products which are the result of processing during enamel development and maturation, and splice variants. LRAP is a 59 residue splice-variant of the full-length protein (Table 1). It consists of the first 33 and the last 26 residues from the parent protein. These two regions contain the only charged residues found in the full-length protein (13 total), and are associated with protein–protein and protein–crystal interactions [45–47]. As such, LRAP serves as an ideal model for quantitating the interaction of amelogenins with HAP, due to its appropriate length for solid phase peptide synthesis, as well as containing the important crystal binding regions.
Using the 13C–15N REDOR dipolar recoupling methodology and labeling schemes implemented with statherin, the structure and orientation of the N- and C-terminus of LRAP bound to HAP were determined. As seen in Table 1, the C-terminus contains most of the charged residues, and would be the likely region to form a strong interaction with HAP. Unlike the highly organized structure found for the HAP-binding region in statherin, this region was found to be largely extended by SSNMR, from L42 to V58 [48,49]. To determine whether or not this region was close enough to the surface to interact, 13C–31P distances from the backbone labeled 13C′ atoms at A46, A49, K54, and V58 to the nearest 31P residue were measured and found to be 7.0 Å, 6.0 Å, 6.5 Å, and 5.8 Å, respectively, positioning the C-terminus close enough to the surface to control HAP formation [48,49]. Distances from the side chains of A46 and K52 to the surface, 8.0 Å and 4.0 Å, respectively, provided further confirmation that this region of the protein is in contact with the surface [49,50]. The combination of the structure and orientation information is consistent with this region of the protein spread out on the surface to optimize electrostatic interactions, very similar to that initially observed by the Reimer group, but quite unlike that of statherin.
To further evaluate the interaction in the C-terminus, the dynamics of the backbone 13C′ were evaluated. In another inconsistency for the expectation of an important binding region, this region was found to be more dynamic than the binding region for statherin. Evidence of significant motion was gathered from analysis of the CSA (Fig. 6), and the T1ρ values which changed from ~20 ms for the frozen sample to 2.5–8.0 ms for the hydrated samples [49], bracketing the motion in the timescale of 10−3 to 10−5 s, similar to the fastest values found for statherin at I11, outside of the N-terminal acidic binding region [33]. At the time, this result was unexpected based on the splayed structure of this region, and the enhanced dynamics relative to statherin. An additional observation is that LRAP and statherin have similar affinity based on the Langmuir isotherm [51–53]. To achieve these apparent inconsistencies in protein–surface interaction, it was hypothesized that LRAP has another region in the protein providing structural stabilization. Since there are charged residues in the N-terminus, and it has been proposed to play a role in HAP binding [54], this region was also investigated.
Fig. 6.

NMR 13C spectra of LRAP of 13C′ labeled L42 (left) and A49 (right), frozen (−80 °C; top), room temperature, same conditions as frozen (middle), and room temperature with better signal to noise. Dynamics are observed in both the significant loss in CP efficiency and the averaging of the CSA, consistent with significant motion in this region of the protein. Reproduced with permission from Ref. [49]. Copyright 2008 CellPress.
The three charged residues in the N-terminus of LRAP are found at pS16, E18 and K24 (Table 1). Using 13C–15N REDOR, the regions from G8–V12, L15–V19, V19–L23 and K24–S28 were investigated, providing a nearly unbroken evaluation of the N-terminus from G12 to S28, with the exception of residues 13 and 14. The serine at residue 16 is phosphorylated in some cases and it is unclear if this plays a physiological role, therefore, the structure and orientation in this region were determined with and without phosphorylation. The N-terminus was found to be more helical than the C-terminus, but still loosely helical, and further away from the surface. It was still close enough to interact with the surface, with the backbone being 8–9 Å from the surface for the unphosphorylated protein. Interestingly, despite its distance from the surface, it was also less mobile than the C-terminal residues, suggesting protein–protein interactions restricting motion. Phosphorylation of S16 (+P) moves each of the backbone residues closer to the surface. This is particularly strong for L15, highlighting the impact of a single charged residue (Fig. 7). Phosphorylation does not significantly change the structure in most of this region, with the exception of the K24S28 region which transitioned from a loose helix or extended structure (5.7 Å) to a structure with significant helical character (4.8 Å; Fig. 8) [55,56]. This region was also nearly helical when it was lyophilized from solution. The observed turn from the surface was first suggested by RosettaSurface analysis to be an important residue [57] and the structural flexibility as a function of the preparation conditions suggests that this may be a critical region for function in this intrinsically disordered protein. Collectively, these results suggest that both the N- and the C-terminus are important in attaching the molecule to the surface. This is supported by independent Neutron Reflectivity data of LRAP bound to COOH-terminated SAMs showing a 3-D fold placing the N and C termini close to the surface. There are differences in the two interaction domains, reflected in both the differences in structure and distance from the surface, suggesting that there are different roles for the two regions that may have to do with additional physiological roles such as protein–protein interactions.
Fig. 7.
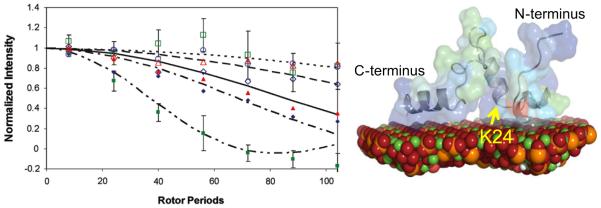
Left: 13C–31P REDOR dephasing curves showing the distance from various residues in the N-terminus to the surface of HAP, ranging from 5.3 (closed green squares) to 9 Å (open green squares), depending on location and phosphorylation state. G8 (open navy diamonds), G8(+P) (closed navy diamonds), L15 (open green squares), L15(+P) (closed green squares), V19 (open red triangles), V19(+P) (closed red triangles), K24 (open blue circles), and K24(+P) (closed blue circles), where +P indicates phosphorylation. Right: The structure derived using RosettaSurface from the SSNMR constraints. Residue K24 may be part of a key structural region, and is indicated. Spinning speed was 4 kHz. Reproduced with permission from Ref. [57]. Copyright 2011 American Chemical Society. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Fig. 8.

Left: The K24S28 region of LRAP is a highly sensitive structural region. This sensitivity can be observed in the 1D spectra which show multiple resonances for the 13C′ of K24 whose relative intensities change as a function of the conditions. Multiple resonances are only observed for this residue in the protein, further evidence of the structural uniqueness of this region. Right, top: The structural sensitivity is quantitatively shown with REDOR studies, with distances ranging from 4.2 Å for the unbound, lypohilized protein (the distance expected for an ideal helix), to 5.7 Å for the bound, unphosphorylated protein, a very extended or random coil structure. Bottom, right: Other conditions such as the Ca2+ concentration when binding, or binding to carbonated HAP instead of HAP, also influence the structure. Reproduced with permission from Ref. [55] and Ref. [56], Copyright 2013 American Chemical Society.
Further studies focused on trying to assess the role of solution conditions in controlling the binding interaction in the N-terminus, and revealed that both the structure and the orientation of LRAP bound to HAP is very stable to changes in pH (5.8, 8.0) and to ionic strength (0.05 M, 0.2 M), relative to the standard binding conditions (pH=7.4, IS=0.15 M, binding surface HAP, protein concentration 300μg/mL) with the most significant change observed for V19L23 at an ionic strength of 0.2 M (from ~6 Å to 8.3 Å) [56]. Because of the high sensitivity of the K24S28 region to phosphorylation, this region was studied as a function of additional variables relevant to enamel formation: excess calcium, the carbonation level of hydroxyapatite and a mutation at T21 (to isoleucine) [55]. This region did show sensitivities to conditions, with the most significant being a tighter helix when bound in the presence of an 8:1 ratio of calcium to protein, or when binding to carbonated apatite (Fig. 8). In the presence of excess calcium, this region was also displaced slightly from the surface compared to standard conditions. The orientation in the K24S28 region changed as a function of the T21I mutation, although the structure was unchanged. Further correlation of growth and inhibition data with the solution conditions is needed to fully evaluate the mechanism of the LRAP-HAP interaction. What is clearly arising from these results is the structural flexibility of the K24S28 region as a function of the changing conditions during enamel development, suggesting a significant role of this region in the interaction mechanism, and also providing evidence that surface induced structural changes are an important component of the biomineralization interface and likely play an important physiological role.
2.3. Two-dimensional approaches for HAP binding proteins
The quantitative structural data for both statherin and LRAP have provided critical insights into a previously unexplored field, however, the almost exclusive use of isolated spin pairs, and the low signal-to-noise results in long experiment times to collect data for a single sample (on the order of 1–2 weeks for a dephasing curve, depending on the distance), and the need for many expensive samples, along with time and solvents for clean-up. Solving protein structures with solution state NMR is done with a single, uniformally 13C, 15N labeled sample. This is possible due to the high resolution achievable with a well-structured protein undergoing isotropic tumbling. Full structural characterization of proteins in the solid state has realized significant advancements in the past 10 years, with 2D- and 3D-techniques providing not only full assignments, but complete structures [58–60]. One of the key advancements in achieving these impressive results has been sample preparation which initially involved the preparation of the micro-crystalline proteins, and while this has subsequently been found not to be essential, sample preparation to result in narrow lines continues to be important to obtaining high quality data. The advances and success in this area for non-biomineralization proteins makes application of these techniques to biomineralization proteins a reality. Depending on the linewidths that are achievable for these proteins interacting with highly charged and heterogeneous surfaces, a partial labeling scheme may need to be employed. However, significant cost and time savings should be realized, even if only a partial labeling scheme is used. Perhaps even more important is that larger proteins can be utilized because the use of multiple labels allows their incorporation via recombinant expression techniques.
There are added challenges in studying biomineralization proteins compared to standard proteins that need to be considered in moving to 2D- and 3D SSNMR techniques. One of the problems is the charged surface with which the protein is interacting. This can result in chemical shifts that do not fall in line with those typically expected for a particular secondary structure, values which are reasonably well defined in solution state or for proteins in the solid state that are not intimately associated with an electron rich surface [61]. Broad lines are also often observed. This may be due to anisotropic magnetic susceptibility, as proposed for statherin [35], or the interaction of the protein with the surface may also have some heterogeneity, where the protein may interact with steps as well as planes, causing structural or interaction dispersion. These details have yet to be completely sorted out, but the net result is reduced resolution, complicating data interpretation of a uniformally labeled protein bound to a surface [14,62]. To approach this problem in initial studies, only select underrepresented residues are labeled to tease out mechanistic details, a valuable approach that is yielding critical insight.
Two-dimensional methods were first implemented on the SN15 peptide of statherin [63]. To achieve higher protein to HAP ratios, Chan and his coworkers co-precipitated SN15 and HAP, a different approach than used in the previous studies by the Drobny group where statherin was bound to preformed HAP crystals. However, the approach is physiologically relevant based on the function of the protein to inhibit both primary and secondary precipitation. The 13C–13C correlation spectrum, with polarization transfer achieved with RFDR, revealed that K6 had two resonances upon binding, suggesting two different structures, while G12 had only one structure (Fig. 9). This suggests structural heterogeneity uniquely at K6 that was not observed in studies of protein bound directly to HAP. Given the different nature of the studies, it is difficult to say if the difference is due to the higher resolution obtained in the second dimension or if it is unique to the sample preparation. Unfortunately, the signal-to-noise of statherin preadsorbed to HAP was inadequate to allow the same analysis. However, the important result is the clear distinction of multiple structures and highlights the power of 2D techniques in distinguishing between different structures much more easily than in 1D spectra.
Fig. 9.

13C–13C two-dimensional techniques applied to statherin’s SN15 peptide labeled at K6 and G12 precipitated from HAP at pH=9 (blue) and lyophilized from solution (red) reveal two unique structures at K6 (left hand resonance in the box) and only one for G12. Reproduced with permission from Ref. [63]. Copyright 2008 American Chemical Society. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The first example of using 2D approaches on a full length biomineralization protein was demonstrated for full length amelogenin [62]. The uniformally labeled protein was investigated initially in both the nanosphere form, a functional form of amelogenin which consists of a self-assembly of 20–100 monomers [2], as well as bound to HAP. Unfortunately, in both sample preparations, the overlap was extensive due to both broad lines as well as the intrinsically disordered nature of the protein. Due to this overlap, a minimal labeling scheme which involved only the lysines was chosen [62]. Lysine is an underrepresented residue in amelogenin, having only three (Table 1), and is also likely to interact with the phosphate groups in the HAP surface, therefore obtaining important insights for the amelogenin-HAP interface were likely with this labeling scheme. The 13C–13C correlation spectrum was collected with either cross polarization (CP) or direct pulse (DP) to prepare the magnetization, followed by dipolar-assisted rotational resonance (DARR) mixing. The nanosphere sample was found to be highly dynamic and unstructured, based on the difficulty of obtaining data even at reduced temperatures and based on the chemical shift values observed (Fig. 10). The immobilized sample was prepared by co-precipitating HAP and amelogenin, a method developed by others and thought to be important in the initiation of crystallization in vivo, and the protein in this case was markedly different. Even at room temperature, high quality spectra were obtained with two lysine resonances observed. These two resonances could be the result of two of the three lysines being in a different environment than the other one, a reasonable hypothesis based on the location of two of the lysines in the C-terminus and one in the N-terminus, regions that have been shown in LRAP to interact differently with the surface. It could also be that all of the lysines in a given protein experience a similar environment, but that the proteins themselves are in two different environments. TEM studies of the crystals, along with the combination of the NMR data in nanospheres and mineralized with HAP suggest that what we are observing is the protein in two different environments, those of an intimate interaction with the surface and an overlayer of protein associated with the crystal–protein complex. Based on the chemical shifts, the lysines in the protein–crystal complex have a β-strand structure and have restricted motion, while those in the protein overlayer have a random coil structure and are very dynamic, similar to that found in the nanosphere. Interestingly, the chemical shifts of the majority peak in the nanosphere sample and the minority peak in the mineralized sample are identical. Further inspection of the nanosphere sample reveals a small contribution from a downfield shifted resonance that has an identical chemical shift to the major peak in the mineralized sample. These observations are consistent with two structures in equilibrium, with the surface interaction stabilizing the β-strand structure. To move this forward, other under-represented residues will be evaluated to provide a more complete understanding of the interface of this critical protein in developing enamel.
Fig. 10.

The 2D 13C–13C correlation spectrum of M180-K, using CP or DP, followed by DARR mixing: Top, in a nanosphere gel using CP, at −35 °C. The poor resolution and signal are all indicative of a highly mobile, unstructured protein. Bottom, bound to calcium phosphate crystals, at 37 °C. CP (red) and DP (blue). The black (β-strand) and green (random coil) traces are shown to distinguish the two structures. Reproduced with permission from Ref. [62]. Copyright 2013 Sage publications. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
2.4. Comparisons of HAP binding proteins
Statherin and LRAP are the two best studied biomineralization proteins to date and provide an opportunity for identifying design principles. In common, they both have structural changes upon binding that have a proposed physiological function. The differences between these two proteins are more numerous. For instance, statherin has a well-defined helix in its single binding region which positions some of the charged residues next to the surface and some away. In contrast, LRAP has two binding regions, neither of which is highly structured. While side chain to surface distances have not been measured for LRAP, the loosely helical or extended structures and fairly constant distance of the backbone from the surface suggests that the protein is oriented to align all of the charged groups near the surface. Statherin has a very rigid interaction with the surface, while LRAP has a significant amount of motion in the binding regions. The differences observed in the interaction are likely directly related to the primary structure which for statherin has five negatively charged residues followed by a positively charged residue. The charged residues in LRAP are more dispersed. For instance, the highest density is in the C-terminus with six charged residues out of nine, with a near balance in charge (three positive and four negative residues). The rest of statherin has only one charged residue, E26, and has a highly Q and P rich region. LRAP is rich in Q and P as well, residues commonly observed in IDP’s [64]. The presence of glutamine residues may be to facilitate interaction with the surface through amide hydrogen bonding. This would likely be a weaker interaction that may facilitate other functional roles of these proteins. The significant differences in these two calcium phosphate binding proteins emphasize the need for the study of more biomineralization proteins, both HAP binding and otherwise, real systems as well as model systems. Only with more data will we be able to extract common design strategies that will allow the development of novel materials that have the exquisite properties of those found in nature.
3. Studies with silicates
The study of amino acids and proteins with silica surfaces has started much more recently, with the majority of the work reported in the past five years. Diatoms are made from silica and exist with an incredible diversity of structure that has been taken advantage of for in vitro preparations [65]. Two distinct advantages exist for studying protein–silica interactions: surface areas of silica can be much higher, up to 1000 m2/g, and structures can be highly ordered. The combination of these two features results in much higher signals, and potentially more homogeneous lineshapes.
Initial studies of silica biomineralization have focused on single amino acids (alanine, methionine or glycine) [66–70] or up to four residue repeats (dianaline [67] and tetraalanine [68]). As with calcium phosphate biominerals, REDOR measurements (15N–29Si, 29Si–15N, and 15N–13C) were used to determine orientation. Detailed amino acid dynamics and disassociation were also evaluated with 1H and 2H NMR methods. Not surprisingly, fundamentally different results are observed for the interaction between amino acids and silica than for calcium phosphates. The backbone is found to interact very specifically with three to four silicon atoms, while the carboxylate group is freely rotating under low and high hydration conditions, suggesting that it plays no role in the interaction. High hydration levels result in disassociation, the kinetics of which has been measured (Fig. 11) [66,67]. In the most recent study, binding of glycine to SBA-15 and MCM-41 was compared [71]. The interaction with MCM-41 was found to be much weaker, determined to be due to an interaction with fewer silanols (Fig. 12). MCM-41 has a smoother surface, fewer Q2 and Q3 silanols, and non-connecting pores. The relative binding strengths suggest that the silanols may also be structured differently, all of which results in a weaker interaction. As studies of larger proteins are undertaken, it will be interesting to see the role of the backbone and other functional groups in associating with the surface.
Fig. 11.

1H (left) and 2H (right) MAS studies of ala-d3 dissolved in water inside SBA-15 after different drying periods. The p value (left) is representative of the water content (H/nm2), with larger values indicating higher water content. The spectra show the dramatic increase in dynamics with increasing water. Reproduced with permission from Ref. [66]. Copyright 2009 American Chemical Society.
Fig. 12.
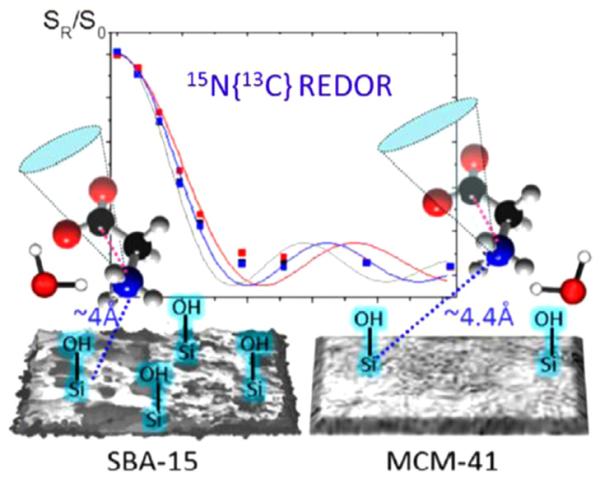
Differences in the interaction of glycine with two types of silica, SBA-15 and MCM-41, were observed using REDOR and dynamic measurements. Reproduced with permission from Ref. [71]. Copyright 2014 American Chemical Society.
More recently, Roehrich and Drobny reported the study of a peptide fragment of silaffin bound to SiO2 prepared by precipitation from solution in the presence of silicic acid [14]. This is the first demonstration of a silica-based protein bound to silica, and they take advantage of the high surface area, relatively narrow lines, and 2D DARR techniques to study selectively labeled samples, with uniformally 13C, 15N labeled residues being introduced three at a time, limiting the number of samples to seven for the 19 amino acid peptide (Table 1; Fig. 13). They saw significant shifts (5 ppm or more) for 11 spins in the backbone and side chains, with the Cγ at R17 and the Cβ at S5 shifting by 10 ppm. These changes are dramatic compared to the more subtle changes observed for LRAP bound to HAP (up to 2 ppm) and suggest significantly different environments when the protein is interacting with SiO2. There wasn’t consistency in the chemical shifts of each type of amino acid (for instance the other serines and arginines did not display these surprising shifts), suggesting a specific interaction that goes beyond mere electrostatic stabilization. Also of interest is that the lysine residues were not the most strongly affected residues based on chemical shifts, despite the observation that for alanine, the interaction with was the primary interaction. While a complete structural analysis of the chemical shift data was also inconclusive, they hypothesize that silaffin forms micells with the C-terminus at the core of the micelle and the N-terminus interacting with the silica surface. More studies will likely follow from this very promising initial study of biomineralization proteins of SiO2 which highlights both the potential of the 2D SSNMR techniques in revealing structural features of surface immobilized proteins, as well as the potential for the silica based biominerals to serve as models for protein–surface interactions.
Fig. 13.
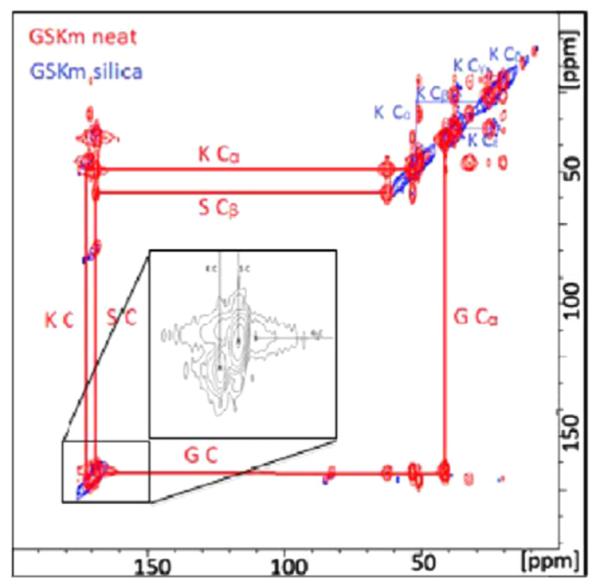
13C–13C DARR spectrum of the R5 silaffin peptide uniformally labeled at G10, S11, and K12 residues. Neat (red) and incorporated into silica (blue). Reproduced with permission from Ref. [14]. Copyright 2013 American Chemical Society. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Studies of the influence of a model peptide on the resulting morphology of silica, and the associated NMR studies have also been reported [72]. A very tight concentration dependence was observed (2–3 mM) to obtain silica nanospheres of 500–800 nm, similar to those observed with the R5 peptide. 1D and 2D SSNMR methods were once again used on doubly labeled samples to investigate structure via chemical shift. Compared to R5, the chemical shift deviations were much smaller (in all cases less than 5 ppm, typically under 1 ppm) from the neat peptide to the peptide in silica, and the peptide maintained a helical structure in both cases. However there was a clustering of leucine residues that had a larger chemical shift than the others, and correlated well with those that would be exposed if the tetrameric complex reported for LK peptides was the structural form of the LK peptide interacting with silica (Fig. 14). While not perfect, this general correlation suggests that the tetrameric structure plays an important role in silica nanosphere formation. The results also allow the authors to speculate that ions are interacting with the protein to assist in the biomineralization process.
Fig. 14.

The interaction of the model LK peptide uses the hydrophobic and hydrophilic regions upon formation of the tetrameric structure to control silica growth. The differences in chemical shift between exposed residues is much larger than for the buried residues for the neat peptide and the silica, as would be expected for such a significant change in environment. Reproduced with permission from Ref. [72]. Copyright 2014 American Chemical Society.
4. Studies with calcium carbonates
The organic matrix controlling the formation of calcium carbonates has been studied in much less detail, where the focus to date has been on the inorganic matrix. In one study, the 13C CPMAS NMR of the chitin matrix, remaining after solubilizing the amorphous calcium carbonate of a crayfish gastrolith, was reported [73]. In relaxation and HETCOR studies of aragonite tablets of nacre from abalone Haliotis laevigata, the organic matrix was found not to interact with the aragonite layer, although it is possible that it is interacting with the amorphous calcium phosphate layer, which was proposed to surround the aragonite tablets [74].
In other studies, coccoliths from Emiliania huxleyi were grown in 13C and 15N enriched media to facilitate NMR studies [75]. In this impressive study on an actual biomineral, the combination of CP and DP 13C–15N and 13C–31P REDOR studies (Fig. 15) suggest that the protein is not interacting closely with the crystalline calcium carbonate sites, based on two observations: the dephasing of the carbonate peak in the DP spectrum (Fig. 15), and the significantly reduced signal intensity of the calcium carbonate resonance in the CP spectrum (not shown). If the protein were in intimate contact and causing the dephasing observed in the 13C–15N REDOR, broader lines would be expected, as well as a significant CP enhancement from the enrichment of protons in the area. The dephasing observed is proposed to be due to anionic N and P inorganic molecules incorporated into the lattice. Further studies of the proteins and other organic components directing calcium carbonate biomineralization would provide an important counterpoint to the calcium phosphate studies and have the potential to significantly broaden our understanding of common, as well as unique, themes for the development of hard tissues across the different inorganic materials.
Fig. 15.
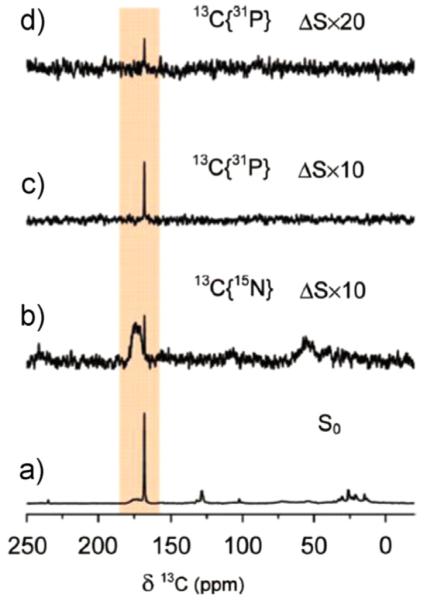
13C DP MAS REDOR at 16 rotor periods (spinning speed 5 kHz) of intact coccolith from E. huxleyi (20 s relaxation delay): (a) So reference spectrum, (b) 13C–15N ΔS spectrum, (c) 13C–31P ΔS spectrum, and (d) 13C–31P ΔS spectrum with 40 s relaxation delay. The sharp calcium carbonate peak (168.6 ppm) dephases due to 31P and 15N, likely due to small inorganic anions and not due to proximity to a protein. Reproduced with permission from Ref. [75]. Copyright 2008 American Chemical Society.
5. Outlook
In summary, SSNMR has had a critical impact in revealing the structure and interaction of the proteins controlling biomineralization. The results of the two most well-studied proteins suggest very different interaction mechanisms, despite the fact that they are both interacting with HAP. When comparing the observations for these proteins with the initial observations made for proteins bound to silica and calcium carbonate, differences are notable. Many postulates can be made as to the source of the different mechanisms, but what is really needed is the study of additional systems at a detailed and quantitative level so that broader rules can be established. Ultimately, the fundamental understanding we learn from natural systems should help to guide and inspire the development of advanced materials with unique properties and the control of surfaces in complex environments such as seawater or the human body. The advancement in our understanding of hard tissues has been phenomenal, and SSNMR has played a critical role in the development of that understanding. However, our understanding is still in its infancy, and some of the more recent advancements position NMR well to continue to progress that understanding as we move forward.
One of the primary techniques that can facilitate the understanding of biomineralization proteins is the 2D- and 3D-techniques that are just beginning to be applied to this class of proteins. Already these techniques are providing unique and important structural insights that would have taken significantly longer or would not have been achieved using the isolated spin pairs utilized for dipolar recoupling methods. As methodology in this area is developed, the biomineralization community will benefit. In the application of these techniques, sample preparation is critical; however, it is essential that care is taken to prepare the sample under conditions that still provide some physiological relevance. Studying biomineralization proteins under physiologically relevant conditions has been a hallmark of implementing SSNMR methodology to these proteins, and trying to maintain that while preparing samples with high resolution will be important. Other SSNMR advancements that have a high likelihood of enhancing our ability to study biomineralization proteins include techniques that enhance signal to noise, such as hyperpolarized xenon [76], or dynamic nuclear polarization (DNP), a technique which has seen significant advancements in recent years, along with the availability of nearly off the shelf instrumentation [77]. Samples that have resonances narrow enough to benefit from enhanced resolution will benefit from ever higher field strengths and ultra-fast MAS.
Computational techniques such as RosettaSurface as well as others [43,78,79] have provided significant advancements in visualizing proteins on the surface as well as providing insight and guiding experimental studies to investigate residues more likely to impact biomineralization processes [32,36,57]. Continuing to develop computational techniques is essential, as their current limitations are significant. For example, the structure and orientation of the 43-amino acid residue statherin bound to HAP could be predicted without using NMR constraints, while the 59-residue LRAP bound to HAP could not [32,57]. Larger biomineralization proteins such as amelogenin are beyond reach with the current methodology, but with experimental constraints, computational methods still provide extremely valuable visualization. Much could be learned from model systems in combined NMR and computational studies, and could supplement studies of real systems, providing avenues for testing certain hypotheses under more controlled conditions. Additional parameters that would be useful for comparison to experiment are controlling pH and ionic strength. As the number of experimentally derived constraints increase, the ability to develop the methods will also increase.
In terms of predicting which residues to study, this has often been empirically chosen, based on the hypothesis that charged residues control the surface interaction. While there is still evidence for this, the NMR evidence for calcium phosphates suggests that this is not a simple picture and the interaction may vary, depending on the primary and secondary structure and likely other structural features that are not yet clearly defined. The NMR data for calcium carbonate proteins also implies a limited interaction of the organic matrix with the crystal [74], further supporting ambiguity in this interaction. An additional development also suggests that looking outside the charged residues might be appropriate. Recent phage display techniques are able to evaluate a large number of sequences and isolate the sequences that interact most strongly. This is performed in a three step process of first determining which are the most strongly binding, screening these several times to narrow the field down to a smaller number of the most tightly binding, and then down selecting those based on a structure prediction study. Results in independent groups suggest that the charged residues are not the strongest to interact with HAP, but that polar, uncharged residues such as histidine, glutamine, tyrosine, and serine provide the strongest interactions [80–82]. As surprising as this is, and while there are places for error in this methodology, what is clear is that the resulting peptides from these studies strongly influence inorganic phase and morphology. These results suggests that we need to continually evaluate our hypotheses when approaching new biomineralization systems and perhaps revisit old systems to further evaluate and understand the protein–inorganic interface.
In addition to computational methods, the coupling of SSNMR with other methods has already been demonstrated to be powerful, and an interdisciplinary approach will likely continue to provide the most insight. Techniques such as SFG, neutron reflectivity, SEM/TEM, and NEXAFS have provided beautiful complements to support or clarify SSNMR data. With a developing field such as the application of SSNMR to the study of biomineralization proteins, this kind of interaction is not only useful, but critical in defining how we interpret results and develop mechanistic understanding. Along these lines, a theme that is becoming more common is the idea of an amorphous precursor phase preceding the crystalline phase or playing a role in the mineralization process [43,83–85]. As already demonstrated in some areas, NMR can be a powerful method for evaluating the crystalline and amorphous phases [16,21,22,24,25,86], in a way that can be missed by other techniques. Reevaluating NMR and computational approaches with these less well defined surfaces may result in different methods to investigate protein–inorganic interfaces.
A final aspect of protein–surface interactions to mention is the role of water. Several studies with statherin suggest a very important role. These include: (1) the N-terminal helix stabilization due to water [33], (2) the similar dynamics of the side chain of glutamic acid although one interacts with the surface and one does not [41], and (3) in demonstrating the displacement of water when statherin binds [87]. The work of Vega and Schmidt have addressed water more directly and rigorously in studies of amino acids interacting with silica, [66–70,88] and there is no doubt that water will play a key role in the interaction mechanism of these highly charged/polar surfaces and highly charged/polar proteins [66–70,88]. Unfortunately, the role of water has not received as much attention as would be appropriate and many questions are outstanding. Questions such as how much water is in the interface (i.e. is there a monolayer, a multilayer, or is water completely displaced), what is the result of removing water that is found in the natural system (i.e. structural change, loss in function), and how does this affect other surface–organic interactions such as soft polymers with hard polymers. Understanding these and other questions relating to the impact of water on the interaction will be critical in the development and control of materials and hard tissues that mimic those found in nature. SSNMR and computational methods will be critical in evaluating this role.
Acknowledgments
This review and the authors work herein was supported by NIH-NIDCR Grant DE-015347. The authors research reviewed in this work was performed at the Pacific Northwest National Laboratory (PNNL), a facility operated by Battelle for the U.S. Department of Energy, with a portion of it performed at the W.R. Wiley Environmental Molecular Sciences Laboratory (EMSL), a national scientific user facility sponsored by the U.S. DOE Biological and Environmental Research program.
Biography

Wendy Shaw received her Ph.D. at the University of Washington, Seattle, WA, under the supervision of Prof. Gary Drobny. Since 2000, she has been at Pacific Northwest National Laboratories in Richland, WA. Her research interests revolve around learning from nature. She studies biomineralization proteins using solid state NMR, physical chemistry methods, neutron reflectivity and solution state NMR. She also develops and studies catalysts for renewable energy applications by developing peptide-derived biomimetic electrocatalysts, studies which involve solution state NMR structure and kinetics studies.
References*
* Article of general interest.
- [1].Lowenstam HA, Weiner S. On Biomineralization. Oxford University Press; New York: 1989. [Google Scholar]
- [2].Moradian-Oldak J, Paine ML, Lei YP, Fincham AG, Snead ML. J. Struct. Biol. 2000;131:27–37. doi: 10.1006/jsbi.2000.4237. [DOI] [PubMed] [Google Scholar]
- [3].Paine ML, Lei Y-P, Dickerson K, Snead ML. J. Biol. Chem. 2002;277:17112–17116. doi: 10.1074/jbc.M110473200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Dunker AK, Brown CJ, Lawson JD, Iakoucheva LM, Obradovic Z. Biochemistry. 2002;41:6573–6582. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- [5].Pines A, Gibby G, Waugh JS. J. Chem. Phys. 1973;59:569–590. [Google Scholar]
- [6].Stejskal EO, Schaefer J. J. Magn. Reson. 1977;28:105–112. [Google Scholar]
- [7].Nielsen NC, Strassoe LA, Nielsen AB. Top. Curr. Chem. 2012;306:1–45. doi: 10.1007/128_2011_129. [DOI] [PubMed] [Google Scholar]
- [8].Gregory DM, Mitchell DJ, Stringer JA, Kiihne S, Shiels JC, Callahan J, Mehta MA, Drobny GP. Chem. Phys. Lett. 1995;246:654–663. [Google Scholar]
- [9].Gullion T, Schaefer J. J. Magn. Reson. 1989;81:196–200. [Google Scholar]
- [10].Carpino LA, Han GY. J. Org. Chem. 1972;37:3404–3409. [Google Scholar]
- [11].Drobny GP, Long JR, Karlsson T, Shaw W, Popham J, Oyler N, Bower P, Stringer J, Gregory D, Mehta M, Stayton PS. Annu. Rev. Phys. Chem. 2003;54:531–571. doi: 10.1146/annurev.physchem.54.011002.103903. [DOI] [PubMed] [Google Scholar]
- [12].Drobny GP, Long JR, Shaw WJ, Cotten M, Stayton PS. Structure and Dynamics of Proteins Adsorbed to Biomaterial Interfaces. Vol. 9. John Wiley & Sons Ltd.; 2002. [Google Scholar]
- [13].Drobny GP, Stayton PS, Long JR, Louie EA, Karlsson T, Popham JM, Oyler NA, Bower PV, Shaw WJ. Structural Studies of Peptides on Biomaterial Surfaces Using Double-quantum Solid-state Nuclear Magnetic Resonance Spectroscopy. CRC Press LLC; 2006. [Google Scholar]
- [14*].Roehrich A, Drobny G. Acc. Chem. Res. 2013;46:2136–2144. doi: 10.1021/ar300321e. (This is the first example of SSNMR studies of a silica derived peptide bound to silica. Unlike proteins bound to HAP, the chemical shift differences of R5 neat and bound were substantial (up to 10 ppm), suggesting significant structural and interaction changes upon binding).
- [15].Wu Y-J, Tsai TWT, Huang S-J, Mou Y, Lin C-J, Chan JCC. Langmuir. 2013;29:11681–11686. doi: 10.1021/la402392b. [DOI] [PubMed] [Google Scholar]
- [16].Tsai TWT, Chou F-C, Tseng Y-H, Chan JCC. Phys. Chem. Chem. Phys. 2010;12:6692–6697. doi: 10.1039/b923338e. [DOI] [PubMed] [Google Scholar]
- [17].Ghindes-Azaria L, Levy E, Keinan-Adamsky K, Goobes G. J. Phys. Chem. C. 2012;116:7442–7449. [Google Scholar]
- [18].Chen W-Y, Yang C-I, Lin C-J, Huang S-J, Chan JCC. J. Phys. Chem. C. 2014;118:12022–12027. [Google Scholar]
- [19].Goobes G. Isr. J. Chem. 2014;54:113–124. [Google Scholar]
- [20].Tsai TWT, Chen W-Y, Tseng Y-H, Chan JCC. Can. J. Chem. 2011;89:885–891. [Google Scholar]
- [21].Tseng Y-H, Mou C-Y, Chan JCC. J. Am. Chem. Soc. 2006;128:6909–6918. doi: 10.1021/ja060336u. [DOI] [PubMed] [Google Scholar]
- [22].Tseng Y-H, Mou Y, Chen P-H, Tsai TWT, Hsieh C-I, Mou C-Y, Chan JCC. Magn. Reson. Chem. 2008;46:330–334. doi: 10.1002/mrc.2096. [DOI] [PubMed] [Google Scholar]
- [23].Tseng Y-H, Tsai Y-L, Tsai TWT, Chao JCH, Lin C-P, Huang S-H, Mou C-Y, Chan JCC. Chem. Mater. 2007;19:6088–6094. [Google Scholar]
- [24].Tseng Y-H, Zhan J, Lin KSK, Mou C-Y, Chan JCC. Solid State Nucl. Magn. Reson. 2004;26:99–104. doi: 10.1016/j.ssnmr.2004.06.002. [DOI] [PubMed] [Google Scholar]
- [25].Tsai TWT, Chan JCC. Annu. Rep. NMR Spectrosc. 2011;73:1–61. [Google Scholar]
- [26].Fernandez VL, Reimer JA, Denn MM. J. Am. Chem. Soc. 1992;114:9634–9642. [Google Scholar]
- [27].Schlesinger DH, Hay DI. J. Biol. Chem. 1977;252:1689–1695. [PubMed] [Google Scholar]
- [28].Raj PA, Johnsson M, Levine MJ, Nancollas GH. J. Biol. Chem. 1992;267:5968–5976. [PubMed] [Google Scholar]
- [29].Stayton PS, Drobny GP, Shaw WJ, Long JR, Gilbert M. Crit. Rev. Oral Biol. Med. 2003;14:370–376. doi: 10.1177/154411130301400507. [DOI] [PubMed] [Google Scholar]
- [30].Goobes G, Goobes R, Shaw WJ, Gibson JM, Long JR, Raghunathan V, Schueler-Furman O, Popham JM, Baker D, Campbell CT, Stayton PS, Drobny GP. Magn. Reson. Chem. 2007;45:S32–S47. doi: 10.1002/mrc.2123. [DOI] [PubMed] [Google Scholar]
- [31].Goobes G, Stayton PS, Drobny GP. Prog. Nucl. Magn. Reson. Spectrosc. 2007;50:71–85. doi: 10.1016/j.pnmrs.2006.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32*].Masica DL, Ash JT, Ndao M, Drobny GP, Gray JJ. Structure (Cambridge, MA, U. S.) 2010;18:1678–1687. doi: 10.1016/j.str.2010.09.013. (This paper represents the first reported collaboration between SSNMR and theory and demonstrates the potential power and time savings of a combined approach).
- [33].Long JR, Shaw WJ, Stayton PS, Drobny GP. Biochemistry. 2001;40:15451–15455. doi: 10.1021/bi010864c. [DOI] [PubMed] [Google Scholar]
- [34].Shaw WJ, Long JR, Campbell AA, Stayton PS, Drobny GP. J. Am. Chem. Soc. 2000;122:7118–7119. [Google Scholar]
- [35].Shaw WJ, Long JR, Dindot JL, Campbell AA, Stayton PS, Drobny GP. J. Am. Chem. Soc. 2000;122:1709–1716. [Google Scholar]
- [36*].Goobes G, Goobes R, Schueler-Furman O, Baker D, Stayton PS, Drobny GP. Proc. Natl. Acad. Sci. U.S.A. 2006;103:16083–16088. doi: 10.1073/pnas.0607193103. (This paper reports the first biomineralization study with a labeling scheme incorporating more than two spins, resulting in multiple intraprotein distances extrapolated from a single sample. Using Rosetta to design the labeling scheme facilitated the measurement of tertiary structure, and demonstrated the power of utilizing computational approaches to guide experimental design).
- [37*].Gibson JM, Popham JM, Raghunathan V, Stayton PS, Drobny GP. J. Am. Chem. Soc. 2006;128:5364–5370. doi: 10.1021/ja056731m.; This excellent fundamental study on the dynamics of the phenyl rings had the surprising result that the phenylalanine further from the binding region was interaction more strongly. In addition to conclusions specific to statherin, it points to the preference for proteins to be near the surface, whether they are strongly interacting or not.
- [38].Weidner T, Dubey M, Breen NF, Ash J, Baio JE, Jaye C, Fischer DA, Drobny GP, Castner DG. J. Am. Chem. Soc. 2012;134:8750–8753. doi: 10.1021/ja301711w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Gibson JM, Raghunathan V, Popham JM, Stayton PS, Drobny GP. J. Am. Chem. Soc. 2005;127:9350–9351. doi: 10.1021/ja050910m. [DOI] [PubMed] [Google Scholar]
- [40*].Ndao M, Ash JT, Stayton PS, Drobny GP. Surf. Sci. 2010;604:L39–L42. doi: 10.1016/j.susc.2010.02.026. (This study used 13C-31P REDOR to analyze the importance of basic residues in binding to HAP, a first of its kind, which shows that two of the three arginine residues in statherin are closely associated to HAP. This is consistent with biophysical studies that show that the basic residues contribute to the affinity of statherin for HAP).
- [41].Ndao M, Ash JT, Breen NF, Goobes G, Stayton PS, Drobny GP. Langmuir. 2009;25:12136–12143. doi: 10.1021/la901647n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Hunter GK. Curr. Opin. Solid State Mater. Sci. 1996;1:430–435. [Google Scholar]
- [43].Hunter GK, O’Young J, Grohe B, Karttunen M, Goldberg HA. Langmuir. 2010;26:18639–18646. doi: 10.1021/la100401r. [DOI] [PubMed] [Google Scholar]
- [44].Masica DL, Gray JJ. Biophys. J. 2009;96:3082–3091. doi: 10.1016/j.bpj.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Moradian-Oldak J, Jimenez I, Maltby D, Fincham AG. Biopolymers. 2001;58:606–616. doi: 10.1002/1097-0282(200106)58:7<606::AID-BIP1034>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- [46].Moradian-Oldak J, Tan J, Fincham AG. Biopolymers. 1998;46:225–238. doi: 10.1002/(SICI)1097-0282(19981005)46:4<225::AID-BIP4>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- [47].Buchko GW, Tarasevich BJ, Bekhazi J, Snead ML, Shaw W. J. Biochem. 2008;47:13215–13222. doi: 10.1021/bi8018288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Shaw WJ, Ferris K. J. Phys. Chem. B. 2008;112:16975–16981. doi: 10.1021/jp808012g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Shaw WJ, Ferris K, Tarasevich B, Larson JL. Biophys. J. 2008;94:3247–3257. doi: 10.1529/biophysj.107.119636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Shaw WJ, Campbell AA, Paine ML, Snead ML. J. Biol Chem. 2004;279:40263–40266. doi: 10.1074/jbc.C400322200. [DOI] [PubMed] [Google Scholar]
- [51].Johnsson M, Richardson CF, Bergey EJ, Levine MJ, Nancollas GH. Arch. Oral Biol. 1991;36:631–636. doi: 10.1016/0003-9969(91)90014-l. [DOI] [PubMed] [Google Scholar]
- [52].Raj PA, Johnsson M, Levine MJ, Nancollas GH. J. Biol Chem. 1992;267:5968. [PubMed] [Google Scholar]
- [53].Bouropoulos N, Moradian-Oldak J. Calcif. Tissue Int. 2003;72:599–603. doi: 10.1007/s00223-002-1099-1. [DOI] [PubMed] [Google Scholar]
- [54].Aoba T, Moreno EC, Kresak M, Tanabe T. J. Dent. Res. 1989;68:1331–1336. doi: 10.1177/00220345890680090901. [DOI] [PubMed] [Google Scholar]
- [55].Lu J.-x., Burton SD, Xu YS, Buchko GW, Shaw WJ. Front. Physiol. 2014;5:1–8. doi: 10.3389/fphys.2014.00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56*].Lu J.-x., Xu YS, Shaw WJ. Biochemistry. 2013;52:21996–22205. doi: 10.1021/bi400071a.; This paper is one of the few times in the biomineralization literature that a single residue is observed to have multiple resonances, indicating two unique structures that are not being averaged at room temperature. It enforces previous assertions that this is an important turn region and provides the proposal of new mechanisms of interaction between proteins and surfaces.
- [57*].Masica DL, Gray JJ, Shaw WJ. J. Phys. Chem. C. 2011 doi: 10.1021/jp202965h. (The second combined study of SSNMR and theory provides the intial insight into the potential importance of residue K24 as a turn region. This study also reveals some of the computational challenges with these complex systems, with the inability to reproduce the SSNMR constraints).
- [58].McDermott AE. Encycl. NMR. 2012;8:4871–4878. [Google Scholar]
- [59].Comellas G, Rienstra CM. Annu. Rev. Biophys. 2013;42:515–536. doi: 10.1146/annurev-biophys-083012-130356. [DOI] [PubMed] [Google Scholar]
- [60].Tang M, Comellas G, Rienstra CM. Acc. Chem. Res. 2013;46:2080–2088. doi: 10.1021/ar4000168. [DOI] [PubMed] [Google Scholar]
- [61].Spera, Bax J. Am. Chem. Soc. 1991;113:5490. [Google Scholar]
- [62*].Lu J, Xu YS, Buchko GW, Shaw WJ. J. Dent. Res. 2013;92:1000–1004. doi: 10.1177/0022034513504929.; This paper is the first example of 2D 13C-13C correlation techniques being applied to a biomineralization protein. The results show a significant increase in structure upon association of amelogenin with HAP, and also demonstrate that 2D correlation techniques can be used on a 20 kDa protein bound to a surface.
- [63*].Chen P-H, Tseng Y-H, Mou Y, Tsai Y-L, Guo S-M, Huang S-J, Yu SSF, Chan JCC. J. Am. Chem. Soc. 2008;130:2862–2868. doi: 10.1021/ja076607y. (This paper is the first report of 2D 13C-13C correlations of a biomineralization derived peptide bound to a surface and demonstrates the potential for applying these techniques to biomineralization systems).
- [64].Tompa P. Trends Biochem. Sci. 2002;27:527–533. doi: 10.1016/s0968-0004(02)02169-2. [DOI] [PubMed] [Google Scholar]
- [65].Dickerson MB, Sandhage KH, Naik RR. Chem. Rev. 2008;108:4935–4978. doi: 10.1021/cr8002328. [DOI] [PubMed] [Google Scholar]
- [66].Amitay-Rosen T, Kababya S, Vega S. J. Phys. Chem. B. 2009;113:6267–6282. doi: 10.1021/jp810572r. [DOI] [PubMed] [Google Scholar]
- [67].Amitay-Rosen T, Vega S. Phys. Chem. Chem. Phys. 2010;12:6763–6773. doi: 10.1039/b924813g. [DOI] [PubMed] [Google Scholar]
- [68].Pizzanelli S, Kababya S, Frydman V, Landau M, Vega S. J. Phys. Chem. B. 2005;109:8029–8039. doi: 10.1021/jp044389e. [DOI] [PubMed] [Google Scholar]
- [69].Ben Shir I, Kababya S, Amitay-Rosen T, Balazs YS, Schmidt A. J. Phys. Chem. B. 2010;114:5989–5996. doi: 10.1021/jp100114v. [DOI] [PubMed] [Google Scholar]
- [70].Ben Shir I, Kababya S, Schmidt A. J. Phys. Chem. C. 2012;116:9691–9702. [Google Scholar]
- [71*].Ben Shir I, Shifi K, Schmidt A. J. Phys. Chem. C. 2014;118:7901–7909. (This paper compares the interaction of glycine with two silica surfaces, SBA-15 and MCM-41. The different interactions in these two model systems highlights the beauty and the challenge of biomineral systems, where seemingly small differences in surface functionalization and morphology significantly alter the interaction mechanism).
- [72].Zane AC, Michelet C, Roehrich AM, Emani PS, Drobny GP. Langmuir. 2014 (Ahead of Print) [Google Scholar]
- [73].Akiva-Tal A, Kababya S, Balazs YS, Glazer L, Berman A, Sagi A, Schmidt A. Proc. Natl. Acad. Sci. U.S.A. 2011;108:14763–14768. doi: 10.1073/pnas.1102608108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Jager C, Colfen H. CrystEngComm. 2007;9:1237–1244. [Google Scholar]
- [75*].Gertman R, Ben Shir I, Kababya S, Schmidt A. J. Am. Chem. Soc. 2008;130:13425–13432. doi: 10.1021/ja803985d. (This ambitious study reports on the protein and mineral of coccoliths grown up in 13C and 15N rich media, a first of its kind. Despite the challenges of studying such a complex, real system, this study is very thorough, analyzing the protein, the inorganic matrix, and ions intercalated into the matrix).
- [76].Smith J, Knagge K, Smith LJ, MacNamara E, Raftery D. J. Magn. Reson. 2002;159:111–125. [Google Scholar]
- [77].Ni Q.z., Daviso E, Can TV, Markhasin E, Jawla SK, Swager TM, Temkin RJ, Herzfeld J, Griffin RG. Acc. Chem. Res. 2013;46 doi: 10.1021/ar300348n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Yang Y, Xu Z, Cui Q, Sahai N. Biomineralization Sourcebook. 2014:265–283. [Google Scholar]
- [79].Friddle RW, Weaver ML, Wierzbicki A, Casey WH, Yoreo JJD. Proc. Natl. Acad. Sci. U.S.A. 2010;107:11–15. doi: 10.1073/pnas.0908205107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Gungormus M, Fong H, Kim IW, Evans JS, Temerler C, Sarikaya M. Biomacromolecules. 2008;9:966–973. doi: 10.1021/bm701037x. [DOI] [PubMed] [Google Scholar]
- [81].Gungormus M, Oren EE, Horst JA, Fong H, Hnilova M, Somerman MJ, Snead ML, Samudrala R, Tamerler C, Sarikaya M. Int. J. Oral Sci. 2012;4:69–77. doi: 10.1038/ijos.2012.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Segvich S, Biswas S, Becker U, Kohn DH. Cells Tissues Organs. 2008. [DOI] [PMC free article] [PubMed]
- [83].Margolis H, Beniash E. Amelogenins: Multifaceted Proteins for Dental and Bone Formation and Repair. 2010:133–142. [Google Scholar]
- [84].Dimasi E, Gower LB, editors. Biomineralization Sourcebook. 2014.
- [85].Gal A, Kahil K, Vidavsky N, Devol RT, Gilbert P, Fratzl P, Weiner S, Adadi L. Advanced Functional Materials. ASAP; 2014. [Google Scholar]
- [86*].Ben Shir I, Kababya S, Schmidt A. Isr. J. Chem. 2014;54:74–85. (This paper compares the interaction of glycine with two silica surfaces, SBA-15 and MCM-41. The different interactions in these two model systems highlights the beauty and the challenge of biomineral systems, where seemingly small differences in surface functionalization and morphology significantly alter the interaction mechanism).
- [87].Goobes R, Goobes G, Shaw WJ, Drobny GP, Campbell CT, Stayton PS. Biochemistry. 2007;46:4725–4733. doi: 10.1021/bi602345a. [DOI] [PubMed] [Google Scholar]
- [88].Ben Shir I, Kababya S, Schmidt A. J. Phys. Chem. C. 2014;118:7901–7909. [Google Scholar]